
Abstract
Background
The human Obg-like ATPase 1 (OLA1) protein has been reported to play an important role in cancer cell proliferation. The molecular mechanism underlying OLA1 regulated oral metastasis is still unknown. We investigated in this study the regulatory role of OLA1 playing in oral squamous cell metastasis.
Results
A series of in vitro assays were performed in the cells with RNAi-mediated knockdown or overexpression to expound the regulatory function of OLA1 in oral cancer. We found that the endogenous level of OLA1 in a highly metastatic oral squamous cell line was significantly lower than that in low metastatic oral cells as well as in normal oral cells. Escalated expression of OLA1 resulted in a reduced ability of metastasis in highly metastatic cells, and enhanced its sensitivity to the paclitaxel treatment. Further analysis of the EMT markers showed that Snail, Slug, N-cadherin were up-expressed significantly. Meanwhile, E-cadherin was significantly down-regulated in the oral cancer cells with OLA1-knocked down, suggesting that OLA1 inactivated EMT process. Furthermore, we found that OLA1 suppressed oral squamous cell metastasis by suppressing the activity of a TGFβ/SMAD2/EMT pathway.
Conclusion
Our data suggests that OLA1 may be developed as a potential target for the treatment of oral cancer metastasis.
Similar content being viewed by others


Background
Oral squamous cell carcinoma (OSCC) is the leading cause of morbidity and mortality in patients with head and neck squamous cell cancer (HNSC). The five-year survival rate for Oral cell carcinoma (OCC) is only about 50% [1, 2]. While, OSCC is a typical oral malignancy ranking at the sixth among all cancer death worldwide, and accounts for 90% of the incidence of OCC [3]. Although the incidence rate has decreased, the outcome has stagnated, and overall survival has increased by only 5% over the past 20 years. An epidemiological study showed that tobacco smoking [4,5,6], alcohol consumption [7, 8] and prevalence of human papilloma virus (HPV) [9] are the major risk factors. In addition, despite deep understanding of surgical techniques, adjuvant therapy, and molecular mechanisms of pathogenesis, the prognosis for patients with advanced cancer or the elderly remains poor [10, 11]. Slightly more than half of the patients were diagnosed with regional or metastatic disease, especially a propensity for lymph node metastasis at diagnosis [12, 13]. Lymph node metastasis has the most significant impact on survival, reducing survival by 50% [11]. Independent of lymph node metastasis, the occurrence of extracapsular spread (ECS) heralds a worse failure rate for regional and distant metastases [14]. The exact molecular mechanism of the pathogenesis of OSCC is not precise yet. It is unmet to understand the molecular mechanism of oral cancer metastasis to develop an effective clinical strategy for therapy of oral cancer.
Increasing evidence indicates that epithelial-mesenchymal transition (EMT) plays a crucial role in the metastasis of cancer [15,16,17,18,19]. Through EMT, epithelial cells losing cell polarity cause morphological alterations and acquire higher interstitial cell phenotypes, such as migration and invasion [20, 21]. EMT involves a series of molecular regulation, including the down-regulation of epithelial cell markers (E-cadherin), transmembrane protein (N-cadherin), transcription factors (Snail, Slug, Zeb-1) [22, 23]. EMT is an important biological process of malignant cell migration and invasion of epithelial origin.
The human Obg-like ATPase 1 (OLA1) is a 45 kDa protein in size and is widely present in the cytoplasm and expressed in most tissues [24]. OLA1 belongs to the YchF subfamily of the Obg-like GTPase family. It has recently been found that YchF homologous proteins have higher binding and hydrolysis efficiencies to ATP than that to GTP [25]. Structural analysis suggests that it is a regulatory protein binding to its downstream effector and exerting its regulatory function through its conformational transition [26]. OLA1 is essential for normal mammalian development and plays a vital role in regulating cell proliferation, partially by inhibiting p21 and organogenesis [27]. Chen et al. showed that OLA1 might be a novel translational GTPase that plays an important role in translation and cell survival as well as cancer development [28]. Zhang et al. showed that OLA1 may play an essential role in promoting breast cancer metastasis [29]. While, Jeyabal et al. reported that OLA1 could inhibit breast cancer cell adhesion and spread in breast cancer [30]. Knockdown of OLA1 in lung adenocarcinoma cells can attenuate the TGF-β-induced EMT process and restore E-cadherin expression [31]. The results of OLA1 expression and functions analysis in metastasis were from different cancer types, which may be the reason for the inconsistency. In summary, the exact regulatory role of OLA1 in cancer is still unknown. Our data in this study showed that OLA1 suppressed oral squamous cell metastasis by inhibition of SMAD2-EMT pathway, suggesting that OLA1 may be developed as a therapeutic target for oral metastasis.
