
Abstract
Background
The high density of tandem repeat sequences (satellites) in nematode genomes and the availability of genome sequences from several species in the group offer a unique opportunity to better understand the evolutionary dynamics and the functional role of these sequences. We take advantage of the previously developed SATFIND program to study the satellites in four Caenorhabditis species and investigate these questions.
Methods
The identification and comparison of satellites is carried out in three steps. First we find all the satellites present in each species with the SATFIND program. Each satellite is defined by its length, number of repeats, and repeat sequence. Only satellites with at least ten repeats are considered. In the second step we build satellite families with a newly developed alignment program. Satellite families are defined by a consensus sequence and the number of satellites in the family. Finally we compare the consensus sequence of satellite families in different species.
Results
We give a catalog of individual satellites in each species. We have also identified satellite families with a related sequence and compare them in different species. We analyze the turnover of satellites: they increased in size through duplications of fragments of 100-300 bases. It appears that in many cases they have undergone an explosive expansion. In C. elegans we have identified a subset of large satellites that have strong affinity for the centromere protein CENP-A. We have also compared our results with those obtained from other species, including one nematode and three mammals.
Conclusions
Most satellite families found in Caenorhabditis are species-specific; in particular those with long repeats. A subset of these satellites may facilitate the formation of kinetochores in mitosis. Other satellite families in C. elegans are either related to Helitron transposons or to meiotic pairing centers.
Similar content being viewed by others


Background
Satellites are tandem repeat sequences present in many eukaryotic genomes. The evolution and biological roles of satellites in different species has recently attracted much attention [1, 2]. Previously we reported that the genome of the worm Caenorhabditis elegans has a large amount of satellites, which represent about 3 % of the genome [3]. Satellites were originally identified by density gradient centrifugation [4]. Recent definitions based on genomic sequences vary across different studies [1, 2]. Here we define satellites as long tandem repeats with at least ten repeats; with each repeat having a length of 10–200 nucleotides. Satellites with fewer repeats are not included, in order to simplify the analysis and get a clear view of the major satellite families. No limit is placed on the total length of individual satellites.
The large number of satellites in nematodes offers the opportunity to study several intriguing features of satellites in more detail, such as their expansion, transposition and elimination from the genome. These questions are strongly related to other features of genome evolution, such as gene duplication and intron turnover, which are very frequent in C. elegans [5–8]. For example, multigene families are subjected to birth and death evolution, with a significant component of neutral change [9, 10].
The satellites of C. elegans show a characteristic distribution of repeat sizes, which suggests that different groups or families may play unique roles in the genome. The number and size distribution of satellites is very different from that observed in mammals, which have a much lower number of satellites with long repeats. In this paper we analyze in detail the different families of satellites present in C. elegans. We compare them with those present in other related Caenorhabditis species. We study in greater detail those satellites which have centromere-like features. As an outside group we study a distant nematode species: Meloidogyne hapla. Several features of satellites in different Meloidogyne species have been previously studied [11, 12], but no attempt has been made to analyze the different families of satellites in either Meloidogyne or in any other nematode species. We have also studied the microsatellite distribution in the Caenorhabditis species, which complement previous detailed studies on microsatellites in diverse species [13, 14]. Some microsatellites have been reported to play a role in gene expression regulation [15]; their variability and complex evolution have been reviewed by Ellegren [16].
Our results contribute to the annotation and interpretation of poorly characterized non-coding regions of the genome [17]. We discuss our results in light of the different hypotheses that have been proposed to explain the expansion and elimination of satellites.
Methods
Genome sequences
We used the genome sequences available in Wormbase [18]: versions WS201 for C. elegans and WS247 for Caenorhabditis briggsae, Caenorhabditis brenneri, Caenorhabditis remanei and M. hapla. The WS247 version of the genome of C. briggsae is of higher quality than the version we used in a previous work [3]; whereas the M. hapla version is identical. The position of satellites in C. elegans is practically the same in the WS201 version of the genome than in the previously employed [3] WS190/ce6, except in chromosome V, where displacements of up to 3 Kb may be found. The use of WS201 was determined by the fact that the CENP-A data we used were obtained from this genome version [19].
Satellite identification
Repetitive sequences were identified with the program SATFIND, which was developed in order to determine the position of long tandem repeats (satellites). SATFIND is available on-line for general use on our website [20]. The underlying algorithm is described in a previous publication [3]. Its source code has been deposited in Dryad [21]. The program determines the localization of clusters of any short sequence of a prefixed size without internal repetitions and repeated a minimum number of times in regions with a fixed size. In this paper we have used the SATFIND program to identify satellites formed by at least ten repeats of any decamer sequence in 2 Kb long regions. Once a satellite is located, the program continues its search along the genome until no further neighboring repeats are detected. In this way repeats of 10–200 nucleotides repeated at least 10 times can be positioned in the genome, with no upper limit for the number of repeats in the satellite. We have analyzed the statistical significance of satellites by computing the expected number of times a pattern of length L will appear by chance n times in a DNA sequence of length N: RE ~ N · (4-L)n. For N = 2000, L = 10 and n = 9 it turns out that RE ~ 10−45; this means that the satellites found using these parameters are far from random.
Most satellites have a regular structure, but there is a significant number which present variations in repeat length and composition. In order to eliminate the most irregular satellites, we have only accepted those which have at least 30 % of their repeats with an identical length. We have analyzed the statistical significance of this choice; given a sequence of length L that is randomly partitioned into n subsequences, the expected number of times that k subsequences will have the same length can be approximated by RE ~ (n k) · L-·(k-1). For n = 10 and L = 2000, the probability to find three sequences (30 %) with the same length is <10−3.
