
Abstract
In the past decade, we have seen the emergence of sequence-based methods to understand chromosome organization. With the confluence of in situ approaches to capture information on looping, topological domains, and larger chromatin compartments, understanding chromatin-driven disease is becoming feasible. Excitingly, recent advances in single molecule imaging with capacity to reconstruct “bulk-cell” features of chromosome conformation have revealed cell-to-cell chromatin structural variation. The fundamental question motivating our analysis of the literature is, can altered chromatin structure drive tumorigenesis? As our community learns more about rare disease, including low mutational frequency cancers, understanding “chromatin-driven” pathology will illuminate the regulatory structures of the genome. We describe recent insights into altered genome architecture in human cancer, highlighting multiple pathways toward disruptions of chromatin structure, including structural variation, noncoding mutations, metabolism, and de novo mutations to architectural regulators themselves. Our analysis of the literature reveals that deregulation of genome structure is characteristic in distinct classes of chromatin-driven tumors. As we begin to integrate the findings from single cell imaging studies and chromatin structural sequencing, we will be able to understand the diversity of cells within a common diagnosis, and begin to define structure–function relationships of the misfolded genome.
Similar content being viewed by others

Background
The sequencing of the human genome [1] has motivated fundamental questions to understand non-coding components of its heritability. The vast majority of human DNA sequences are located outside the exon regions of the genome, or “exome”. This leads to the question, is there selective pressure to retain large non-coding regions as physical scaffolding, to provide regulation for genic regions? Methods to sequence protein-genome interactions in trans and long-distance cis-chromatin interactions have revealed insights into regulatory functions of non-coding regions through comprehensive mapping [2].
The versatility of high-throughput genome sequencing has enabled mapping of “one-to-all” chromatin interactions with a single “viewpoint” (4C) [3], or with several “viewpoints” (5C) [4]. The sequencing of “all-to-all” chromatin interactions (Hi-C) [5] has been refined with in situ approaches to better preserve native chromatin structure [6]. The protein-centric versions of these chromatin sequencing technologies (ChIA-PET, HiChIP, AQuA-HiCHIP) now enable precise quantitative examination of how distinct regulatory factors mediate loops [7,8,9]. One of the key findings from chromatin sequencing is that the cancer genome is structurally distinct from human reference genomes.
We describe evidence for altered chromosome folding in cancer in the context of chromatin interaction domains, and chromosome structural variation in malignancies. Studies of chromatin loops, topologically associating domains (TADs), chromatin compartments, and structural variation (SV) provide evidence for this finding by revealing key elements of altered genome structure in cancer. Thus, we examine genome structure–function relationships in human malignancy, with a focus on alterations in chromatin interaction domains.
Main text
Chromatin loops in cancer
In Hi-C data, chromatin loops appear as punctate regions of heightened interactions relative to neighboring chromatin [10, 11]. While cis-chromatin sequencing methods including Hi-C enable detection of chromatin loops and long-range interactions, more recent methods, including HiChIP [7, 9], TrAC-looping [12], and Capture-HiC (capture-C) [13], focus on high-resolution sequencing of shorter-range loops at kilobase resolution with increased accuracy. Each of these recent methods attains high proportions of paired-end tags, or PETs that are useful for defining chromatin interactions, including functional enhancer-promoter interactions.
Substituting the micrococcal nuclease (MNase) enzyme for other cutting enzymes increases resolution in sequencing shorter-range chromatin domains. Excitingly, 3C-based methods and micrococcal nuclease (MNase) have converged in recent methods for high-resolution chromatin structural sequencing, including Micro-C [14] and Micro-capture-C (MCC) [15]. The more recent MCC method resolves proximal enhancer-promoter contacts, within several kb, which has remained challenging for Hi-C at standard sequencing depth. This presents new opportunities to systematically examine altered short-range chromatin interactions in human cancers.
Moreover, the higher-resolution chromatin interactions observed in MCC and Micro-C, also provide context for defining transcription factor (TF) binding sites within chromatin loops. This enables new approaches to understand TF-driven childhood malignancies such as the chimeric oncoproteins that drive rhabdomyosarcoma (RMS) [16] and Ewing sarcoma (EWS) [17]. We anticipate exciting advances in the years ahead in the precise determination of localization of oncogenic TF-chimeras in the context of chromatin looping.
Recent studies in RMS, a rare pediatric soft tissue cancer, have revealed context-specific roles for chromatin looping. In the RMS subtype driven from TF-chimeras, termed fusion-positive (FP-RMS), there is evidence that the PAX3-FOXO1 oncoprotein has pioneer activity [16]. The intrinsic ability of this chimeric TF to alter repressive chromatin states enables a network of chromatin interactions in FP-RMS, including looping at the MYOD1 and SOX8 gene loci to promote positive autoregulation of tumor-specific gene activation [18]. The clinical molecule entinostat, which inhibits the function of histone-H3 deacetylases, systematically alters chromatin looping in FP-RMS preceding myogenic differentiation and loss of tumor proliferation [9, 19]. With spike-in quantification of HiChIP (AQuA-HiChIP), we observed that entinostat treatment has immediate-early effects to augment chromatin looping in FP-RMS, deregulating gene expression [9, 18]. In another major subtype of RMS, termed fusion-negative (FN-RMS), chromatin looping stabilizes expression of the pseudo-oncogene SNAI2 [20]. The essential TF, MYOD, drives RMS in each major subtype, through induced gene expression and through chromatin organization, observed through HiChIP [18, 20]. The clinical RAS inhibitor, trametinib, inhibits ERK activity and suppresses expression of SNAI2, promoting FN-RMS tumor differentiation [20, 21]. Determining the chromatin architectural functions of RAS activity in FN-RMS will be of high interest. Taken together, there are distinct parallels between MYOD associated looping events in FP-RMS and FN-RMS, each of which can be altered with clinical or pre-clinical molecules. Studies to determine the precise regulatory influences of pioneer TFs on chromatin looping in sarcomas and other childhood tumors will likely illuminate general principles of chromatin domain dysregulation in aggressive cancers.
