
Abstract
Background
Bacteria of the order Rickettsiales (Alphaproteobacteria) are obligate intracellular parasites that infect species from virtually every major eukaryotic lineage. Several rickettsial genera harbor species that are significant emerging and re-emerging pathogens of humans. As species of Rickettsiales are associated with an extremely diverse host range, a better understanding of the historical associations between these bacteria and their hosts will provide important information on their evolutionary trajectories and, particularly, their potential emergence as pathogens.
Results
Nine species of Rickettsiales (two in the genus Rickettsia, three in the genus Anaplasma, and four in the genus Ehrlichia) were identified in two species of hard ticks (Dermacentor nuttalli and Hyalomma asiaticum) from two geographic regions in Xinjiang through genetic analyses of 16S rRNA, gltA, and groEL gene sequences. Notably, two lineages of Ehrlichia and one lineage of Anaplasma were distinct from any known Rickettsiales, suggesting the presence of potentially novel species in ticks in Xinjiang. Our phylogenetic analyses revealed some topological differences between the phylogenies of the bacteria and their vectors, which led us to marginally reject a model of exclusive bacteria-vector co-divergence.
Conclusions
Ticks are an important natural reservoir of many diverse species of Rickettsiales. In this work, we identified a single tick species that harbors multiple species of Rickettsiales, and uncovered extensive genetic diversity of these bacteria in two tick species from Xinjiang. Both bacteria-vector co-divergence and cross-species transmission appear to have played important roles in Rickettsiales evolution.
Similar content being viewed by others


Background
Bacteria of the order Rickettsiales are obligate intracellular parasites of eukaryotes. While some symbionts are known (for example, many Wolbachia species), most described species of Rickettsiales are best known as human pathogens that cause several diseases, including rickettsioses, anaplasmosis, and ehrichiosis [1]. Historically, rickettsial agents have been important causes of human morbidity and mortality, including R. prowazekii that caused several million deaths in the USSR [2], and it is estimated that Orientia tsutsugamushi is currently responsible for approximately one million cases of scrub typhus per year [2],[3]. The discovery of new pathogenic species or their associated diseases has attracted attention to the Rickettsiales as pathogens [4]-[8]. As their arthropod vectors often live at high densities and in close proximity to domestic animals and humans, Rickettsiales will continue to pose a risk for transmission to humans. Hence, the identification and characterization of novel Rickettsiales is of importance for both animal and human health.
The number of novel Rickettsiales associated with protists, arthropods, and mammals has increased rapidly through the application of molecular detection and phylogenetics [4]-[6],[9],[10]. Remarkably, analysis of the Trichoplax adhaerens genome also reveals novel species in the order Rickettsiales (for example, [11]). At present, this order contains three established families (Rickettsiaceae, Anaplasmataceae, and Holosporaceae) and one proposed family (Candidatus Midichloriaceae) [8],[11]-[14]. Additionally, some unclassified species warrant further attention to determine their phylogenetic and systematic positions [8],[11]. The intra- and inter-species genetic diversity and evolutionary relationships within genera of Rickettsiales bacteria have been characterized using 16S rRNA gene (rrs) sequences, especially in the case of those bacteria causing animal and human disease [5],[6],[9],[14],[15]. However, relatively little is known about their potential for cross-species transmission and emergence.
Compared with other zoonotic or vector-borne bacteria, Rickettsiales are associated with a more extremely diverse host range, including protists, hydra, annelids, arthropods, vertebrates, and even plants [5],[8],[15],[16]. While some Rickettsiales are specific to particular vectors and hosts [16],[17], others experience host-switching or regularly cycle between different hosts, typically a mammal (e.g. rodents, cattle and humans) and a blood-feeding arthropod (e.g. fleas, mites and ticks) [5],[16],[17]. However, the evolutionary associations between Rickettsiales and their hosts are not well understood [6],[16],[18],[19]. In particular, it is unclear whether Rickettsiales most often evolve by long-term bacteria-host co-divergence or cross-species transmission [20],[21]. As most emerging infectious diseases in humans are caused by spillover from animal hosts or vectors, a better understanding of the evolutionary relationships among Rickettsiales bacteria could provide important information on the likelihood of their emergence as agents of disease.
Xinjiang (one of five autonomous regions of China) is located in the northwestern part of China, and borders Russia, Mongolia, Kazakhstan, Kyrgyzstan, Tajikistan, Afghanistan, Pakistan and India (Additional file 1: Figure S1) and is one of the nation’s major grazing areas. Several important tick-borne diseases are endemic in Xinjiang [22]. The main aim of this study was to explore the diversity of Rickettsiales in Xinjiang, China, where their presence has only previously been shown by serological data [23],[24]. Accordingly, we screened ticks and identified bacteria by sequencing and analyzing three genes; rrs, citrate synthase (gltA), and heat shock protein (groEL). With these data in hand we explored key aspects of Rickettsiales biodiversity and evolution.
Results
Collection of ticks and detection of Rickettsiales bacterial DNA
In the spring of 2011, a total of 2062 adult ticks were collected from domestic animals (sheep and cattle) and grasslands in the border areas of the Bole and Tacheng regions of Xinjiang Uygur Autonomous Region, China (Additional file 1: Figure S1). The numbers, species, and geographic distributions of the adult ticks collected are shown in Table 1. After morphological examination and sequence analysis of mitochondrial 18S and 12S rDNA sequences as described previously [25], only Dermacentor nuttalli and Hyalomma asiaticum were found in Xinjiang.