We have further limited our study to satellites with repeats shorter than 200 bases, since there are very few satellites with longer repeats in Caenorhabditis. Some repeat sequences were compared with the satellites embedded in the C. elegans Helitron sequences obtained from Repbase [22]. Helitrons are a special class of DNA transposons, which are associated with different satellite families in C. elegans, as we will show below.
Satellite sequence comparisons
To compare satellites we have used Malig, a progressive multiple sequence alignment algorithm, which we have developed to align satellite repeats and identify families with a related sequence. Its source code has been deposited in Dryad [23]. As a progressive method, Malig first computes the similarity score between all pairs of sequences using a dynamic programming algorithm [24]. The program considers reverse sequences as well, normalizes the alignment score to the maximum possible value and selects the cycle permutation with the highest score. Then the progressive multi-alignment is applied to the matrix of pairwise alignment scores. The process finishes when the score is smaller than a similarity threshold (input parameter) which we set to 0.6.
To calculate the statistical significance of the initial pairwise alignments, we have generated a set of random sequences with the same length distribution than satellites and equivalent GC content (30 % A and T; 20 % G and C) and performed pairwise alignments as described previously. We have used the random sequence pairwise score distribution to set up a score threshold, for which the observed probability in random alignments (p-value) is <10−4. As the p-value depends on the length of the alignment, we have used the linear regression (−0.2837’ · length of alignment + 66.0155)/100 to adjust it to different satellite lengths. We have applied this method to the detection of satellites with a common origin in different species.
Each family within a species is characterized by the abbreviated name of the species and three values, eg Cele_Fam_a_b_c. The order in the list of families is given by “a”, starting with those families with the largest number of members. The second value “b” gives the size of the repeat; “c” gives the number of members in the family. For simplicity we have omitted c throughout the text. Unique satellites appear at the end of the list, as families with a single member. We also define families as “large families” if they have ten or more members. The consensus sequence of the repeat is calculated taking into account the circularly permuted sequence of all repeats. To compare satellites from different species we have used the consensus sequence from each satellite family.
Quantification of CENP-A affinity
In order to determine the sequence affinity for CENP-A, we used the file 2446-OD00079_HCP3_N2_MXEMB_1.wig, available from modENCODE [25]. Several centromere specification arrays, obtained with different rabbit polyclonal antibodies [19] are available at Wormbase [18]; all of them give a similar distribution of CENP-A affinity. We transformed the logarithm values in the wig file to their natural number value, in order to calculate average affinity values for the regions covered by the satellites. All average affinity values in our work are given as natural numbers. We have arbitrarily divided the satellites in two equal groups, with either a high or low CENP-A affinity. We have defined the limit between the two groups by the value of the median, which is 0.922.
Results
General satellite features in C. elegans
We identified 1,779 satellites, using a threshold of a minimum of ten repeats, each with 10–200 bases (Additional file 1). Over half of the satellites are shorter than 1 Kb and only 21 % are longer than 2Kb. A list of the longest satellites is given in Table 1. Satellites may be classified as a function of the size of its repeats, (Fig. 1 and Table 2); the distribution of repeat sizes is clearly non-random. In C. elegans there is a negligible amount of satellites with repeat sizes between 50–58 and 110–160 bases. Such distribution suggests that satellites with different repeat sizes might have different origins and/or functions. The distribution of repeat sizes is similar in all chromosomes (Additional file 2: Figure S1); with the exception of the X chromosome, which has fewer satellites. The satellites do not have a uniform distribution throughout chromosomes, they are less frequent in the center (Fig. 2 and Additional file 2: Figure S2). This fact was previously reported when the whole genome sequence was published [26]. Unlike in other species, recombination in nematodes mainly occurs in the gene-poor arms of the chromosomes, which have a higher density of satellites and other types of noncoding DNA [27].

Repeat size distribution of satellites in the genomes of Caenorhabditis. The number of satellites found is represented as a function of their repeat size. For repeats longer than 60 bases the data have been merged in bins of 5 bases. The sixteen bases repeat in C. briggsae is very abundant; the total number of these satellites is indicated

Detailed distribution of satellites in chromosome I of C. elegans. The ordinate corresponds to the size of individual satellites. Larger symbols indicate longer repeats: Black triangles, 127–224 bases; dark blue squares, 56–102; red squares, 37–46: red diamonds, 32–36; blue dots, 19–31; orange squares 13–18; orange dots, 11–12. Only a few short satellites are found in the central 4 Mb of the chromosome
We observe that, as a function of repeat size, no clear pattern of distribution of satellites along the chromosomes is apparent. This is represented well in chromosome I, where repeats of different sizes may occupy any region in the chromosome (Fig. 2). Mutational dynamics of satellites may include frequent insertions and deletions. We have compared the genomic positions of the satellites in C. elegans to available copy number variation from twelve strains [28], but found no significant enrichment or depletion, as shown in Additional file 3: Table S1.
Short repeat satellite families in C. elegans
The satellites can be grouped into families, which share a related repeat sequence. In C. elegans we have detected a total of 364 families, each with a different number of members (Table 3). Most families are present in all chromosomes, whereas a few of them appear only in one or two chromosomes (Additional file 3: Table S2). In this section we describe the major 25 families of satellites with short repeats (less than 50 bases), which represent 64.2 % of all satellites.