The motifs of TFs influence regulatory chromatin looping in human cancer. Recent studies reveal that highly penetrant noncoding genetic variants have the potential to affect chromatin interactions. Massively parallel TF-motif binding assays coupled to sequencing have revealed the specificities for disease-causing DNA mutations that alter the ability of TFs to recognize their motifs [22]. A capture-C study in human breast cancer demonstrated that TF-motif pairs were altered at regulatory loci encoding disease SNPs [23]. These regulatory SNPs associated with altered chromatin loops in breast cancer were associated with pioneer factors (FOXA1, GATA3) and estrogen receptor (Fig. 1A,B) [24]. Moreover, altered chromatin looping was found to occur at loci encoding major oncogenes (MYC) and tumor suppressors (CDKN2A). Conceptually, these findings motivate examining altered enhancer interactions in cancers. Analyzing CTCF binding sites in human cancers reveals recurrent mutations and deletions of these motifs in leukemia (T-ALL), esophageal tumors, and liver cancer [25].
Experimental evaluation of many of these CTCF motif alterations with a method called ChIA-PET [25], which enables sequencing protein-centric loops, indicates functional consequences for chromatin interactions. Key loci encoding genes required for tumor proliferation are found in regions associated with altered loop anchor sites. Observations of noncoding variants in human cancers motivated systematic analyses of altered chromatin loops in cancer cell lines, using Hi-C and ChIA-PET [26]. Similar to the Baxter et al. study [23], Snyder and co-workers observed cell-type specific pioneer factors at chromatin loop anchors (e.g., PU.1), and enrichments for penetrant noncoding disease variants at loops [26]. In a recent study on subtype-specific chromatin states in bladder cancer, the pioneer factors FOXA1 and GATA3 were each found to serve as “loop anchors” [27]. Providing further conceptual links between pioneer factor function and 3D genome structure, a recent report demonstrated that GATA3 gene expression levels can alter chromatin architecture in leukemia, and that polymorphisms in GATA3’s intronic regulatory sequences could impact its expression [28]. These studies motivate hypotheses that noncoding cancer mutations might be disrupting chromatin loop structures, and altering binding of tissue-specific TFs or pioneer factors at loop anchors (Fig. 1B).
Recent evidence indicates that chromatin domains can be targeted by clinical [11] or pre-clinical [9, 29] chemotherapeutic strategies. New insights into the regulatory roles of topoisomerases have revealed the potential of the clinical molecule etoposide to covalently disrupt chromatin domains [11]. The conceptual advance highlighted by a chemotherapeutic agent targeting torsional stress associated with cis chromatin interactions suggests that altered chromatin domains might serve dual roles as drivers and vulnerabilities in human cancer. This leads to the question, what are the characteristics of chromatin contact domains in cancer? We explore this question, in the context of driving alterations in key gene classes, and structural variation in cancer genomes.
Structural variation and chromatin domains
Recent comparative studies in whole genome sequencing (WGS), chromatin sequencing, and imaging methods, have revealed that Hi-C, especially in combination with whole genome sequencing, can be extremely powerful in identifying structural variation (SV) [30, 31]. From studies of SV and chromatin architecture (reviewed, [32]), it is becoming clear that Hi-C represents an efficient approach for de novo detection of SVs in cancer genomes (Fig. 1C,D,E). The impact of these studies will be transforming in several key areas. New insights into how focal deletions, inversions, and translocations are systematically altering the regulatory functions of enhancers or insulators will provide connections between gene regulation and structural variation. Additionally, topological context for copy number variation (CNV) and gene-fusion events in cancer will reveal how alterations reside within chromatin domains. Examining the effects of SVs on the non-coding genome as well as the impact on coding regions will continue to illuminate epigenetic mechanisms driving tumors.
A recent study has revealed that in leukemia genomes, SV modifies the proximity of the BCL11B gene locus and its enhancer, thereby driving its expression in progenitor cells [33]. The authors mapped HiChIP data from leukemia samples onto patient-specific reference genomes to account for the SV present. The recurrent translocations impacting the BCL11B gene locus were found to frequently involve transposition of enhancer elements that produced functional consequences in gene expression. The study also revealed enhancer-specific CNV (enhancer amplification) affecting BCL11B gene regulation. Thus, through structural repositioning, or amplification of enhancers, leukemia gene regulation is systematically altered. Recently, shallow Hi-C approaches have helped define SVs leading to ETV6-RUNX1 gene fusion events in leukemia, and have revealed new patterns of potential chromothripsis (a series of multiple catastrophic chromosomal rearrangements) [34, 35]. It is of note that lower resolution methods and exome-focused methods like SNP arrays or RNA-seq, may not efficiently capture information regarding chromothripsis, while 3D chromatin sequencing may be more efficient for identifying these patterns of SVs. We anticipate further utility of spike-in normalized chromatin architectural sequencing in the context of chromosomal imbalances (e.g., aneuploidy), which occur in as much as 90% of human tumors [36]. Studies of childhood cancers which rarely exhibit signatures of high mutational frequencies but often display signs of chromothripsis [37, 38] may benefit from these new approaches.
In diffuse intrinsic pontine glioma (DIPG), CNV affecting tumor-specific gene expression of TCF12 and amplification of its enhancer have been observed in Hi-C studies [39]. In another recent study, enhancers subject to SV were shown to drive expression of MYC in lymphoma, through translocation events [40]. In bladder cancers, where GATA3 and FOXA1 may have characteristic altered gene expression, Hi-C has been used elegantly to detect patterns of CNV and SV [27]. In hematologic tumors, and solid tumors, SV-induced enhancer transposition can regulate the expression of oncogenic drivers through proximity. The increased usage of low depth Hi-C or HiChIP to elucidate patterns of SV or altered enhancer function will be impactful across the clinical and basic sciences.