A total of 388 tick pools (1862 ticks) were investigated in this study, 314 of which were from Bole and 74 from Tacheng. PCR was performed to detect Rickettsiales DNA based on rrs. PCR products of the expected size were amplified from 50 tick pools from Bole and 37 from Tacheng. Genetic analyses of these sequences indicated that all products belonged to Rickettisales (see below).
Genetic analysis of bacterial DNA sequences
The rrs, gltA, and groEL gene sequences amplified from the Rickettsiales DNA-positive tick-pool samples were sequenced (sequences are described in detail in Additional file 2: Table S1). Genetic analyses indicated that all sequences recovered from ticks from Xinjiang shared strong similarities with those from species of Anaplasma, Ehrlichia, and Rickettsia (with percentages greater than 97%, 97.9% and 98.8%, respectively, in the rrs gene), and hence within the standard reference values used for assignments to these genera (i.e. above 96%, 97.6% and 97.2% with the genus Anaplasma, Ehrlichia, and Rickettsia SFG group, respectively) [26]. Hence, these bacterial groups circulate in Dermacentor and Hyalomma ticks in Xinjiang (Additional file 2: Table S1). A PCR based on individual tick samples confirmed the likelihood that in each case the three genes of each identified species from tick pool are from a single bacterial species.
Phylogenetic relationships between newly identified and known Rickettsiales
To determine the phylogenetic relationships among the Rickettsiales bacteria identified here and those described previously, we estimated phylogenetic trees based on the rrs, gltA, and groEL genes using ML and Bayesian methods, all of which produced similar topologies. In agreement with previous studies [8],[11], all Rickettsiales bacteria including those identified in this study were classified into four well-supported monophyletic groups in the rrs trees (Figure 1), corresponding to the families Holosporaceae, Rickettsiaceae, Candidatus Midichloriaceae, and Anaplasmataceae. The family Rickettsiaceae comprises two genera – Orientia and Rickettsia – while the family Anaplasmataceae contains the genera Neorickettsia, Wolbachia, Ehrlichia, Anaplasma, and Candidatus genus Neoehrlichia. As the gltA gene of O. tsutsugamushi strains is a pseudogene, the ML and Bayesian trees based on gltA gene sequences did not include O. tsutsugamushi, although this did not change the topological positions of the other taxa.

Phylogenetic trees based on partial Rickettsiales rrs sequences using Bayesian (MrBayes and ML (PhyML methods. Numbers at each branch indicate posterior probabilities for the Bayesian (left and bootstrap values for the ML (right trees. The ML tree is shown here.
Within the genus Rickettsia, the bacteria (R. raoultii BL029-1, R. raoultii BL029-2, R. raoultii TC249-10, and R. raoultii TC250-11) identified in ticks from the Bole and Tacheng regions were closely related to the species R. raoultii carried by Dermacentor spp. and R. pumilio ticks [27] in the rrs tree (Figure 1, Additional file 3: Figure S2a), while the sequences (R. slovaca TC250-17) recovered from ticks from the Tacheng region had a closer evolutionary relationship with R. slovaca isolated from Dermacentor spp. [28]. Hence, at least two species of Rickettsia circulate in ticks from the Bole and Tacheng regions of Xinjiang. Similar clustering patterns were observed in the trees inferred from groEL and gltA sequences (Figures 2, 3, and Additional file 3: Figure S2bc).

Phylogenetic trees based on the parital coding region of citrate synthase gene ( gltA of order Rickettsiales bacteria using the Bayesian and ML methods. The figure description follows that in Figure 1.

Phylogenetic trees based on the parital coding region of heat shock protein gene ( groEL of order Rickettsiales bacteria using the Bayesian and ML methods. The figure description follows that in Figure 1.
Within the genus Ehrlichia, the sequences (Ehrlichia sp. TC251-2, Ehrlichia sp. TC249-2, and Ehrlichia sp. TC248-16) recovered from D. nuttalli ticks from Tacheng clustered in the rrs tree with E. ewingii carried by A. americanum and D. variabilis ticks [29],[30] (Figure 1, Additional file 3: Figure S2d). These sequences also clustered together in both gltA and groEL trees (Figures 2, 3, and Additional file 3: Figure S2ef), but were distinct from E. ewingii, suggesting that they represent a potential new species of Ehrlichia in ticks from Tacheng region. The bacterial sequences (Ehrlichia sp. BL157-9, Ehrlichia sp. BL157-4, and Ehrlichia sp. BL157-6) identified in H. asiaticum ticks from Bole clustered in rrs trees together with the Ehrlichia. sp. ERm58 sequences identified in Rhipicephalus muhsamae ticks [31] and Ehrlichia. sp. Fujian identified in R. microplus ticks from China [32]. However, the evolutionary relationships of these five bacterial sequences were not well resolved in the rrs trees. Interestingly, they shared a relatively close evolutionary relationship with Ehrlichia. sp. ERm58 in the gltA tree, but were a distinct lineage in the groEL tree, suggesting the possible presence of a new variant of Ehrlichia. sp. ERm58 in ticks from Bole. The bacterial sequences (Ehrlichia sp. BL116-7 and Ehrlichia sp. BL116-8) recovered from H. asiaticum ticks from Bole formed a distinct lineage in the rrs tree, and showed a relatively close relationship with E. canis primarily transmitted by R. sanguineus and D. variabilis[33],[34]. They also formed a distinct lineage in both gltA and groEL trees, possibly indicative of a new species.