Cele_Fam_1_35 is the largest family of satellites (213 members). It is practically absent in the X chromosome, where only one satellite of this family was found. In the autosomes it is only found in the terminal regions; it is completely absent in a central region of about 7–10 Mb. Its consensus repeat has an internal repeated motif of eight bases and two palindromic regions of thirteen bases. This satellite is only present in C. elegans, no related satellites have been detected in the other species we have studied. A combination of this repeat with the related Cele_Fam_9_43 has been previously described as MINISAT1 [22].
Cele_Fam_2_12 corresponds to telomeric sequences, which in this species forms 203 short satellites distributed throughout the genome, as previously described [26]. Interspersed telomere sequences have also been found in other species, such as some fish [29]. Chromosome X is an exception, since telomere sequences only appear at its canonical position: the ends of the chromosome.
Another large group of satellites are associated with Helitron transposons, which have been described in many species [30]. This group has 345 members distributed in six families: Cele_Fam_17_20, 3_34 and 20_37 have related sequences, whereas Cele_Fam_4_11, 5_15 and 7_40 are unrelated. Two of these families (Cele_Fam_5_15 and 3_34) had already been described in the pioneering work of Naclerio et al. [31]. Usually Helitrons only contain satellites of one or two families, but a larger number may be present. For example the Helitron Y4_CE located in chromosome I (starting at position 7868158) contains satellites of Cele_Fam_4_11, 5_15, 3_34 and 7_40. Satellites related to Cele_Fam_7_40 are also found in the other Caenorhabditis, as we will describe below.
Another two families, Cele_Fam_26_31 and Cele_Fam_13_32, are associated with terminal repeat sequences, related to the meiotic pairing centers described by various authors [32–34].
Most of the other families in this group are neither related among themselves nor with the other families. No apparent general features can be ascribed to any of them. They have between nine and forty eight members.
Analysis of the sequence of different satellite families revealed that they usually increase in size through the duplication of fragments of about 100–300 bases. We have focused on one family (Cele_Fam_24_19), to illustrate the mechanisms of expansion of satellites (Additional file 3: Tables S4 and S5) In this family all individual satellites present a mixture of repeats (ATTT)nAATTAAT, with either three or four ATTT units. This repeat sequence is completely absent in all other members of Caenorhabditis. From the distribution of point mutations and repeat sizes, it appears that the ancestral satellite was rather short and contained very few repeats. In a second step it was copied to other locations in the genome. Later the satellites increased in size through the duplication of fragments (Additional file 3: Table S5).
Centromere-like satellites
The centromeres found in most monocentric species are associated with satellites with relatively long repeats, typically around 170 base pairs [35–38]. In the case of C. elegans there are 47 satellites with a similar repeat size, in the range of 151–226 base pairs (Additional file 3: Table S3). Most satellites in this group either have unique sequences or form families with a small number of members; the largest is Cele_Fam_34_163 with seven members.
The centromere is enriched in several proteins, a key one being CENP-A. We used the recently generated mapping of CENP-A on the C. elegans genome [18] to determine the affinity of different satellite sequences for this protein. The CENP-A domains were found to be distributed evenly in regions of the genome with low affinity and a very variable size (median 10–12 Kb). About five thousand CENP-A domains were identified, as deduced from the average domain size and genome coverage. Thus the genome may be considered as a mosaic, with about five thousand regions of 10–12 Kb which have affinity for CENP-A and are separated by regions of a similar size with low affinity for CENP-A. In agreement with the results of Gassmann et al. [18], we find that satellites are randomly distributed in domains of either low or high affinity for CENP-A. However all the longest satellites (>7 Kb) with a long repeat (>40 bases) have a high affinity for CENP-A (Table 1). Additionally, over 90 % of the long satellites (>2Kb) with repeats longer than 160 bases (Additional file 3: Table S3) are in regions of high CENP-A affinity (p-value = 4.376 · 10−9, Fisher’s Exact Test).
We have also compared the position of all the satellites we have detected with the 708 positions with CENP-A affinity reported by Steiner and Henikoff [39]. We found that only two of their positions are found within a satellite. Thus there is no correlation between both results.
A unique feature of the centromere-like satellites is their rather uniform distribution throughout chromosomes (Additional file 3: Table S3), whereas most other repeated regions tend to be located at the ends of chromosomes (Additional file 2: Figure S2). We have also found that all chromosomes have satellites with high CENP-A affinity and repeat size > 150.
The satellites in Caenorhabditis
Next we compared the satellites of C. elegans with those present in other Caenorhabditis: C. briggsae (Cbrig), C. remanei (Crema) and C. brenneri (Cbren). The evolutionary relationship of these species is given in Fig. 3. The genome data presently available allow a clear view of the main features of satellites, although the genomes have not yet been fully assembled. The distribution of repeat sizes (Fig. 1) shows that all species are rich in satellites. The number of satellites identified is largest in C. briggsae and lowest in M. hapla, although satellite density is always greater than ten satellites per Mb (Table 2). Satellites with short repeats (11–20 bases) predominate; C. elegans stands out with the largest number of satellites with repeats of 30–40 bases, which are not frequent in the other species. The size distribution of the satellites in C. elegans is significantly different to the size distribution in the other species (Wilcoxon-Mann–Whitney test p-value < 10−5).

Phylogenetic relationship of the indicated species. Branch lengths were derived from RNA polymerase genes [70]
We generated satellite families in the different Caenorhabditis species. We frequently detected the presence of AAAA (or TTTT) tracts and common palindromes in the consensus sequence of many of these families. A summary of the size distribution of satellite families is given in Table 3. The main features of each species can be summarized as follows:
In C. briggsae we have localized 2,788 satellites, 2,352 of which can be grouped in 24 large families with more than 9 members each. This species stands out by having a large number of satellites (1,943) with a consensus repeat of 16 bases, most of which are octamer repeats. The two largest families have a similar consensus repeat AAWYTCAG. There are only two large families with a consensus repeat longer than 30 bases. There are 41 satellites with repeats longer than 150 bases. Surprisingly we only detected two short telomere repeats; apparently these repeats were not positioned in the available genome sequence.