Results
The endogenous level of OLA1 in OSCC and oral cell lines
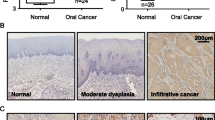
To understand the endogenous levels of OLA1 in oral normal and its paired tumor tissues, the OLA1 RNA sequencing data from Gene Expression Profiling Interactive Analysis (GEPIA) was analyzed [32]. We found that OLA1 is highly expressed in most of cancer, and have no difference between Head and Neck squamous cell carcinoma (HNSC) and its paired normal tissues (Figs. 1a, S1). To further investigate the OLA1 expression in OSCC, we analyzed gene expression omnibus (GEO) Datasets (GSE140707) with three tumorous and adjacent tissues from OSCC sufferers and found that OLA1 mRNA was downregulated in tumors (Fig. 1b). Immunohistochemical analysis in clinical oral tumor tissues further proved that OLA1 expression was also lower in tumors than the adjacent tissues (Fig. 1c). In order to investigate the role of OLA1 in oral cancer cells, five oral squamous cell lines were chosen to detect the endogenous level of OLA1. Our results also validated that OLA1 mRNA had no significant difference in five OSCC cell lines (Fig. 1d). We speculated that OLA1 might undergo post-translational modification. Therefore, we performed SUMO (small ubiquitin-related modifier) modification, but the results were negative. Interestingly, the endogenous level of the OLA1 protein in oral cancer cell lines was also significantly lower than that in normal oral cells, as shown in Fig. 1e. To understand whether OLA1 may be associated with oral cancer metastasis, the endogenous level of OLA1 in metastatic oral cancer cell line was analyzed. We found that OLA1 expression in metastasis cell line UM-1 was significantly lower than the carcinoma in situ cell line UM-2, suggesting a negative role OLA1 playing in oral cancer metastasis. To study the effect of OLA1 on the proliferation of oral cancer cells, silenced OLA1 assays were performed (Fig. 1f) and found that there was no significant effect observed on oral cancer cell proliferation (Fig. 1g, h), which is consistent with another report [28].

The endogenous level of OLA1 in OSCC and oral cell lines. a The average expression level of OLA1 in patients HNSC in TCGA and GTEx oral cancer dataset. T = Tumor, N = Normal, num = Numbers. b OLA1 RPKM in OSCC from GEO140707, N = 3 paried tissues. c OLA1 expression in clinical OSCC (Red arrow) and the adjacent tissues (Black arrow) n = 5. mRNA (d) and protein (e) level in five oral cell lines. f OLA1 expression in OLA1-siRNA cells. Top panel: Fluorescent photos of cells transfected with OLA1 siRNA for 24 h. Middle panel: OLA1 protein expression in indicated cells. Bottom panel: OLA1 mRNA expression in indicated cells. g and h Cell proliferation curve was drawn in indicated cells. vs. Control, *P < 0.05, ns = no significance
Dysregulation of OLA1 affected the ability of metastasis in oral cancer cells
To determine whether OLA1 can regulate the strength of metastasis in oral cancer cells, OLA1 activity was silenced by small interfering OLA1 RNA (siR-OLA1). We found that the wound healing ability of oral cancer cells was much higher in OLA1 silenced oral cells as compared to control (Fig. 2a). The metastatic ability of oral cancer cells was also much higher in the OLA1 silenced oral cancer cells than the control cells (Fig. 2b). These data indicated that knocked-down OLA1 in UM-1 and UM-2 enhanced cell migrative ability. To further characterize the regulatory role of OLA1 in oral cancer metastasis, two oral cancer cell lines were established with either OLA1-overexpressed (OLA1OE) in UM-1, or OLA1 knocked down in UM-2 (shOLA1) (Fig. 2c). UM-1 OLA1OE cells showed a glomerate growth morphologically, while UM-2 shOLA1 cells showed an elongated fibroblast-like morphology (Fig. 2d). This phenomenon was coincident with the initial stage of the EMT process. Invasion and metastasis of oral cancer cells were also evaluated by Transwell and wound healing assays, respectively. The wound healing rate in the UM-1 OLA1OE cells was slower than control (Fig. 2e), and the numbers of the metastatic OLA1OE cells were less than the Vector cells (Fig. 2f). The results in the UM-2 shOLA1 cells showed the opposite way to that in the UM-1 OLA1OE cells. These data suggested that OLA1 might play a negative role in the metastasis of oral cancer.