Within the genus Anaplasma, the bacterial sequences (A. ovis TC249-5, A. ovis TC248-1, and A. ovis TC251-9) recovered from D. nuttalli ticks from Tacheng were closely related to A. ovis bacteria in Dermacentor spp. and Rhipicephalus spp. [9] in the rrs, gltA, and groEL trees (Figures 1, 2, 3, and Additional file 3: S2ghi). In the rrs and groEL trees the bacterial sequences (Anaplasma sp. BL126-13 and Anaplasma sp. TC250-2) identified in H. asiaticum ticks from Bole and D. nuttalli ticks from Tacheng clustered together and showed a close relationship with the species A. bovis, which is predominantly vectored by Amblyomma spp., Rhipicephalus spp., and Hyalomma spp. [9]. As A. bovis gltA sequences are not available, the sequence (Anaplasma sp. BL126-13) formed a distinct lineage. Remarkably, the sequences (Anaplasma sp.BL102-7, Anaplasma sp.BL099-6, and Anaplasma sp.BL11) recovered from H. asiaticum ticks from Bole were divergent from any known Anaplasma bacteria (percentage similarity > 1.6% for rrs, > 43.6% for gltA, and > 23.2% for groEL). They formed a distinct lineage in all three phylogenetic trees, suggesting the presence of a new Anaplasma species in ticks. Finally, it was notable that different clustering patterns of Anaplasma bacteria were observed in the trees estimated using the groEL and gltA gene sequences. Additional work is needed to determine whether these differences are due to recombination.
Evolutionary association between Rickettsiales bacteria and their vectors
In agreement with the recent studies [8],[11], almost all known species of the family Holosporaceae, which are the most divergent group in the order, are associated with protists (Figure 4 and Additional file 4: Figure S3), except one found in prairie dog flea [35]. The most divergent species within the family Rickettsiaceae are also predominantly associated with protists (Diophrys appendiculata, Haplosporidium sp etc.), and occupy the most divergent position in the phylogeny of vectors. Several exceptions include the uncharacterized species detected from Hydra oligactis and the leech Torix tagoi. All other Rickettsia or Rickettsia-like species were found in arthropods, with the majority found in ticks and a few in insects. The unclassified species, potentially a new family, are associated with protists as well as Hydra vulgaris[36]. Like the family Rickettsiaceae, bacteria from the family Candidatus Midichloriaceae are associated with a wide range of hosts, from protists to a variety of animals, including ticks [11],[14]. Although the bacteria of the family Anaplasmataceae are not found in protists, the earliest appearance is of bacteria in the genus Neorickettsia found within Trematoda or aquatic insects [37]. All known bacteria from the genera Anaplasma and Ehrlichia are found within ticks.

Molecular clock (BEAST phylogeny of the order Rickettsiales based on parital 16S rRNA gene sequences. The inferred aquatic/terrestrial traits were mapped onto the phylogeny with supporting values for each trait indicated on internal nodes.
The evolutionary association between these species and their corresponding vectors was further evaluated with a co-phylogeny analysis. For the tick-only data sets, the null hypotheses of no co-divergence could not be rejected, although only marginally so (P > 0.084, Additional file 5: Table S2). Analyses based on overall data sets yielded a similar conclusion, with a P value that was closer to 0.05 (P = 0.064).
Inferred ancestral habitat for the Rickettsiales
Interestingly, our phylogenetic analysis of possible ancestral character states on these data gave support to an aquatic origin for the families Rickettsiaceae, Anaplasmataceae, Holosporaceae and Candidatus Midichloriaceae with strong support values of 1.00, 0.80, 1.00 and 1.00, respectively. In addition, this analysis suggested that there were at least five independent adaptations to terrestrial animals: (1) within the family Holosporaceae, (2) within the genus Rickettsia, (3) within the currently known species of Orientia circulating in mites, (4) within the ancestral lineage that diverged into the genera Wolbachia, Anaplasma, and Ehrlichia, and (5) within the Candidatus Midichloriaceae, which is associated with a wide range of hosts.
Discussion
Serological studies provided the earliest evidence for the presence of the bacteria of the genera Anaplasma, Ehrlichia, and Rickettsia in ticks from Xinjiang area of China [23],[24]. Since this initial work, only a small number of molecular epidemiological studies have been performed, mostly on Rickettsiales and limited to partial sequences of a single gene [38]. By sequencing and analyzing the bacterial sequences of complete length rrs, gltA, and groEL genes, we identified at least nine species of bacteria belonging to the Rickettsia, Anaplasma and Ehrlichia genera of Rickettsiales, indicating extensive genetic diversity of Rickettsiales in the two primary species of ticks in Xinjiang. Given that at least 39 species of ticks are present in Xinjiang [39], it is likely that additional tick-associated Rickettsiales circulating in this region will be discovered in the future.
Human cases of infection by Rickettsiales, leading to lymphadenopathy caused by a spotted fever group (SFG) Rickettsia, were documented in the 1980s in the Bole region of Xinjiang [40]. Serological analyses of the strains isolated from patients and ticks suggested that R. sibirica might be the etiological agent [41],[42]. In this study, phylogenetic analyses of bacterial sequences recovered from ticks indicated the presence of R. raoultii in Bole region and R. slovaca in Tacheng, species which were previously found in other regions of Xinjiang [43]. As R. slovaca is known to be associated with lymphadenopathy and R. raoultii with similar disease [27],[44], our results suggest there is potential risk to humans by species of Rickettsiales detected in Xinjiang, which clearly warrants additional investigation.