The satellites of C. brenneri have a similar size distribution to those of C. briggsae (Fig. 1), although the total number of satellites is lower (1,643). This is mainly due to the absence of any large family of octamer repeats. It has 31 large families and only two of them are in the 30–160 bases range. It has 103 satellites with repeats longer than 150 bases.
Satellite families in C. remanei are of very different sizes (Fig. 1). It is the species with the largest number of satellites with long repeats, with 292 satellites with repeats longer than 150 bases.
Comparison of satellite families in Caenorhabditis
We compared the satellite families from the different Caenorhabditis species, using all against all pairwise alignments. We adjusted the score threshold to a random expectation (p-value) < 10−4. Once the homologues in different families had been identified, we built super-families, using a progressive multiple alignment approach. Only seven super-families include satellites from all four Caenorhabditis species, whereas 1,046 families are species-specific (75.3 % of the total number of families; Fig. 4 and Additional file 2: Figure S3). At the individual species level the number of species-specific families ranges from 67.6 % for C. briggsae to 78.3 % for C. elegans. Inspection of the results showed that most super-families only involved small families with a few satellites each (Additional file 3: Tables S2 and S6). We found several families with short repeats which had related families in other species, whereas all large families with repeats longer than 22 bases were species- specific, with the exception of two families in C. brenneri (10_24 and 3_28). Since there are 38 large families with repeats longer than 22 bases, 94.7 % of them are species-specific.

Venn diagram showing species-specific and shared satellite families between and among the four Caenorhabditis species studied in this paper
We were surprised that none of the Helitron families in C. elegans had any related families in the other species. We thus compared the sequence of all satellite families with the standard repeats of Helitrons and found that only Cele_Fam_7_40 had related families in the other species (Fig. 5). All of them are approximate multiples of a basic palindromic decamer sequence AGAANNTTCT.

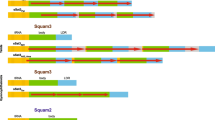
Comparison of Helitron related repeats found in different Caenorhabditis. The internally repeated palindromic sequence (AGAANNTTCT) is shown in bold. The different motifs are separated by dots
Another peculiar case is the Cele_Fam_11_14 which has an (ACTACAA)2 repeat. We detected this motif as one of the most abundant sequences in C. elegans [34], where it is mainly found in chromosomes I and II. Strikingly, the distribution of this motif is similar in chromosomes I of C. elegans and C. briggsae (Additional file 2: Figure S4). However it does not form any satellite in C. briggsae. The significance of this evolutionary conservation is not clear.
Comparison with M. hapla
We have compared the satellite families found in M. hapla with those of all Caenorhabditis (Additional files 1 and 4). We found that only 24 families of M. hapla satellites had some relation with Caenorhabditis satellite families (Additional file 3: Table S7). All the related M. hapla families had very few members; only three of them had more than three members. The longest repeat had 23 bases. Inspection of the sequences shows that most of them have long repeats of As or Ts. No clear evolutionary significance can be attributed to any of the repeats we have found. In summary these observations indicate that, as expected, the set of satellites in this species is largely unrelated to the set in Caenorhabditis.
Microsatellites in Caenorhabditis
For comparison we have also studied the microsatellite distribution in the different Caenorhabditis species (Additional file 3: Table S8). For reasons given elsewhere [14], we have chosen a minimum length of 24 bases in order to define a microsatellite. We have focused on repeats of 1–3 bases, as the number of microsatellites with longer repeats (4–6 bases) is quite small. As with satellites, the distribution of microsatellites is remarkably different in each species. For example the genomes of C. briggsae and C. brenneri contain many AG and AAG microsatellites, which are less abundant in the other two species. Thus, at the microsatellite level, these two species appear to be more closely related than any of them to C. remanei. M. hapla stands out for its low content in microsatellites. Surprisingly, in spite of its low CG content, it has very few A and AT repeats, but a large amount of random A,T sequences (W24 in Additional file 3: Table S8). A similar pattern is found when shorter microsatellites are studied [40].
Satellites in mammalian genomes
Satellites have been extensively studied in humans [41, 42]. However each study uses different parameters to define satellites. For a fair comparison with the Caenorhabditis data, we applied the same algorithm to detect satellites in three different mammalian genomes. In addition, we have not considered the centromeric alpha satellites, mostly restricted to the centromeres. It is clear that the relative number of satellites with longer repeats is significantly lower in the mammalian species, when compared with C. elegans, especially for satellites with repeats in the 100–169 size range (Table 2).
We have determined the distribution of repeat sizes in human satellites (Additional file 2: Figure S5). Only a few scattered satellites are found in the size range 100–169 bases. Satellites with repeats 49–50 and 84 bases stand out by having a comparatively higher number of members. The main 49–50 satellite has 85.7 % CG and corresponds to part of the very abundant SVA transposon. The 84 satellite corresponds to the repeated motif of 28 amino acids, typically found in Zn-finger proteins. It is found mainly in chromosome 19 (100 cases). It is also present in mouse and rat, where it had been previously described as MMSAT4 and RNSAT1, which contain two identical 84 repeats [22]. The presence of a large cluster of genes encoding Zinc-finger proteins in those species had already been described by Castresana et al. [43].