The developmental consequences of SVs on altered chromatin domains can be severe, with altered gene expression patterns resulting from de novo TAD formation (“neo-TADs”), TAD-fusion events, and altered boundaries (reviewed [32]) (Fig. 1A,C,E). In comparative studies of cancer 3D genomes, Yue and colleagues uncovered SVs which alter chromatin interactions in prostate, breast, gastric, tumors and hematologic tumors [41]. Recurrent alterations in cis-chromatin interactions were observed at loci encoding the pioneer factor FOXA1 (prostate cancer), the cell cycle gene CDK12 (breast cancer), and the RAB36 gene (leukemia). Interestingly, RAB36 is frequently observed within a conjoined chromatin contact domain resulting from SV. Yue and colleagues observed that RAB36 gene expression was associated with poorer patient outcomes, linking SV-mediated chromatin domain alterations with disease etiology. We propose that the developmental alterations in gene expression patterns derived from SV-altered chromatin domains are highly relevant in human cancers, and we anticipate exciting advances to in this area in coming years.
There is evidence that SVs occurring in human cancers are frequently more complex than in other tissues, and this has implications for cis-chromatin interactions. A recent comprehensive analysis of SV in human cancer observed recurrent enhancer-deletions for loci encoding tumor suppressive genes, and recurrent de novo TAD formation enabling oncogene expression[30]. Interestingly SVs are also a strikingly common feature across the spectrum of human tumors, but many Mb-scale SVs are challenging to define with short-read sequencing alone [42]. However, short-read genome sequencing data could be used to construct subtype-specific reference genomes, which would allow more accurate SV identification using 3D sequencing data. It is of note that the overall frequency of SV occurrence is positively associated with the accessibility of local chromatin states in cancers, suggesting that euchromatin might be predisposed to these alterations.
In distinct cancers, there are common “SV pathways” toward recurrent fusion-oncogene events, while a diversity of SV types may result in amplification of common oncogenes or losses of major tumor suppressors [42]. Increasing evidence supports an association between unique cancer types and idiosyncratic SV patterns, linked to altered chromatin domains [43]. One important aspect of this, is that distinct tumors might have recurrent alterations in chromatin domain boundaries, linking SVs to gene mis-regulation including deletions, interchromosomal rearrangements and intrachromosomal variation (Fig. 1C,D,E). Understanding the major chromatin architectural drivers of human cancers will require integrating SVs in the context of repurposing transcriptional regulatory elements and domains. We anticipate definitions of hallmarks of architectural drivers of cancer as we learn increasingly about the recurrent patterns of domain alterations induced from SVs.
Cancer metabolism and cis-chromatin interactions
Increasing evidence has revealed chromatin structural phenotypes driven from recurrent cancer mutations in genes encoding metabolic regulators. Two major classes of metabolic mutation that each alter the Krebs cycle are highly penetrant in human tumors. In each case, toxic accumulations of metabolites result in differentially methylated regions (DMRs) across the genome. However, the SDH class and IDH class of oncogenic mutations rely on distinct mechanisms to induce their convergent effects on the epigenetic state of the cell. One particular class of these penetrant mutations renders the SDH-family enzymes catalytically deficient, which results in accumulation of succinate before it can be processed. High levels of succinate can inhibit several classes of demethylase enzymes, including TET-family and JMJD-family demethylases, thus increasing methyl-CpG content [44], augmenting chromatin succinylation [45], and increased histone H3K9-methylation [46]. Moreover, recent studies suggest that aberrant succinylation levels may also augment the placement of H3K4me3 at loci encoding cell-type specific regulatory genes [47].
Connecting altered CpG methylation and altered tumor metabolism, a recent report revealed DNA hypermethylation at CpG islands in IDH-driven gliomas [48]. Importantly, CTCF binding sites were associated with these DMRs. With evidence that CTCF binding anchors genomic looping, [49] these observations motivated chromatin structural studies. Strikingly, approximately half of the DMRs occurring at CTCF sites overlapped with chromatin loop anchors [44]. Key chromatin contact domains were disrupted, included at the FGF4 locus, and KIT insulator elements. The altered DMRs at these loci resulted in deregulated gene expression for these two GIST drivers. These could be targeted as vulnerabilities with clinical FGFR4, and KIT inhibitors.
A related class of metabolic cancer mutations in the IDH enzyme has also been reported as a driver in leukemias and gliomas [50]. Similar to SDH mutations, IDH mutations induce de novo DMRs through accumulations of metabolites, α-ketoglutarate and most notably 2-hydroxyglutarate, that can inhibit TET-family enzymes and histone demethylases. Interestingly, gene pairs spanning TAD boundary junctions are highly sensitive to IDH mutational status, suggesting that altered CTCF binding may be associated with sensitized DMRs [48]. The IDH glioma insulator-loss mechanism results from methylation-sensitive defects in genomic binding of CTCF, enabling aberrant chromatin domains to drive gene oncogene expression, including PDGFRA [48]. Studies of chromatin structure in IDH/SDH-mutant tumors highlight that while altering chromatin domain structures can have subtle or context-specific effects on transcription [10], identifying key alterations in tumor-specific gene expression can lead to targetable vulnerabilities.
While metabolic products can alter chromatin structure–function relationships through enzymatic processes, there is evidence that non-enzymatic processes link metabolic outputs and chromatin structure as well. With new insights from non-enzymatic covalent histone modifications (NECMs) [51, 52], there are additional opportunities to (1) expand the scope of known chromatin PTMs, and (2) interrogate the recently discovered metabolic drivers of NECMs to ask if they have instructive effects on chromatin structure. Examples include evidence for histone glycation [53], histone acylation [54], and histone lipidation [55]. With evidence of altered metabolism [56] and oxidative stress [57] in human cancers, we anticipate exciting advances in the coming years to conceptually relate non-enzymatic histone PTMs with genome structure.