Currently, the genus Ehrlichia contains five species and more than five unclassified genetic variants [9]. To date, only one study reports the presence of antibodies against E. chaffeensis in the ruminants from Xinjiang [23]. In this study, at least four species of Ehrlichia were discovered circulating in ticks in Xinjiang. Although the rrs tree could not provide resolution between newly discovered bacteria and previously characterized Ehrlichia species (Figure 1), the genetic separation is more obvious in the gltA and groEL genes, where the Ehrlichia sequences were clearly divided into four lineages. Remarkably, the sequences (Ehrlichia sp. BL116-7 and Ehrlichia sp. BL116-8) recovered from ticks from Bole were quite distinct from any known Ehrlichia spp. Thus, our data suggest that there are novel clades of Ehrlichia in Xinjiang ticks.
The genus Anaplasma includes six species [9]. Through analysis of a short fragment of rrs, A. phagocytophilum in H. asiaticum and sheep were recently found in other parts of Xinjiang [24],[39]. In this study, the bacteria (Anaplasma sp.TC249-5, Anaplasma sp.TC248-1, and Anaplasma sp.TC251-9) detected in ticks from Tacheng were closely related to A. ovis carried by Dermacentor spp. and Rhipicephalus spp. ticks in the rrs, gltA, and groEL trees, with 99.8%, 99.2%, and 99.7% nucleotide similarity, respectively, thereby indicating the presence of A. ovis in ticks from Tacheng region. As for Anaplasma sp. BL126-13 and Anaplasma sp. TC250-2 recovered in ticks from Bole and Tacheng, respectively, a closer relationship with A. bovis (98.9% and 86.1%) was observed in both rrs and groEL trees, suggesting that A. bovis circulates in ticks in Bole and Tacheng regions. Finally, the bacterial sequences (Anaplasma sp.BL102-7, Anaplasma sp.BL099-6, and Anaplasma sp.BL099-11) recovered from ticks form a lineage distinct from any known Anaplasma, suggesting a novel species circulating in ticks in this region.
It is important to note that bacterial endosymbionts are known to be abundant in tick species, although many are considered to be harmless to humans [45]. Further research is needed to confirm whether the sequences detected in this research are indeed from novel Rickettsiales species, and whether these species are endosymbiotic or potentially pathogenic.
An interesting observation of this study is that the phylogenetic analysis of this sample of sequences suggests that Rickettsiales may have originated in aquatic environments, with five adaptive shifts from an aquatic to terrestrial habitat. It had previously been suggested that the common ancestor of Rickettsiales was free-living, and that the transition to an intracellular lifestyle occurred 525-775 million years ago [6]. Interestingly, the genome of R. bellii includes many genes that are characteristic of amoebal symbionts [46], and it was suggested that the ancestors of Rickettsia could have used amoebae (or related protozoa) as hosts, from which further adaptation to terrestrial organisms, including ticks, occurred. For bacteria of the family Candidatus Midichloriaceae, aquatic/environmental protists likely have served as evolutionary reservoirs, from which one or more lineages evolved with the capacity to infect metazoans [11],[14]. In sum, all these data support the notion that aquatic/environmental protists played an important role in the evolution of the Rickettsiales [8],[11],[14],[16].
Our analyses also revealed that, although there is clearly congruence between the bacteria and vector/host trees, a model of exclusive bacteria-vector co-divergence can be rejected, albeit marginally. Hence, the biodiversity of the Rickettsiales must also reflect, at least in part, the occurrence of cross-species transmission. Host associations encompass free-living extracellular, facultative intracellular, and obligate intracellular (endosymbiotic) species, with the latter often exhibiting reductive genome evolution [21]. All of these lifestyles are accompanied by infecting new host species. It is possible that co-divergence occurred in the early stage of Rickettsiales evolution. This is apparent in the phylogeny (Additional file 4: Figure S3), in which species sampled from protists formed basal lineages to all Rickettsia, and that Neorickettsia (Trematoda/aquatic insect-associated) formed a basal lineage to Wolbachia, Anaplasma, and Ehrlichia. Since some Rickettsiales bacteria are endosymbiotic, their vertical transmission style may, to some degree, provide a mechanistic basis to the occurrence of co-divergence. However, it is clear that more data are needed to determine the precise evolutionary association between these bacteria and their vectors.
Finally, the diversity of tick-associated Rickettsiales is particularly noteworthy because both Anaplasma and Ehrlichia genera are tick-specific. In addition, the family Rickettsia has significant diversity associated with ticks, and some species of the family Candidatus Midichloriaceae are also found in ticks. For these tick-borne groups, the bacteria could be directly transmitted from aquatic protists to ticks. Alternatively, ticks could acquire microbes from other terrestrial organisms through cross-species transmission. Nevertheless, distinguishing between these two pathways is beyond the scope of this study and requires data from bacteria characterized from a variety of other organisms.
Conclusions
Our screen for Rickettsiales bacteria in two tick species of Xinjiang revealed nine species, of which some Ehrlichia and Anaplasma species were distinct from any known Rickettsiales. Our phylogenetic analyses indicated that both co-divergence and cross-species transmission were responsible for the current evolutionary diversity of the Rickettsiales.