Discussion
Satellites in related species
Our results show that 75.3 % of the satellite families are species-specific. We also find that there is a significant difference when we consider the 38 satellite families which have more than nine members and their repeat size is longer than 22 bases; in this case 94 % of the satellite families are species-specific. A striking example is Cele_Fam_1_35, the largest family in C. elegans, which has no related satellites in any other species. The identification of satellites with a common origin has been performed using a rigorous statistical framework and it is improbable that we have missed related satellites.
According to the library hypothesis [44], the ancestor of Caenorhabditis should already contain most satellites, which would develop at variable degrees in different species. Although we do find satellites which are conserved across different species (Fig. 4 and Additional file 2: Figure S3), most satellite families are clearly species-specific. It appears therefore that the library hypothesis cannot be generalized to all satellites.
Our results also indicate that the precursor of Caenorhabditis probably had many satellites which were lost during evolution, since we have not detected any conserved large satellite family shared by all or some species. This lack of conservation does not allow reconstructing the phylogenetic relationships between the species, except for C. elegans, which shares fewer satellites with the other Caenorhabditis species (Additional file 2: Figure S3), in agreement with its position in the phylogenetic tree (Figure 3).
Comparison with mammalian satellites
The distribution of satellites in mammalian species is very different from that found in Caenorhabditis and nematodes in general (Table 2 and Additional file 2: Figure S4). The very low frequency of satellites with long repeats is particularly striking; on average there is only one satellite in every 90 Mb of the human genome, compared to one in every 1.15 Mb in C. elegans. Probably more satellites with long repeats will be discovered when the heterochromatic regions of acrocentric chromosomes become available. In our study we have excluded the alpha satellite, with a repeat of 171 bases. It occupies several Mb in the centromeric region of each human chromosome, but is practically absent in other chromosomal regions. We have only detected six satellites with this repeat in human genome regions outside the centromeres. It may be that scattered satellites with long repeats are eliminated, because they interfere with the centromeric role of alpha satellites. The wide distribution of satellites with long repeats in Caenorhabditis might be related to their holocentric structure, a question which we will discuss in more detail in the next section.
Centromere-like satellites
There is an ongoing debate on the location of centromeres in holocentric chromosomes. It is generally accepted that centromeres should be found in regions which contain the CENP-A histone. The results of Gassmann et al. [19] suggest that centromeres are randomly positioned on the mitotic chromosomes. Recently Steiner and Henikoff [39] have presented an alternative model. They find strong evidence for localized individual CENP-A proteins, but they find 708 preferred positions, whereas in mitosis there are only 50–100 kinetochores in each half spindle [45, 46]. As described in the results section, the sites of localization reported by the aforementioned authors [39] do not coincide with satellite positions; they may be related to the position of CENP-A in interphase. The details of CENP-A synthesis and deposition on chromatin during the cell cycle are not known in C. elegans. The complexity of these processes in other species has been recently reviewed by Catania and Allshire [47]. In particular it has been shown that CENP-A undergoes important changes during the cell cycle [48]. Its deposition on centromeric DNA is also related to transcription [49]. Furthermore, CENP-A plays a general role on chromosome condensation [50] and in double-strand break repair [51]. Thus the studies mentioned above on CENP-A localization are not conclusive, since they have been carried out in whole embryos, not in mitotic cells. Further studies should be done on isolated mitotic cells.
Our results suggest an alternative model for point centromeres: the centromere-like satellites which we have described may accumulate CENP-A and promote kinetochore formation during mitosis. They have sequence features similar to species with monocentric chromosomes [35, 38] and are also found in regions with high CENP-A affinity (Table 1 and Additional file 3: Table S3). Satellites with a similar repeat size have also been found in the other species we have studied (Fig. 1 and Table 2). In favor of the model we suggest, it should be noted that proteins similar to all those involved in mitosis in monocentric species are also found in C. elegans [52]. Furthermore holocentric plants are known to have dispersed satellites with a similar repeat size [53] which is 178 base pairs in the case of Luzula nivea [54]. Finally it is interesting to note the unique case of holocentric insects, which do not need CENP-A for mitosis [55].
Origin and expansion of satellites
The sequences of satellite repeats from different families are clearly different. The first step in the birth of a satellite should be one duplication event, so in principle any sequence may become a satellite. However, it is likely that satellite expansion is favored by local sequence features, such as the short A-tracts or palindromes [56], which are frequently found in many satellites.
The satellite expansion process may be different depending on the repeat size. Shorter repeats usually give rise to shorter satellites. They may expand by replication slippage, as it has been suggested for microsatellites [16]. In the case of the Cele_Fam_24_19 (which we have described in detail in Additional file 3: Table S5), it appears that the ancestral satellite was rather short. Later it was copied into other points in the genome and increased in size. Such increases often took place with segments of 100–300 bases in length (Fig. 6). This size corresponds to the minimal size of homology for recombination to occur, as found in several species [57, 58].

Model for the expansion of satellites. Each repeat is indicated by a rectangle. A relatively small satellite (three repeats in the figure) is transposed to another location. In the course of time it may accumulate some point mutations, indicated by the letters drawn inside the rectangles. Later the satellite increases in size. This process can be followed by the distribution of point mutations (Additional file 3: Table S5)
Satellites with long repeats should expand by different mechanisms. Repeat expansion may occur at either the mitotic or meiotic level. Errors may take place associated with either DNA unequal crossing-over or duplication, including the repair of double strand breaks [59]. Various models have been discussed by different authors [1, 43, 60]. An alternative mechanism is expansion of satellites through Okazaki fragments, which usually have an approximate size of 200 bases. This possibility has been investigated by Shah et al. [61]; an Okazaki fragment may be duplicated during DNA synthesis. Upon association with the forward strand, a bulge would be formed which could be later repaired. For this process to be effective, the individuality of the repeats should be preserved, so that satellites accurately conserve the size of their repeats. This could be due to some feature of the satellite repeats, such as a palindromic region, which would determine the exact size of the Okazaki fragments involved.