Imaging and chromatin structure
Bulk-cell genomics has revealed internally consistent principles for contact domains, compartments, and loops. However, single cell imaging sometimes yields distinct or complementary answers to the questions of genome organization. While there is a diffraction-limiting feature in traditional imaging experiments on the order of the visible wavelengths of light (~ 200 nm diffraction limit), 3D-STORM imaging approaches 20 nm resolution [58]. This increased resolution enables characterization of fine chromatin structural features in single cells. Where traditional sequencing-based methods are better equipped for detection of paired cis-chromatin interactions, imaging-based methods can capture multi-locus interactions. With 3D-STORM based studies, Zhuang and co-workers examined the cohesion-dependence of domain organization in single cells. With rapid RAD21 depletion [10], the authors observed a statistical retention of chromatin domain structures, suggesting that cohesin plays a primary role in noise-reduction for coherence of contact domain maintenance in bulk cell populations [59]. Similar results, revealing cohesin-independence for contact domains, have been observed with measurements of “globularity” of chromatin domains with super-resolution imaging and cryo-EM [60]. In recent work from Cavalli and colleagues, super resolution microscopy enabled definition of significantly decreased intra-TAD chromatin interactions in the absence of cohesin complexes [61]. Thus, results from bulk cell chromatin sequencing and single cell super resolution microscopy each suggest roles for cohesin function in chromatin interactions within TADs. Also of note was the finding that CTCF loss enables increased inter-TAD chromatin interactivity in single cells [61]. Thus, while stereotypic TAD architecture defined in bulk cell Hi-C and next-generation imaging might differ, key fundamental properties of cohesin and CTCF are conserved at the single cell level [10, 49, 61].
In recent advances, Boettiger and co-workers have reported reconstruction of chromatin interaction domains from high resolution imaging, optical reconstruction of chromatin architecture (ORCA), providing new insights about TAD function and transcription during development [62]. With ORCA, it was found that developmental gene transcription correlates well with chromatin domain formation. ORCA thus helps overcome challenges in characterizing associations between chromatin domain formation and nascent RNA transcription that are problematic for bulk cell sequencing approaches [10]. The resolution of ORCA enables quantification of interaction distances within and across domains. While results from ORCA indicate that active chromatin compartments are correlated with RNA transcription, promoter-enhancer proximity within a single cell is not strongly predictive of transcriptional state. One possible explanation is that within repressed chromatin regions, long-range contacts can still occur [63, 64] while inappropriate enhancer loops might not be productive for initiation [62]. The early results from ORCA in developing drosophila embryos also integrate conceptually with observations from pluripotent cells [65], where domain boundaries are highly sensitive to CTCF positioning and H3K27me3. Understanding locus-specific contexts for CTCF function in single cells, as an insulator for repressive and active chromatin domains will be of high interest. Disruption of heterochromatin is a common feature observed in high resolution imaging studies modeling human cancer progression. ORCA has also revealed dependencies for spatial HOX gene de-repression in recent reports of loss-of-function mutations in mammalian SWI/SNF chromatin remodeling complexes [66]. These studies provide important context for mechanisms of loss of epigenetic tumor suppression with altered SWI/SNF complexes. In recent studies in conditional tumor mouse models, Xu and colleagues observed chromatin restructuring during the course of tumor progression [67]. Consistent with other studies [68], H3K9me3 was observed at DNA repeat elements in the chromatin fiber, while tumor progression resulted in systematic loss of chromatin compaction and altered folding at these regions.
Defining the fundamental connections between deposition of heterochromatin marks and chromatin folding in human cancer will be impactful in coming years. Moreover, applications of ORCA and 3-D STORM imaging to understand conserved properties of chromatin domains in cancers will be highly impactful, as these methods can take into account cellular heterogeneity in human tumors, as well as defining RNA transcription in the precise cellular context in which the chromatin structure is measured. Moreover, integrations with single cell Hi-C (sc-HiC) [69,70,71] and high-resolution imaging will be impactful. With sc-HiC, it is possible to determine genome structural components in the context of developmental and cell-cycle transitions [71]. It will be exciting to see the synergy between single cell imaging and chromatin sequencing approaches in the coming years in the context of human cancers.
Cohesinopathies
An important epigenetic pathway towards architectural dysregulation is the mutation of genes encoding major drivers of genome structure, cohesin complexes [10]. The Cancer Genome Atlas (TCGA) sequencing efforts have revealed recurrent cohesin subunit mutations in cancers of the blood system [72]. Of these mutations, penetrance can occur with alterations to the cohesin “motor” subunit (RAD21), for genes encoding structural or scaffolding subunits (e.g., SMC1A, SMC3), and for the genes encoding associated STAG1/2 proteins. The class of hematologic “cohesinopathy” connects basic studies of chromatin looping [10, 73], with tumor biology and studies of the differentiation blockade in human malignancy (Fig. 1F,G). The genetics of cohesinopathy provide evidence for RAD21, STAG2, SMC1A, and SMC3 as driver mutations in acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and pre-leukemic states (myelodysplastic syndrome; MDS) [74].
While cohesin mutations are frequently loss-of-function, it is of high interest to characterize rare sub-classes of cohesinopathy resulting in gain-of-function function rather than haploinsufficiency or total loss (as for X-linked STAG2, SMC1A mutations). Of note, cohesinopathy mutations have high variant allele frequency, and are considered as founder- or driver-events in leukemic tumor evolution [74,75,76]. Understanding precise mechanisms for chromatin structural dysregulation as early events in tumor evolution will be impactful. Early reports established that STAG2 or SMC3 mutations occur in MDS or de novo AML, supporting the role of cohesin loss in early, driving events in leukemogenesis [77]. With noted roles of requirements of cohesin for chromosomal organization in cell division [78], the functional consequences of early alterations in these complexes in leukemias are not mechanistically linked to significant SV or genome instability [79]. Excitingly, recent studies have also implicated STAG2 alterations in chromatin structural phenotypes in EWS [80, 81].