Methods
Tick sampling
During May 2011, ticks were collected from the Bole and Tacheng regions of Xinjiang Uygur Autonomous Region, China (Additional file 1: Figure S1). Ticks were directly obtained from domestic animals and grassland. All ticks were first identified morphologically by light microscopy and then verified by analyzing molecular markers [25]. The identified ticks were pooled into groups of 8 to 20 according to species and geographic origin, and stored at -70°C for subsequent screening for Rickettsiales bacterial DNA.
DNA extraction, PCR and Sequencing
After washing twice with phosphate-buffered saline, ticks from each pool (or a single tick) were homogenized with a mortar and pestle in 1 mL (or 0.5 mL for individual tick) of phosphate-buffered saline solution. After homogenization, the suspension was incubated at 4°C for 1 h and centrifuged at 2,500 g for 5 min. The upper fraction was collected. DNA was then extracted from individual tick or tick pools with the DNeasy Tissue Kit according to the manufacturer’s instructions, and then subjected to PCR for amplification of bacterial gene sequences and tick mitochondrial rRNA genes (Qiagen, Valencia, CA, USA).
Rickettsial DNA was detected using PCR using primers fD1 and rD1 [47], which amplify a partial fragment (1.5 K) of Rickettsiales rrs. A negative control (distilled water) instead of tick DNA template in the PCR master mix, as well as a positive control (DNA from E. chaffeensis) were included in each test. To amplify gene sequences from samples positive for bacterial DNA, primers were designed based on conserved regions of complete rrs, gltA, and groEL gene sequences from the Rickettsiales spp. To determine whether the three gene sequences amplified from a pool of ticks were derived from a single tick sample, de-pool screening experiments were conducted. The three genes were screened from samples of new individual ticks. Finally, tick mitochondrial 12S and 18S rRNA genes were amplified as described previously [25].
The PCR products were purified with the Agarose Gel DNA Purification kit (TaKaRa, Dalian, China) according to the manufacturer’s recommendations. Purified DNA fragments were cloned into the pMD19-T vector (TaKaRa, Dalian, China), with the vector subsequently transformed into JM109-competent cells. At least 20 clones from each positive tick pool were selected for sequencing. DNA sequencing was performed using Applied Biosystems 377 gene sequencers at Shanghai Sangon Biological Engineering Technology and Services Co., Ltd. (Shanghai, China).
Sequence data and genetic analyses
DNA sequences of the three bacterial genes (Additional file 6: Table S3) were aligned using ClustalW (default parameters) implemented in the MEGA program, version 5.2 [48]. The following data sets were then used in the evolutionary analysis: (i) a 1,243 bp rrs alignment (N = 110 sequences); (ii) a 466 bp gltA gene alignment (N = 79); and (iii) a 720 bp groEL gene alignment (N = 80). The 18S rRNA genes of the vectors (approximately 1700 bp, Additional file 7: Table S4) were aligned by R-coffee [49] with reference to rRNA predicted secondary structures. Finally, the sequences recovered in this study were named according to their relatedness with known bacteria, geographic origins, and sample numbers.
Phylogenetic analyses
Phylogenetic trees were estimated using the Maximum Likelihood (ML) method implemented in the PhyML program (version 3) [50]. The General Time Reversible (GTR) nucleotide substitution model with a gamma (Γ)-distribution model of among-site rate variation and a proportion of invariable sites (i.e. the GTR + Γ + I substitution model) was utilized. Phylogenetic trees were also inferred using the Bayesian method implemented in MrBayes v3.2 [51]. The same substitution model was employed as described above. When using MrBayes v3.2, three hot chains and one cold Markov chain Monte Carlo (MCMC) were used, with trees and parameters sampled every 100 generations. A 25% burn-in was enforced for all analyses. Estimated sample sizes >200 for every model and search parameter were considered as indicators of adequate sampling of posterior distributions.
To infer the direction of evolutionary change within the order Rickettsiales (Additional file 6: Table S3, Additional file 8: Table S5), we inferred a molecular clock (i.e. rooted) phylogenetic tree using the BEAST software package (version 1.7.5) [52] assuming the GTR + Γ + I substitution model. This analysis also utilized the Yule process coalescent model. The MCMC chain was run for 108 generations to ensure convergence. Statistical support for individual nodes was reflected in posterior probability values. Using the rooting determined in the BEAST tree, we then employed ML [53] and Bayesian [54] methods implemented in the Mesquite package [55] to tentatively reconstruct the evolution of habitat among the Rickettsiales by treating “aquatic vs. terrestrial” habitats as discrete character states and mapping their occurrence onto the phylogenies.
Analysis of co-divergence events
We tested the hypothesis of bacterial-host co-divergence using the ParaFit method [56] as implemented in the COPYCAT software package [57], which compares the patristic distance matrices derived from the bacteria and vector phylogenies. For this analysis we prepared three tick-only data sets including (i) tick-associated Rickettsia, (ii) Anaplasma, and (iii) Ehrlichia, as well as an overall data set including all Rickettsiales. The bacterial genetic distance matrices were derived from the rrs trees inferred by both BEAST and ML methods, while the vector genetic distance matrices were derived from the 18S rRNA gene trees generated using BEAST as described above. Significance testing was based of 9,999 randomizations of the association matrices. Additionally, to illustrate the association between bacteria (Additional file 6: Table S3, Additional file 8: Table S5) and their vectors, a tanglegram was generated by matching each bacterial species (or group) to their associated vectors using TreeMap 3.0 [58].