Dysregulation of OLA1 affected the ability of metastasis in oral cancer cells. a Wound healing assay to examine the effect of down regulating OLA1 on UM-1 and UM-2 cells (n = 3). b Transwell assay to investigate the impact of downregulating OLA1 on metastatic ability in UM-1 and UM-2 cells (n = 3). c Construction of stable cell lines with UM-1 (YFP-OLA1OE) plasmid or lentiviral vector in UM-2 (GFP-shOLA1). Top panel: Fluorescent photos of indicated cells. Bottom panel: OLA1 expression in relatived cells. d Morphology of cells (200X) after OLA1 plasmid transfection or TGFβ1 (1 ng/mL) induction. e Wound healing assay to examine the effect of upregulating OLA1 on UM-1 and downregulating OLA1 on UM-2 cells (n = 3). f Transwell assay to examine the effect of upregulating OLA1 on UM-1 and downregulating OLA1 on UM-2 cells (n = 3). vs. Control, *P < 0.05, **P < 0.01, ***P < 0.001
OLA1 regulates oral cancer metastasis through TGFβ/Smad2/Smad4 mediated EMT pathway
EMT is a biological transformation process that can make polarized epithelial cells gradually present a mesenchymal cell phenotype, which is manifested in enhanced migration ability, invasiveness, and enhanced anti-apoptotic ability. To understand the molecular mechanism underlying OLA1-mediated metastasis of oral cancer cells, biomarkers in the EMT process were analyzed. The results showed that decreased expression of OLA1 in the UM-1 siR-OLA1 or UM-2 shOLA1 cells increased the expression of CDH2 (N-cadherin), SNAI1 (Snail), SNAI2 (Slug), and VIM (**P < 0.01, ***P < 0.001), and decremental CDH1 (E-cadherin) (Fig. 3a, b). On the other hand, expression of SNAI1(Snail) and SNAI2 (Slug), was significantly decreased in UM-1-OLA1OE cells, and increased CDH1 (*P < 0.05, ***P < 0.001) (Fig. 3c). Western Blotting assay showed that escalated OLA1 expression in UM-1 OLA1OE cells decreased N-cadherin, Snail, Slug, and incremental E-cadherin. While in UM-2 shOLA1 cell had the contrary results (Fig. 3g), suggesting that OLA1 might regulate EMT process negatively. To further investigate the molecular mechanisms of OLA1 in the regulation of EMT resulting in metastasis of OSCC, biomarkers in TGF-β/Smad pathway including TGFB1, TGFB2, SMAD2 and SMAD4 were characterized in the following cell lines UM-1, UM-1-siR-OLA1,UM-1-OLA1OE, UM-2 and UM-2-shOLA1 cells. The results showed that OLA1 knockdown increased TGFB1, SMAD2 and SMAD4 expression (Fig. 3d, e). Inversely, OLA1 overexpression decreased TGFB1 and SMAD4 expression (Fig. 3f). Western Blotting verified the results of mRNA level of TGF/SMAD axis markers. Meanwhile, knocking down OLA1 could promote the phosphorylation level of SMAD2 to active TGFβ/SMAD pathway, indicating that TGF-β/Smad was inhibited in UM-1-OLA1OE cells respectively (P < 0.05) (Fig. 3h). In order to further prove that OLA1 regulates the TGFβ/SMAD2 pathway, cellular morphology was obtained after treated with TGFβ(1 ng/mL) in UM-1 OLA1OE cells or transfected siR-SMAD2 in UM-2 shOLA1 cells. TGFβ reactivated the mesenchymal phenotype of UM-1 OLA1OE cells, while silencing SMDA2 reversed the mesenchymal phenotype of UM-2 shOLA1 cells (Fig. 2d). Furtherly, UM-2 shOLA1 siR-SMAD2 cells were constructed to study the role of SMAD2 in OLA1 involvement in tumor migration. Western Blotting assay showed that we successfully silenced SMAD2 in UM-2 shOLA1 cells (Fig. 3i). Western blotting assay showed that silenced SMAD2 increased OLA1 expression in UM-2 cells (Fig. 3j). Wound healing test showed that SMAD2 silencing could rescue enhanced metastasis induced by knocking down OLA1 in UM-2 cells (Fig. 3k). Similarly, Transwell assay showed that, the number of migrating cells was reinhibited again after silenced SMAD2 in UM-2 shNC or shOLA1 cells, and back to the UM-2 shNC level or even lower (Fig. 3l). These results indicated that OLA1 overexpression weakened the EMT phenotype through the inhibition of the TGF-β/SMAD2/SMAD4 pathway in oral cancer cell lines.