In the case of very long satellites, duplication of a long stretch containing many repeats may take place in a way similar to gene duplication, which is very common in C. elegans [8]. Several processes such as unequal crossing-over, DNA transposition and retro-transposition may be involved [62].
Transposition and turnover
It is clear that some satellites in C. elegans have been propagated throughout the genome by Helitron transposons, although other transposition mechanisms may also be active. For example, in the case of Cele_Fam_24_19, some satellites are found associated with Helitron transposons (Additional file 3: Table S4). Other mechanisms of transposition in C. elegans and other species have been reviewed by Huang et al. [63]. The observation that in the silkworm (a holocentric insect) most Helitron families experienced a single burst of expansion in the past two million years [64] is of particular interest; if transposons in Caenorhabditis have a similar life span, bursts of transposon-associated satellite expansions may explain the large differences in the satellite families present in each Caenorhabditis species. Individual families may have appeared on different occasions during the evolution of Caenorhabditis.
It would be of great interest to determine the extent to which the satellites vary among different C. elegans strains. In this sense full genome sequencing and assembly of different wild-type C. elegans strains [65] will be very helpful, in particular since a whole genome sequence of the Hawaiian strain has been recently released [66]. The results of Maydan et al. [28], which we have analyzed (Additional file 3: Table S1), provide a first step in this direction.
Satellite elimination
In all Caenorhabditis there are many widespread families of satellites. In the case of C. elegans there are twelve families with more than thirty members distributed over all chromosomes (Additional file 3: Table S2). Individual satellites may be either eliminated by excision or may accumulate mutations and lose its repetitive nature. The repair of double strand breaks [59] may also contribute to either partial or total elimination of satellites. As an example we present some sequences which may correspond to degraded Cele_Fam_24_19 satellites (shown at the end of Additional file 3: Table S5). An intriguing case is the short motif ACTACAA, found in Cele_Fam_11_14 and as clusters of isolated motifs in other species, as in C. briggsae (Additional file 2: Figure S3). It is not clear if this motif either corresponds to degraded ancestral satellites or if it has some unknown function.
Finally we should mention that the elimination of satellites by random drift may not be sufficiently effective to result in the complete disappearance of large satellite families, which may have hundreds of members: an additional mechanism might be required. It is possible that satellite elimination by excision may occur in a concerted manner, similar to the programmed DNA elimination process described in some nematodes and in many other organisms [67]. Unfortunately the molecular mechanisms involved in DNA elimination are not known. It is possible that once the genetic load of some satellites increases, a process of specific DNA elimination is triggered during meiosis. Our data suggest that many ancestral satellites disappeared, while every species developed new families, in a seemingly random fashion. Note that this mechanism differs from the DNA elimination processes previously described, in which there is a selective loss of genomic regions. Elimination of specific satellites may be triggered by particular chromatin structures, resulting from repetitive sequences. A possible mechanism could involve extrachromosomal DNA circles, which have been found to originate from satellite regions of plants [68] and yeast [69]. The presence of palindromic regions would facilitate their formation.
Conclusions
-
1.
Each Caenorhabditis species studied contains a large number of satellites. About 90 % of the satellites in each species can be grouped into families of related satellites with similar repeats. A few of these families contain several hundred related satellites.
-
2.
In C. elegans we have defined a subset of large satellites that have strong affinity for CENP-A. These satellites may facilitate the formation of kinetochores in mitosis.
-
3.
Other satellite families in C. elegans are either related to Helitron transposons or to meiotic pairing centers.
-
4.
The main satellite families found in different Caenorhabditis species appear to be unrelated. In particular there are some extremely large families of satellites which are species-specific. They have undergone an explosive expansion, perhaps because they have acquired a yet unknown function.
-
5.
Our analysis of the internal structure of satellites demonstrates that they expand by the duplication of segments of about 100–300 bases.
Availability of supporting data
The data sets supporting the results of this article are available in its additional files. The source code for the programs used has been deposited in the Dryad Data Repository (http://datadryad.org).
References
Richard GF, Kerrest A, Dujon B. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol Mol Biol Rev. 2008;72:686–727.
Garrido-Ramos MA. Repetitive DNA: Karger Medical and Scientific Publishers; 2012.
Subirana JA, Messeguer X. A satellite explosion in the genome of holocentric nematodes. PLoS One. 2013;8, e62221.
Sulston JE, Brenner S. The DNA of Caenorhabditis elegans. Genetics. 1974;77:95–104.
Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, et al. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 2003;1, E45.
Katju V, Lynch M. On the formation of novel genes by duplication in the Caenorhabditis elegans genome. Mol Biol Evol. 2006;23:1056–67.
Cutter AD, Dey A, Murray RL. Evolution of the Caenorhabditis elegans genome. Mol Biol Evol. 2009;26:1199–234.
Lipinski KJ, Farslow JC, Fitzpatrick KA, Lynch M, Katju V, Bergthorsson U. High spontaneous rate of gene duplication in Caenorhabditis elegans. Curr Biol. 2011;21:306–10.
Nei M. The new mutation theory of phenotypic evolution. Proc Natl Acad Sci U S A. 2007;104:12235–42.
Katju V, Bergthorsson U. Copy-number changes in evolution: rates, fitness effects and adaptive significance. Frontiers in genetics. 2013;4:273.