Additional files
References
Rickettsial Diseases. 2007, Informa Healthcare, London
Kelly DJ, Richards AL, Temenak JJ, Strickman D, Dasch GA: The past and present threat of rickettsial diseases to military medicine and international public health. Clin Infect Dis. 2002, 34: s145-s169.
Tamura A, Ohashi N, Urakami H, Miyamura S: Classification of Rickettsia tsutsugamushi in a new genus, Orientia gen. nov., as Orientia tsutsugamushi comb. nov. Int J Syst Bacteriol. 1995, 45: 589-591.
Zhang L, Liu Y, Ni D, Li Q, Yu Y, Yu XJ, Wan K, Li D, Liang G, Jiang X, Jing H, Run J, Luan M, Fu X, Zhang J, Yang W, Wang Y, Dumler JS, Feng Z, Ren J, Xu J: Nosocomial transmission of human granulocytic anaplasmosis in China. JAMA. 2008, 300: 2263-2270.
Weinert LA, Werren JH, Aebi A, Stone GN, Jiggins FM: Evolution and diversity of Rickettsia bacteria. BMC Biol. 2009, 7: 6-
Merhej V, Raoult D: Rickettsial evolution in the light of comparative genomics. Biol Rev Camb Philos Soc. 2011, 86: 379-405.
Pritt BS, Sloan LM, Johnson DK, Munderloh UG, Paskewitz SM, McElroy KM, McFadden JD, Binnicker MJ, Neitzel DF, Liu G, Nicholson WL, Nelson CM, Franson JJ, Martin SA, Cunningham SA, Steward CR, Bogumill K, Bjorgaard ME, Davis JP, McQuiston JH, Warshauer DM, Wilhelm MP, Patel R, Trivedi VA, Eremeeva ME: Emergence of a new pathogenic Ehrlichia species, Wisconsin and Minnesota, 2009. N Engl J Med. 2011, 365: 422-429.
Gillespie JJ, Nordberg EK, Azad AF, Sobral BW: Phylogeny and Comparative Genomics: The Shifting Landscape in the Genomics Era. Intracellular Pathogens II: Rickettsiales. Edited by: Azad AF, Palmer GH. 2012, American Society of Microbiology Press, Herndon, Virginia, US
Rar V, Golovljova I: Anaplasma, Ehrlichia, and “Candidatus Neoehrlichia” bacteria: pathogenicity, biodiversity, and molecular genetic characteristics, a review. Infect Genet Evol. 2011, 11: 1842-1861.
Vaughan JA, Tkach VV, Greiman SE: Neorickettsial endosymbionts of the digenea: diversity, transmission and distribution. Adv Parasitol. 2012, 79: 253-297.
Driscoll T, Gillespie JJ, Nordberg EK, Azad AF, Sobral BW: Bacterial DNA sifted from the Trichoplax adhaerens Animalia: Placozoa genome project reveals a putative rickettsial endosymbiont. Genome Biol Evol. 2013, 5: 621-645.
Brenner DJ, O’Connor SP, Winkler HH, Steigerwalt AG: Proposals to unify the genera Bartonella and Rochalimaea, with descriptions of Bartonella quintana comb. nov., Bartonella vinsonii comb. nov., Bartonella henselae comb. nov., and Bartonella elizabethae comb. nov., and to remove the family Bartonellaceae from the order Rickettsiales. Int J Syst Bacteriol. 1993, 43: 777-786.
Dumler JS, Barbet AF, Bekker CP, Dasch GA, Palmer GH, Ray SC, Rikihisa Y, Rurangirwa FR: Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales: unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia and Ehrlichia with Neorickettsia, descriptions of six new species combinations and designation of Ehrlichia equi and ‘HGE agent’ as subjective synonyms of Ehrlichia phagocytophila. Int J Syst Evol Microbiol. 2001, 51: 2145-2165.
Montagna M, Sassera D, Epis S, Bazzocchi C, Vannini C, Lo N, Sacchi L, Fukatsu T, Petroni G, Bandi C: “Candidatus Midichloriaceae” fam. nov. Rickettsiales, an ecologically widespread clade of intracellular Alphaproteobacteria. Appl Environ Microbiol. 2013, 79: 3241-3248.
Perlman SJ, Hunter MS, Zchori-Fein E: The emerging diversity of Rickettsia. Proc Biol Sci. 2006, 273 (1598): 2097-2106.
Darby AC, Cho NH, Fuxelius HH, Westberg J, Andersson SG: Intracellular pathogens go extreme: genome evolution in the Rickettsiales. Trends Genet. 2007, 23: 511-520.
Renvoisé A, Merhej V, Georgiades K, Raoult D: Intracellular Rickettsiales: Insights into manipulators of eukaryotic cells. Trends Mol Med. 2011, 17: 573-583.
Gillespie JJ, Beier MS, Rahman MS, Ammerman NC, Shallom JM: Plasmids and rickettsial evolution: insight from Rickettsia felis. PLoS One. 2007, 2: e266-
Walker DH, Ismail N: Emerging and re-emerging rickettsioses: endothelial cell infection and early disease events. Nat Rev Microbiol. 2008, 6: 375-386.