It is of high interest that cohesin mutations are often mutually exclusive with TP53 mutations in AML. This bears similarity to the mutually exclusive relationship between mutations in mammalian SWI/SNF and PTEN or TP53 mutations in human cancers [82]. This mutual exclusivity, or lack of cooccurrence suggests roles for cohesin complexes as major tumor suppressors [83]. To understand the precise roles of cohesin in myeloid malignancy, Levine and co-workers developed conditional alleles for subunits STAG1 or STAG2 under the control of the Mx1 promoter [84]. They observed that the loss of STAG2 protein results in the expansion of undifferentiated leukemic progenitor cells in mouse models. The authors asked questions about the dependencies on STAG2 and STAG1 for leukemic transcriptional programs, and found key dysregulated genes, despite an overall low occurrence of statistically altered gene expression. With STAG2 conditional deletion, there are fewer than 200 statistically altered transcripts, which agrees with previous reports decoupling the role of cohesin function from RNAP2 [10]. However, of the altered transcriptional targets, the overall effect is reminiscent of losses in myeloid differentiation and gains in genes associated with leukemic stemness. Integrating DNA accessibility with these findings, the authors observe losses in pioneer factor PU.1 motifs, alterations in key TAD-boundaries, and altered CTCF motif densities concomitant with STAG2 loss.
Recent studies have also implicated STAG2 loss in lower chromatin contact frequencies within TADs and loops (cf., Fig. 1F,G) [85]. In HiChIP experiments, the authors find that STAG2 loss confers decreased chromatin looping associated with loci encoding leukemia drivers. De novo or altered chromatin looping in a STAG2-deficient background induces relative upregulation of key genes within the HOXA1-HOXA7 region of the HOX gene cluster and general losses of expression of HOXA9-HOXA13. Interestingly there is evidence from several studies regarding compensatory STAG1 activity in STAG2-deficient leukemia, which might result in altered cohesin processivity and a shift from smaller chromatin domains to larger domains. Further studies will be critical to understand the compensatory roles of STAG1/2, and mechanisms of STAG2-mediated maintenance of contact domains for transcription. With recurrent cohesin mutations as drivers of altered chromatin architecture in CML, AML, and EWS, it will be of high interest to understand the commonalities and distinctions in genome structure–function relationships in these tumors.

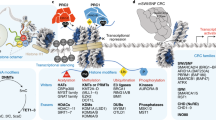
Major chromatin structural attributes of cancer. A Disease variants are associated with chromatin interactions. We illustrate non-coding mutations affecting CTCF binding sites in the context of weakened TADs, neo-TADs, and TAD boundaries. CTCF HiChIP data visualized (from [85]) with annotations for how these structural elements may be altered in tumors. B Tissue-specific pioneer transcription factors at loop anchors. We illustrate chromatin loop domains, visualized (from [85]) with CTCF HiChIP, and annotations for how transcription factors would occupy the termini of loop domains. C Structural variants can alter chromatin domains. We illustrate how deletions alter the visualization of TADs and chromatin domains. Data visualization is from IMR90 cells [6]. D Illustration of interchromosomal rearrangements revealed in in Hi-C experiments, with interactions spanning chromosome 10 and chromosome 16 from GM12878 cells, visualized from available Hi-C data [6]. E Intrachromosomal structural variation with rearrangements occurring within a chromosome, viewed from HiChIP experiments in AML cells, focused on chromosome 13 [85]. F Mammalian cohesin complexes are illustrated with the major subunits SMC1, SMC3, RAD21, STAG2 shown (left). Chromatin domains are shown in AML cells with wild-type cohesion complexes (right; chromosome 7), visualized from available data [85]. G Cohesin mutations affect chromatin interactions. Cohesin loss can occur through alterations in the major subunits, as shown (left). The result of Cohesin mutations on chromatin interactions is the substantial loss of TAD-level interactions, as visualized from available data on AML cells with STAG2 loss (right; chromosome 7, [85])
Conclusions
We have examined four major areas of architectural dysregulation in the context of human cancer. These common structural tumor drivers are (1) frequent noncoding mutations at chromatin loop anchors and domain insulators, (2) altered TF binding at sites of chromatin interaction, (3) structural variation resulting in domain redistricting, and (4) mutations in cohesin and metabolic genes, upon which chromatin structure is heavily reliant. In each case, further work will be required to establish causality of the chromatin architecture in tumorigenesis. New technology to enable sequencing of altered chromatin domains in human cancer (e.g., long-read sequencing, Hi-C, AQuA-HiChIP) and next-generation imaging of chromatin domains (e.g., 3D-STORM, ORCA) will allow for integration of 3D sequencing and microscopy to define common structural drivers. We anticipate that connections between chromatin structural alterations and patient outcomes will ultimately influence clinical decision making. For example, for low mutational burden tumors with high SV illuminated through 3D genomics, radiation therapy may not be the most efficacious strategy [86]. We look forward to many exciting advances in the coming years with increased integration of single cell imaging approaches and 3D chromatin sequencing to understand chromatin structure in cancer, and to separate cause from consequence in altered chromatin domains.
Availability of data and materials
Not applicable.
Abbreviations
- 3C:
-
Chromosome conformation capture
- RMS:
-
Rhabdomyosarcoma
- EWS:
-
Ewing sarcoma
- FP-RMS:
-
Fusion-positive rhabdomyosarcoma
- FN-RMS:
-
Fusion-negative rhabdomyosarcoma
- AML:
-
Acute myeloid leukemia
- CML:
-
Chronic myeloid leukemia
- MNase:
-
Micrococcal nuclease
- SV:
-
Structural variation
- ORCA:
-
Optical reconstruction of chromatin architecture
- cryo-EM:
-
Cryo-electron microscopy
- TF:
-
Transcription factor
- cohesinopathy:
-
Tumor resulting from mutations in genes encoding cohesin subunits
- SNP:
-
Single nucleotide polymorphism
- DMR:
-
Differentially methylated region
References
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921.