Mestrovic N, Castagnone-Sereno P, Plohl M. Interplay of selective pressure and stochastic events directs evolution of the MEL172 satellite DNA library in root-knot nematodes. Mol Biol Evol. 2006;23:2316–25.
Mestrovic N, Pavlek M, Car A, Castagnone-Sereno P, Abad P, Plohl M. Conserved DNA Motifs, Including the CENP-B Box-like, Are Possible Promoters of Satellite DNA Array Rearrangements in Nematodes. PLoS One. 2013;8, e67328.
Toth G, Gaspari Z, Jurka J. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 2000;10:967–81.
Subirana JA, Messeguer X. Structural families of genomic microsatellites. Gene. 2008;408:124–32.
Gemayel R, Vinces MD, Legendre M, Verstrepen KJ. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet. 2010;44:445–77.
Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435–45.
Alexander RP, Fang G, Rozowsky J, Snyder M, Gerstein MB. Annotating non-coding regions of the genome. Nat Rev Genet. 2010;11:559–71.
Wormbase. [http://www.wormbase.org].
Gassmann R, Rechtsteiner A, Yuen KW, Muroyama A, Egelhofer T, Gaydos L, et al. An inverse relationship to germline transcription defines centromeric chromatin in C. elegans. Nature. 2012;484:534–7.
Algorithmics and Genetics Group website [http://alggen.lsi.upc.edu]
SATFIND. [http://dx.doi.org/10.5061/dryad.h5s2q].
Genetic Information Research Institute. [http://www.girinst.org].
MALIG. [http://dx.doi.org/10.5061/dryad.h5s2q].
Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–53.
modENCODE. [https://github.com/modENCODEDCC/tools/blob/master/liftover_everything_scripts/07_liftover.sh].
Consortium CS. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282:2012–8.
Barnes TM, Kohara Y, Coulson A, Hekimi S. Meiotic recombination, noncoding DNA and genomic organization in Caenorhabditis elegans. Genetics. 1995;141:159–79.
Maydan JS, Lorch A, Edgley ML, Flibotte S, Moerman DG. Copy number variation in the genomes of twelve natural isolates of Caenorhabditis elegans. BMC Genomics. 2010;11:62.
Cioffi MB, Bertollo LA. Chromosomal distribution and evolution of repetitive DNAs in fish. Genome Dyn. 2012;7:197–221.
Kapitonov VV, Jurka J. Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci U S A. 2001;98:8714–9.
Naclerio G, Cangiano G, Coulson A, Levitt A, Ruvolo V, La Volpe A. Molecular and genomic organization of clusters of repetitive DNA sequences in Caenorhabditis elegans. J Mol Biol. 1992;226:159–68.
Sanford C, Perry MD. Asymmetrically distributed oligonucleotide repeats in the Caenorhabditis elegans genome sequence that map to regions important for meiotic chromosome segregation. Nucleic Acids Res. 2001;29:2920–6.
Phillips CM, Meng X, Zhang L, Chretien JH, Urnov FD, Dernburg AF. Identification of chromosome sequence motifs that mediate meiotic pairing and synapsis in C. elegans. Nat Cell Biol. 2009;11:934–42.
Subirana JA, Messeguer X. The most frequent short sequences in non-coding DNA. Nucleic Acids Res. 2010;38:1172–81.
Wang G, Zhang X, Jin W. An overview of plant centromeres. J Genet Genomics. 2009;36:529–37.
Melters DP, Bradnam KR, Young HA, Telis N, May MR, Ruby JG, et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013;14:R10.
Plohl M, Mestrovic N, Mravinac B. Centromere identity from the DNA point of view. Chromosoma. 2014;123:313–25.
Lermontova I, Sandmann M, Demidov D. Centromeres and kinetochores of Brassicaceae. Chromosome Res. 2014;22:135–52.
Steiner FA, Henikoff S. Holocentromeres are dispersed point centromeres localized at transcription factor hotspots. eLife. 2014;3, e02025.
Castagnone-Sereno P, Danchin EG, Deleury E, Guillemaud T, Malausa T, Abad P. Genome-wide survey and analysis of microsatellites in nematodes, with a focus on the plant-parasitic species Meloidogyne incognita. BMC Genomics. 2010;11:598.
Warburton PE, Hasson D, Guillem F, Lescale C, Jin X, Abrusan G. Analysis of the largest tandemly repeated DNA families in the human genome. BMC Genomics. 2008;9:533.
Hayden KE, Strome ED, Merrett SL, Lee HR, Rudd MK, Willard HF. Sequences associated with centromere competency in the human genome. Mol Cell Biol. 2013;33:763–72.
Castresana J, Guigó R, Albà MM. Clustering of genes coding for DNA binding proteins in a region of atypical evolution of the human genome. J Mol Evol. 2004;59:72–9.
Plohl M, Mestrovic N, Mravinac B. Satellite DNA evolution. Genome Dyn. 2012;7:126–52.
Albertson DG, Thomson JN. The kinetochores of Caenorhabditis elegans. Chromosoma. 1982;86:409–28.
O'Toole ET, McDonald KL, Mantler J, McIntosh JR, Hyman AA, Muller-Reichert T. Morphologically distinct microtubule ends in the mitotic centrosome of Caenorhabditis elegans. J Cell Biol. 2003;163:451–6.
Catania S, Allshire RC. Anarchic centromeres: deciphering order from apparent chaos. Curr Opin Cell Biol. 2014;26:41–50.
Yu Z, Zhou X, Wang W, Deng W, Fang J, Hu H, et al. Dynamic phosphorylation of CENP-A at Ser68 orchestrates its cell-cycle-dependent deposition at centromeres. Dev Cell. 2015;32:68–81.