Azad AF, Beard CB: Rickettsial pathogens and their arthropod vectors. Emerg Infect Dis. 1984, 4: 179-186.
Toft C, Andersson SG: Evolutionary microbial genomics: Insights into bacterial host adaptation. Nat Rev Genet. 2010, 11: 465-475.
Wu XB, Na RH, Wei SS, Zhu JS, Peng HJ: Distribution of tick-borne diseases in China. Parasit Vectors. 2013, 6: 119-
Chahan B, Jian Z, Xuan X, Sato Y, Kabeya H, Tuchiya K, Itamoto K, Okuda M, Mikami T, Maruyama S, Inokuma H: Serological evidence of infection of Anaplasma and Ehrlichia in domestic animals in Xinjiang Uygur Autonomous Region area, China. Vet Parasitol. 2005, 134: 273-278.
Sun X, Zhang GL, Liu XM, Zhao Y, Zheng Z: Investigation of tick species and tick-borne pathogens in Hoxud county of Xinjiang Uyghur Autonomous Region, China. Chin J Vector Biology Control. 2013, 24: 5-7.
Lu X, Lin XD, Wang JB, Qin XC, Tian JH, Guo WP, Fan FN, Shao R, Xu J, Zhang YZ: Molecular survey of hard ticks in endemic areas of tick-borne diseases in China. Ticks Tick Borne Dis. 2013, 4: 288-296.
Garrity GM, Winters M, Kuo AW, Searles DB: Taxonomic Outline of the Prokaryotes. Bergey’s Manual of Systematic Bacteriology, 2nd Edn, Volume II Part C. 2002, Springer-Verlag, New York, US
Mediannikov O, Matsumoto K, Samoylenko I, Drancourt M, Roux V, Rydkina E, Davoust B, Tarasevich I, Brouqui P, Fournier PE: Rickettsia raoultii sp. nov., a spotted fever group rickettsia associated with Dermacentor ticks in Europe and Russia. Int J Syst Evol Microbiol. 2008, 58: 1635-1639.
Beati L, Finidori JP, Raoult D: First isolation of Rickettsia slovaca from Dermacentor marginatus in France. Am J Trop Med Hyg. 1993, 48: 257-268.
Anziani OS, Ewing SA, Barker RW: Experimental transmission of a granulocytic form of the tribe Ehrlichieae by Dermacentor variabilis and Amblyomma americanum to dogs. Am J Vet Res. 1990, 51: 929-931.
Steiert JG, Gilfoy F: Infection rates of Amblyomma americanum and Dermacentor variabilis by Ehrlichia chaffeensis and Ehrlichia ewingii in southwest Missouri. Vector Borne Zoonotic Dis. 2002, 2: 53-60.
Parola P, Inokuma H, Camicas JL, Brouqui P, Raoult D: Detection and identification of spotted fever group Rickettsiae and Ehrlichiae in African ticks. Emerg Infect Dis. 2001, 7: 1014-1017.
Jiang BG, Cao WC, Niu JJ, Wang JX, Li HM, Sun Y, Yang H, Richadus JH, Habbema JD: Detection and identification of Ehrlichia species in Rhipicephalus Boophilus microplus ticks in cattle from Xiamen, China. Vector Borne Zoonotic Dis. 2011, 11: 325-
Groves MG, Dennis GL, Amyx HL, Huxsoll DL: Transmission of Ehrlichia canis to dogs by ticks Rhipicephalus sanguineus. Am J Vet Res. 1975, 36: 937-940.
Johnson EM, Ewing SA, Barker RW, Fox JC, Crow DW, Kocan KM: Experimental transmission of Ehrlichia canis Rickettsiales: Ehrlichieae by Dermacentor variabilis Acari: Ixodidae. Vet Parasitol. 1998, 74: 277-288.
Jones RT, McCormick KF, Martin AP: Bacterial communities of Bartonella-positive fleas: Diversity and community assembly patterns. Appl Environ Microbiol. 2008, 74: 1667-1670.
Fraune S, Bosch TCG: Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proc Natl Acad Sci. 2007, 10: 13146-13151.
Madigan JE, Pusterla N, Johnson E, Chae JS, Pusterla JB, Derock E, Lawler SP: Transmission of Ehrlichia risticii, the agent of Potomac horse fever, using naturally infected aquatic insects and helminth vectors: preliminary report. Equine Vet J. 2000, 32: 275-279.
Huang L, Duan XD, Meng QL, Li R, Zhao QL: Detection of Anaplasma phagocytophilum among sheep in Shihezi, Xinjiang Uyghur Autonomous Region, China and analysis of its 16S rRNA gene sequences. Chin J Vector Biology Control. 2013, 24: 141-143.
Chen Z, Yang X, Bu F, Yang X, Yang X, Liu J: Ticks Acari: Ixodoidea: Argasidae, Ixodidae of China. Exp Appl Acarol. 2010, 51: 393-404.
Fan MY, Walker DH, Yu SR, Liu QH: Epidemiology and ecology of rickettsial diseases in the People’s Republic of China. Rev Infect Dis. 1987, 9: 823-840.
Fan MY, Jiang YX, Wang LC, Wang JG, Lin YF: Isolation and characterization of tick-borne spotted fever group rickettsia from a patient in Jinghe county of Xinjiang. Chin J Zoonoses. 1985, 2: 9-
Fan MY, Wang JG, Jiang YX, Zong DG, Lenz B: Isolation of a spotted fever group rickettsia from a patient and related ecologic investigations in Xinjiang Uygur Autonomous Region of China. J Clin Microbiol. 1987, 25: 628-632.