Consortium EP, Snyder MP, Gingeras TR, Moore JE, Weng Z, Gerstein MB, Ren B, Hardison RC, Stamatoyannopoulos JA, Graveley BR, et al. Perspectives on ENCODE. Nature. 2020;583(7818):693–8.
Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet. 2006;38(11):1348–54.
Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485(7398):381–5.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–93.
Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–80.
Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, Chang HY. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods. 2016;13(11):919–22.
Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, Weintraub AS, Schujiers J, Lee TI, Zhao K, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159(2):374–87.
Gryder BE, Khan J, Stanton BZ. Measurement of differential chromatin interactions with absolute quantification of architecture (AQuA-HiChIP). Nat Protoc. 2020;15(3):1209–36.
Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. Cohesin loss eliminates all loop domains. Cell. 2017;171(2):305-320 e324.
Canela A, Maman Y, Jung S, Wong N, Callen E, Day A, Kieffer-Kwon KR, Pekowska A, Zhang H, Rao SSP, et al. Genome organization drives chromosome fragility. Cell. 2017;170(3):507-521 e518.
Lai B, Tang Q, Jin W, Hu G, Wangsa D, Cui K, Stanton BZ, Ren G, Ding Y, Zhao M, et al. Trac-looping measures genome structure and chromatin accessibility. Nat Methods. 2018;15(9):741–7.
Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, De Gobbi M, Taylor S, Gibbons R, Higgs DR. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat Genet. 2014;46(2):205–12.
Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell. 2015;162(1):108–19.
Hua P, Badat M, Hanssen LLP, Hentges LD, Crump N, Downes DJ, Jeziorska DM, Oudelaar AM, Schwessinger R, Taylor S, et al. Defining genome architecture at base-pair resolution. Nature. 2021;595(7865):125.
Sunkel BD, Wang M, LaHaye S, Kelly BJ, Fitch JR, Barr FG, White P, Stanton BZ. Evidence of pioneer factor activity of an oncogenic fusion transcription factor. iScience. 2021;24(8):102867.
Boone MA, Taslim C, Crow JC, Selich-Anderson J, Byrum AK, Showpnil IA, Sunkel BD, Wang M, Stanton BZ, Theisen ER, et al. The FLI portion of EWS/FLI contributes a transcriptional regulatory function that is distinct and separable from its DNA-binding function in Ewing sarcoma. Oncogene. 2021;40(29):4759–69.
Gryder BE, Pomella S, Sayers C, Wu XS, Song Y, Chiarella AM, Bagchi S, Chou HC, Sinniah RS, Walton A, et al. Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat Genet. 2019;51(12):1714–22.
Laubscher D, Gryder BE, Sunkel BD, Andresson T, Wachtel M, Das S, Roschitzki B, Wolski W, Wu XS, Chou HC, et al. BAF complexes drive proliferation and block myogenic differentiation in fusion-positive rhabdomyosarcoma. Nat Commun. 2021;12(1):6924.
Pomella S, Sreenivas P, Gryder BE, Wang L, Milewski D, Cassandri M, Baxi K, Hensch NR, Carcarino E, Song Y, et al. Interaction between SNAI2 and MYOD enhances oncogenesis and suppresses differentiation in Fusion Negative Rhabdomyosarcoma. Nat Commun. 2021;12(1):192.
Yohe ME, Gryder BE, Shern JF, Song YK, Chou HC, Sindiri S, Mendoza A, Patidar R, Zhang X, Guha R, et al. MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci Transl Med. 2018;10(448):eaan4470.
Yan J, Qiu Y, Ribeiro Dos Santos AM, Yin Y, Li YE, Vinckier N, NariaiBenaglio NP, Raman A, Li X, et al. Systematic analysis of binding of transcription factors to noncoding variants. Nature. 2021;591(7848):147–51.
Baxter JS, Leavy OC, Dryden NH, Maguire S, Johnson N, Fedele V, Simigdala N, Martin LA, Andrews S, Wingett SW, et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nat Commun. 2018;9(1):1028.
Zaret KS. Pioneer transcription factors initiating gene network changes. Annu Rev Genet. 2020;54:367–85.
Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351(6280):1454–8.
Grubert F, Srivas R, Spacek DV, Kasowski M, Ruiz-Velasco M, Sinnott-Armstrong N, Greenside P, Narasimha A, Liu Q, Geller B, et al. Landscape of cohesin-mediated chromatin loops in the human genome. Nature. 2020;583(7818):737–43.
Iyyanki T, Zhang B, Wang Q, Hou Y, Jin Q, Xu J, Yang H, Liu T, Wang X, Song F, et al. Subtype-associated epigenomic landscape and 3D genome structure in bladder cancer. Genome Biol. 2021;22(1):105.
Yang H, Zhang H, Luan Y, Liu T, Yang W, Roberts KG, Qian MX, Zhang B, Yang W, Perez-Andreu V, et al. Noncoding genetic variation in GATA3 increases acute lymphoblastic leukemia risk through local and global changes in chromatin conformation. Nat Genet. 2022;54(2):170–9.
Tallan A, Stanton BZ. Inducible protein degradation to understand genome architecture. Biochemistry. 2021;60(31):2387–96.
Dixon JR, Xu J, Dileep V, Zhan Y, Song F, Le VT, Yardimci GG, Chakraborty A, Bann DV, Wang Y, et al. Integrative detection and analysis of structural variation in cancer genomes. Nat Genet. 2018;50(10):1388–98.
Wang X, Luan Y, Yue F. EagleC: A deep-learning framework for detecting a full range of structural variations from bulk and single-cell contact maps. Sci Adv. 2022;8(24):eabn9215.
Spielmann M, Lupianez DG, Mundlos S. Structural variation in the 3D genome. Nat Rev Genet. 2018;19(7):453–67.
Montefiori LE, Bendig S, Gu Z, Chen X, Polonen P, Ma X, Murison A, Zeng A, Garcia-Prat L, Dickerson K, et al. Enhancer hijacking drives oncogenic BCL11B expression in lineage ambiguous stem cell leukemia. Cancer Discov. 2021;11(11):2846.