Catania S, Pidoux AL, Allshire RC. Sequence features and transcriptional stalling within centromere DNA promote establishment of CENP-A chromatin. PLoS Genet. 2015;11, e1004986.
Maddox PS, Portier N, Desai A, Oegema K. Molecular analysis of mitotic chromosome condensation using a quantitative time-resolved fluorescence microscopy assay. Proc Natl Acad Sci U S A. 2006;103:15097–102.
Zeitlin SG, Baker NM, Chapados BR, Soutoglou E, Wang JY, Berns MW, et al. Double-strand DNA breaks recruit the centromeric histone CENP-A. Proc Natl Acad Sci U S A. 2009;106:15762–7.
Kitagawa R. Key players in chromosome segregation in Caenorhabditis elegans. Front Biosci (Landmark Ed). 2009;14:1529–57.
Collet C, Westerman M. Interspecies comparison of the highly-repeated DNA of Australasian Luzula (Juncaceae). Genetica. 1987;74:95–103.
Haizel T, Lim YK, Leitch AR, Moore G. Molecular analysis of holocentric centromeres of Luzula species. Cytogenet Genome Res. 2005;109:134–43.
Drinnenberg IA, deYoung D, Henikoff S, Malik HS. Recurrent loss of CenH3 is associated with independent transitions to holocentricity in insects. eLife. 2014;3. doi: 10.7554/eLife.03676.
Northam MR, Moore EA, Mertz TM, Binz SK, Stith CM, Stepchenkova EI, et al. DNA polymerases zeta and Rev1 mediate error-prone bypass of non-B DNA structures. Nucleic Acids Res. 2014;42:290–306.
Rubnitz J, Subramani S. The minimum amount of homology required for homologous recombination in mammalian cells. Mol Cell Biol. 1984;4:2253–8.
Opperman R, Emmanuel E, Levy AA. The effect of sequence divergence on recombination between direct repeats in Arabidopsis. Genetics. 2004;168:2207–15.
Onozawa M, Zhang Z, Kim YJ, Goldberg L, Varga T, Bergsagel PL, et al. Repair of DNA double-strand breaks by templated nucleotide sequence insertions derived from distant regions of the genome. Proc Natl Acad Sci U S A. 2014;111:7729–34.
Kim JC, Mirkin SM. The balancing act of DNA repeat expansions. Curr Opin Genet Dev. 2013;23:280–8.
Shah KA, Shishkin AA, Voineagu I, Pavlov YI, Shcherbakova PV, Mirkin SM. Role of DNA polymerases in repeat-mediated genome instability. Cell Rep. 2012;2:1088–95.
Hahn MW. Distinguishing among evolutionary models for the maintenance of gene duplicates. J Hered. 2009;100:605–17.
Huang CR, Burns KH, Boeke JD. Active transposition in genomes. Annu Rev Genet. 2012;46:651–75.
Han MJ, Shen YH, Xu MS, Liang HY, Zhang HH, Zhang Z. Identification and evolution of the silkworm helitrons and their contribution to transcripts. DNA Res. 2013;20:471–84.
Thompson O, Edgley M, Strasbourger P, Flibotte S, Ewing B, Adair R, et al. The million mutation project: a new approach to genetics in Caenorhabditis elegans. Genome Res. 2013;23:1749–62.
Thompson OA, Snoek LB, Nijveen H, Sterken MG, Volkers RJM, Branchley R, et al. Remarkably Different Regions Punctuate the Genome Assembly of the Caenorhabditis elegans Hawaiian Strain CB4856. Genetics. 2015;200:975–89.
Wang J, Davis RE. Programmed DNA elimination in multicellular organisms. Curr Opin Genet Dev. 2014;27:26–34.
Navrátilová A, Koblizková A, Macas J. Survey of extrachromosomal circular DNA derived from plant satellite repeats. BMC Plant Biol. 2008;8:90.
Møller HD, Parsons L, Jørgensen TS, Botstein D, Regenberg B. Extrachromosomal circular DNA is common in yeast. Proc Natl Acad Sci U S A. 2015;112:E3114–22.
Kiontke KC, Felix MA, Ailion M, Rockman MV, Braendle C, et al. A phylogeny and molecular barcodes for Caenorhabditis, with numerous new species from rotting fruits. BMC Evol Biol. 2011;11:339.
Acknowledgments
We are thankful to Dr. Lourdes Campos for valuable suggestions. Also to Messrs. W. Blevins and F. Acosta-Reyes for help in the preparation of the manuscript and figures. This work was funded by Ministerio de Economía y Competitividad (TIN2010-21062-C02-01, TIN2013-45732-C4-3-P, BFU2012-36820 and 2009–10380) with additional Feder funds.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
JAS conceived the study, carried out calculations, analyzed the data and drafted the paper; MMA carried out calculations and analyzed data; XM developed new software and supervised the calculations. All authors read, corrected and approved the final manuscript.
Authors’ information
Not applicable.
Additional files
Additional file 1:
Files containing the description of all satellites and their families in several formats (31 files). (ZIP 1068 kb)
Additional file 2:
Supplementary Figures. (PDF 568 kb)
Additional file 3:
Supplementary Tables. (PDF 802 kb)
Additional file 4:
Satellite families in Caenorhabditis and in M. hapla . (PDF 268 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Subirana, J.A., Albà, M.M. & Messeguer, X. High evolutionary turnover of satellite families in Caenorhabditis . BMC Evol Biol 15, 218 (2015). https://doi.org/10.1186/s12862-015-0495-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-015-0495-x