Tian ZC, Liu GY, Shen H, Xie JR, Luo J, Tian MY: First report on the occurrence of Rickettsia slovaca and Rickettsia raoultii in Dermacentor silvarum in China. Parasit Vectors. 2012, 5: 19-
Raoult D, Lakos A, Fenollar F, Beytout J, Brouqui P, Fournier PE: Spotless rickettsiosis caused by Rickettsia slovaca and associated with Dermacentor ticks. Clin Infect Dis. 2002, 34: 1331-1336.
Ahantarig A, Trinachartvanit W, Baimai V, Grubhoffer L: Hard ticks and their bacterial endosymbionts (or would be pathogens). Folia Microbiol (Praha). 2013, 58: 419-428.
Ogata H, La Scola B, Audic S, Renesto P, Blanc G, Robert C, Fournier PE, Claverie JM, Raoult D: Genome sequence of Rickettsia bellii illuminates the role of amoebae in gene exchanges between intracellular pathogens. PLoS Genet. 2006, 2: e76-
Weisburg WG, Barns SM, Pelletier DA, Lane DJ: 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991, 173: 697-703.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739.
Moretti S, Wilm A, Higgins DG, Xenarios I, Notredame C: R-Coffee: a web server for accurately aligning noncoding RNA sequences. Nucleic Acids Res. 2008, 36 (Web Server issue): W10-W13.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O: New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010, 59: 307-321.
Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001, 17: 754-755.
Drummond AJ, Suchard MA, Xie D, Rambaut A: Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012, 29: 1969-1973.
Pagel M: The maximum likelihood approach to reconstructing ancestral character states of discrete characters on phylogenies. Syst Biol. 1999, 48: 612-622.
Huelsenbeck JP, Nielsen R, Bollback JP: Stochastic mapping of morphological characters. Syst Biol. 2003, 52: 131-158.
Maddison WP, Maddison DR: Mesquite: a modular system for evolutionary analysis. Version 2.75. 2011,., [http://mesquiteproject.org]
Legendre P, Desdevises Y, Bazin E: A statistical test for host-parasite coevolution. Syst Biol. 2002, 51: 217-234.
Meier-Kolthoff JP, Auch AF, Huson DH, Göker M: COPYCAT: cophylogenetic analysis tool. Bioinformatics. 2007, 23: 898-900.
Jackson AP, Charleston MA: A cophylogenetic perspective of RNA-virus evolution. Mol Biol Evol. 2004, 21: 45-57.
Acknowledgements
This study was supported by National Natural Science Foundation of China (Grants 81290343, 81273014), State Key Laboratory for Infection Disease Prevention and Control, the Priority Project on Infectious Disease Control and Prevention (Grant 2012ZX10004215), Mega Project of Research on the Prevention and Control of HIV/AIDS, Viral Hepatitis Infectious Diseases (Grant 2011ZX10004-001, 2013ZX10004-101). ECH is supported by a National Health and Medical Research Council Australia Fellowship. JJG acknowledges support from National Institute of Health/National Institute of Allergy and Infectious Diseases grants R01AI017828 and R01AI59118 awarded to Abdu F. Azad (University of Maryland, School of Medicine). JSD is supported in part through grants R01AI44102 and R21AI096062 from the National Institutes of Allergy and Infectious Diseases.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YZZ conceived the research project; YZZ, YJK, XND, MHC, YX, WMF, YJG, and BP collected the samples, GYZ, YJK, and XPC performed research; YJK, GYZ, and MS analyzed the data; YZZ, YJK, MS, ECH, JJG, and SJD wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12862_2014_167_MOESM1_ESM.jpeg
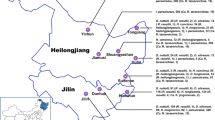
Additional file 1: Figure S1.: Map of sampling locations in Xinjiang province, China. The Bole and Tacheng regions are labeled by the red dots. (JPEG 254 KB)
12862_2014_167_MOESM3_ESM.pdf
Additional file 3: Figure S2.: Detailed ML phylogenetic trees based on the sequences of Rickettsiales rrs (a, d, g) , gltA (b, e, h), and groEL (c, f, i) genes. The numbers at each branch indicate bootstrap values. (PDF 289 KB)
12862_2014_167_MOESM4_ESM.jpeg
Additional file 4: Figure S3.: Tanglegram of Rickettsiales bacteria and their hosts. The bacterial tree on the left panel of the figure was inferred based on rrs using BEAST and ML (PhyML) methods, while the vector tree on the right panel of the figure was inferred based on 18S rRNA sequences. Each bacterial species (or group) was linked to their associated vectors. In the bacterial tree different genera are distinguished by different colors. The BEAST tree is shown here. (JPEG 791 KB)
12862_2014_167_MOESM7_ESM.doc
Additional file 7: Table S4.: Sequences of the 18S rRNA gene of the vector species used in the phylogenetic analysis. (DOC 54 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Kang, YJ., Diao, XN., Zhao, GY. et al. Extensive diversity of Rickettsiales bacteria in two species of ticks from China and the evolution of the Rickettsiales. BMC Evol Biol 14, 167 (2014). https://doi.org/10.1186/s12862-014-0167-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-014-0167-2