Mallard C, Johnston MJ, Bobyn A, Nikolic A, Argiropoulos B, Chan JA, Guilcher GMT, Gallo M. Hi-C detects genomic structural variants in peripheral blood of pediatric leukemia patients. Cold Spring Harb Mol Case Stud. 2022;8(1):a006157.
Voronina N, Wong JKL, Hubschmann D, Hlevnjak M, Uhrig S, Heilig CE, Horak P, Kreutzfeldt S, Mock A, Stenzinger A, et al. The landscape of chromothripsis across adult cancer types. Nat Commun. 2020;11(1):2320.
Vasudevan A, Schukken KM, Sausville EL, Girish V, Adebambo OA, Sheltzer JM. Aneuploidy as a promoter and suppressor of malignant growth. Nat Rev Cancer. 2021;21(2):89–103.
Drews RM, Hernando B, Tarabichi M, Haase K, Lesluyes T, Smith PS, Morrill Gavarro L, Couturier DL, Liu L, Schneider M, et al. A pan-cancer compendium of chromosomal instability. Nature. 2022;606(7916):976–83.
Steele CD, Abbasi A, Islam SMA, Bowes AL, Khandekar A, Haase K, Hames-Fathi S, Ajayi D, Verfaillie A, Dhami P, et al. Signatures of copy number alterations in human cancer. Nature. 2022;606(7916):984–91.
Wang J, Huang TY, Hou Y, Bartom E, Lu X, Shilatifard A, Yue F, Saratsis A. Epigenomic landscape and 3D genome structure in pediatric high-grade glioma. Sci Adv. 2021;7(23):eabg4126.
Sungalee S, Liu Y, Lambuta RA, Katanayeva N, Donaldson Collier M, Tavernari D, Roulland S, Ciriello G, Oricchio E. Histone acetylation dynamics modulates chromatin conformation and allele-specific interactions at oncogenic loci. Nat Genet. 2021;53(5):650–62.
Wang X, Xu J, Zhang B, Hou Y, Song F, Lyu H, Yue F. Genome-wide detection of enhancer-hijacking events from chromatin interaction data in rearranged genomes. Nat Methods. 2021;18(6):661–8.
Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, Khurana E, Waszak S, Korbel JO, Haber JE, et al. Patterns of somatic structural variation in human cancer genomes. Nature. 2020;578(7793):112–21.
Akdemir KC, Le VT, Chandran S, Li Y, Verhaak RG, Beroukhim R, Campbell PJ, Chin L, Dixon JR, Futreal PA, et al. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat Genet. 2020;52(3):294–305.
Flavahan WA, Drier Y, Johnstone SE, Hemming ML, Tarjan DR, Hegazi E, Shareef SJ, Javed NM, Raut CP, Eschle BK, et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature. 2019;575(7781):229–33.
Smestad J, Erber L, Chen Y, Maher LJ 3rd. Chromatin succinylation correlates with active gene expression and is perturbed by defective TCA cycle metabolism. iScience. 2018;2:63–675.
Sulkowski PL, Oeck S, Dow J, Economos NG, Mirfakhraie L, Liu Y, Noronha K, Bao X, Li J, Shuch BM, et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature. 2020;582(7813):586–91.
Chen X, Sunkel B, Wang M, Kang S, Wang T, Gnanaprakasam JNR, Liu L, Cassel TA, Scott DA, Munoz-Cabello AM, et al. Succinate dehydrogenase/complex II is critical for metabolic and epigenetic regulation of T cell proliferation and inflammation. Sci Immunol. 2022;7(70):eabm8161.
Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, Bernstein BE. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529(7584):110–4.
Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017;169(5):930-944 e922.
Turcan S, Makarov V, Taranda J, Wang Y, Fabius AWM, Wu W, Zheng Y, El-Amine N, Haddock S, Nanjangud G, et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat Genet. 2018;50(1):62–72.
Faulkner S, Maksimovic I, David Y. A chemical field guide to histone nonenzymatic modifications. Curr Opin Chem Biol. 2021;63:180–7.
Maksimovic I, David Y. Non-enzymatic covalent modifications as a new chapter in the histone code. Trends Biochem Sci. 2021;46(9):718–30.
Maksimovic I, Zheng Q, Trujillo MN, Galligan JJ, David Y. An azidoribose probe to track ketoamine adducts in histone ribose glycation. J Am Chem Soc. 2020;142(22):9999–10007.
Simithy J, Sidoli S, Yuan ZF, Coradin M, Bhanu NV, Marchione DM, Klein BJ, Bazilevsky GA, McCullough CE, Magin RS, et al. Characterization of histone acylations links chromatin modifications with metabolism. Nat Commun. 2017;8(1):1141.
Galligan JJ, Rose KL, Beavers WN, Hill S, Tallman KA, Tansey WP, Marnett LJ. Stable histone adduction by 4-oxo-2-nonenal: a potential link between oxidative stress and epigenetics. J Am Chem Soc. 2014;136(34):11864–6.
Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519–30.
Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative stress in cancer. Cancer Cell. 2020;38(2):167–97.
Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu CT, Zhuang X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016;529(7586):418–22.
Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, Zhuang X. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science. 2018;362(6413):eaau1783.
Miron E, Oldenkamp R, Brown JM, Pinto DMS, Xu CS, Faria AR, Shaban HA, Rhodes JDP, Innocent C, de Ornellas S, et al. Chromatin arranges in chains of mesoscale domains with nanoscale functional topography independent of cohesin. Sci Adv. 2020;6(39):eaba8811.
Szabo Q, Donjon A, Jerkovic I, Papadopoulos GL, Cheutin T, Bonev B, Nora EP, Bruneau BG, Bantignies F, Cavalli G. Regulation of single-cell genome organization into TADs and chromatin nanodomains. Nat Genet. 2020;52(11):1151–7.
Mateo LJ, Murphy SE, Hafner A, Cinquini IS, Walker CA, Boettiger AN. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature. 2019;568(7750):49–54.
Schoenfelder S, Sugar R, Dimond A, Javierre BM, Armstrong H, Mifsud B, Dimitrova E, Matheson L, Tavares-Cadete F, Furlan-Magaril M, et al. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat Genet. 2015;47(10):1179–86.
Rhodes JDP, Feldmann A, Hernandez-Rodriguez B, Diaz N, Brown JM, Fursova NA, Blackledge NP, Prathapan P, Dobrinic P, Huseyin MK, et al. Cohesin disrupts polycomb-dependent chromosome interactions in embryonic stem cells. Cell Rep. 2020;30(3):820-835 e810.
Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, Reinberg D. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science. 2015;347(6225):1017–21.
Weber CM, Hafner A, Kirkland JG, Braun SMG, Stanton BZ, Boettiger AN, Crabtree GR. mSWI/SNF promotes Polycomb repression both directly and through genome-wide redistribution. Nat Struct Mol Biol. 2021;28(6):501–11.
Xu J, Ma H, Ma H, Jiang W, Mela CA, Duan M, Zhao S, Gao C, Hahm ER, Lardo SM, et al. Super-resolution imaging reveals the evolution of higher-order chromatin folding in early carcinogenesis. Nat Commun. 2020;11(1):1899.
Cooper S, Dienstbier M, Hassan R, Schermelleh L, Sharif J, Blackledge NP, De Marco V, Elderkin S, Koseki H, Klose R, et al. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep. 2014;7(5):1456–70.
Tan L, Xing D, Chang CH, Li H, Xie XS. Three-dimensional genome structures of single diploid human cells. Science. 2018;361(6405):924–8.
Ramani V, Deng X, Qiu R, Gunderson KL, Steemers FJ, Disteche CM, Noble WS, Duan Z, Shendure J. Massively multiplex single-cell Hi-C. Nat Methods. 2017;14(3):263–6.
Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502(7469):59–64.
Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74.
Vian L, Pekowska A, Rao SSP, Kieffer-Kwon KR, Jung S, Baranello L, Huang SC, El Khattabi L, Dose M, Pruett N, et al. The energetics and physiological impact of cohesin extrusion. Cell. 2018;173(5):1165-1178 e1120.
Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, Yoshida K, Okuno Y, Bando M, Nakato R, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet. 2013;45(10):1232–7.
Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A. 2014;111(7):2548–53.
Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–10.
Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, Larson DE, McLellan MD, Dooling D, Abbott R, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366(12):1090–8.
Gruber S, Arumugam P, Katou Y, Kuglitsch D, Helmhart W, Shirahige K, Nasmyth K. Evidence that loading of cohesin onto chromosomes involves opening of its SMC hinge. Cell. 2006;127(3):523–37.
Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–78.
Adane B, Alexe G, Seong BKA, Lu D, Hwang EE, Hnisz D, Lareau CA, Ross L, Lin S, Dela Cruz FS, et al. STAG2 loss rewires oncogenic and developmental programs to promote metastasis in Ewing sarcoma. Cancer Cell. 2021;39(6):827-844 e810.
Surdez D, Zaidi S, Grossetete S, Laud-Duval K, Ferre AS, Mous L, Vourc’h T, Tirode F, Pierron G, Raynal V, et al. STAG2 mutations alter CTCF-anchored loop extrusion, reduce cis-regulatory interactions and EWSR1-FLI1 activity in Ewing sarcoma. Cancer Cell. 2021;39(6):810-826 e819.
Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45(6):592–601.
Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, Miller CA, Niu B, McLellan MD, Dees ND, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013;27(6):1275–82.
Viny AD, Bowman RL, Liu Y, Lavallee VP, Eisman SE, Xiao W, Durham BH, Navitski A, Park J, Braunstein S, et al. Cohesin members Stag1 and stag2 display distinct roles in chromatin accessibility and topological control of HSC self-renewal and differentiation. Cell Stem Cell. 2019;25(5):682-696 e688.
Smith JS, Lappin KM, Craig SG, Liberante FG, Crean CM, McDade SS, Thompson A, Mills KI, Savage KI. Chronic loss of STAG2 leads to altered chromatin structure contributing to de-regulated transcription in AML. J Transl Med. 2020;18(1):339.
Thariat J, Chevalier F, Orbach D, Ollivier L, Marcy PY, Corradini N, Beddok A, Foray N, Bougeard G. Avoidance or adaptation of radiotherapy in patients with cancer with Li-Fraumeni and heritable TP53-related cancer syndromes. Lancet Oncol. 2021;22(12):e562–74.
Acknowledgements
We would like to apologize if space constraints limited our ability to discuss any contributions to this exciting area. We thank all members of Stanton lab for helpful discussion, and special thanks also to Robert and Pamela Wolf. We acknowledge the AWRI Graphics Support Resource for assistance with the figures. We are grateful to funding from the St. Baldrick’s Foundation (B.Z.S.), The Mark Foundation for Cancer Research (B.Z.S.), the Andrew McDonough B+ Foundation (B.Z.S.), CancerFree Kids Foundation (B.Z.S.), and Nationwide Children’s Hospital for support.
Funding
St. Baldrick’s Foundation (B.Z.S.), The Mark Foundation for Cancer Research (B.Z.S.), the Andrew McDonough B + Foundation (B.Z.S.), CancerFree Kids Foundation (B.Z.S.), and Nationwide Children’s Hospital. We are grateful for each of these funding sources for contributing to the fundamental concept of the study and the driving question of whether altered chromatin structure can drive tumorigenesis.
Author information
Authors and Affiliations
Contributions
B.Z.S., M.W., and B.D.S. conceived of the conceptual basis for this Review. B.Z.S., M.W., and WCR wrote and edited the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
There are no competing interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, M., Sunkel, B.D., Ray, W.C. et al. Chromatin structure in cancer. BMC Mol and Cell Biol 23, 35 (2022). https://doi.org/10.1186/s12860-022-00433-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12860-022-00433-6