OLA1 regulates oral cancer metastasis through Smad2/Smad4 mediated EMT pathway. a mRNA fold change level of EMT markers between UM-1 siR-OLA1 cell and siR-NC cell. mRNA fold change level of EMT marker in UM-2 OLA1 knock down cells (b) and UM-1 OLA1 overexpression cells (c). d mRNA fold change level of TGFβ/SMDA2 pathway markers between UM-1 siR-OLA1 cells and siR-NC cells. mRNA fold change level of TGFβ/SMDA2 pathway markers in UM-2 OLA1 knock down cells (e) and UM-1 OLA1 overexpression cells (f). g Protein level of EMT marker in UM-1 OLA1 overexpression cells and UM-2 OLA1 knock down cells. h Protein level of TGFβ/SMDA2 pathway markers in UM-1 OLA1 overexpression cells and UM-2 OLA1 knock down cells. i Western blotting assay showed SMAD2 was silenced by siR-SMAD2 in UM-2 shOLA1 cells. j Western blotting assay showed that silenced SMAD2 decreased OLA1 expression in UM-2 cells. k Wound healing assay showed SMAD2 silenced could rescue enhanced metastasis induced by knocking down OLA1 in UM-2 cells (n = 3). l Transwell assay to examine the effect of OLA1 on metastasis by smad2 in UM-2 cells (n = 3). vs. Control, *P < 0.05, **P < 0.01, ***P < 0.001
Reduced OLA1 expression is related to paclitaxel-induced cancer cell metastasis
It has been reported in the literature that EMT transformation of cells does not necessarily lead to the occurrence of metastasis, but it does affect the chemoresistance of tumors [33]. This article also investigated whether the OLA1 medicated EMT process leading to tumor metastasis is related to tumor drug resistance. Some chemotherapeutic drugs, while killing cancer cells, also promote the metastasis of cancer cells [34,35,36]. To characterize the regulatory effect of OLA1 on the oral cancer metastasis induced by paclitaxel (PTX), UM-1 cells were treated with PTX (100 nM) in a time course of 0–8 days. We found that the increased concentration of the drug suppressed the expression of OLA1 (Fig. 4a). Besides, overexpression of OLA1 can increase sensitivity to PTX in UM-1 cells (Fig. 4b, c). These evidences indicated that OLA1 may increase the sensitivity of cells to drugs by reducing the migration of oral cancer cells, thus playing an anti-tumor role. Flow cytometry was used to detect the necrosis and apoptosis of cells after incubation with 20 μM PTX for 24 h. UM-1-OLA1OE cells increased the late apoptosis rate of the cells and the overall mortality rate after treated with PTX. Some of the early apoptotic cells turned to necrosis instead of late apoptosis (Fig. 4d). It showed that OLA1 could increase the sensitivity of oral cancer cells to PTX. The results are consistent with the MTT experiment. Similarly, Knockdown of OLA1 greatly decreased the apoptosis of UM-2 cells by PTX. However, the continued silence of SMAD2 promoted the killing of UM-2 shOLA1 cells by PTX (Fig. 4e). These results indicate that OLA1 enhances the sensitivity of cells to drugs by inhibiting the TGF/Smad axis.

Reduced OLA1 expression is related to paclitaxel-induced cancer cell metastasis. a The OLA1 expression in UM-1 cell treated with 100 nM PTX for 0–8 days. b and c Cell viability assay showed that OLA1 overexpression enhanced the PTX sensitivity in UM-1 cells (n = 3). d Cell flow cytometry was used to determine the effect of overexpression of OLA1 on paclitaxel-induced (20 μM) cell necrosis and apoptosis (n = 2). e Cell flow cytometry showed the effect of OLA1 on paclitaxel-induced (20 μM) cell necrosis and apoptosis through SMAD2 (n = 2). f Mechanism of OLA1 inhibits metastasis of OSCC. vs. Control, *P < 0.05, **P < 0.01, ***P < 0.001
Mechanism of OLA1 inhibits metastasis of OSCC
OLA1 is a metabolic enzyme that has both ATPase and GTPase activity, and it participates in many important physiological processes. In this article, we found that OLA1 inhibits the metastasis of OSCC cells through the TGFβ/SMAD2/SMAD4 axis (Fig. 4f). Briefly, OLA1 inhibits the expression of TGFβ1, thereby inhibiting the phosphorylation of SMAD2, resulting in a decrease in the expression of SMAD4, inhibiting the expression of Snail and Slug, the important transcription factors of EMT, increasing E-cadherin, and achieving inhibition of the EMT pathway, which ultimately leads to a decrease in the metastatic ability of tumor cells.
Discussion
The expected curative rates for one-third of patients with OSCC diagnosed at stage I / II is about 80% (stage I) and 65% (stage II), respectively [37]. However, most cases of OSCC are diagnosed as an advanced disease (III or IV) with the 5-year survival rate of less than 50% [38], and a curative rate of 30% [37]. The survival rate of patients with metastatic disease is about 4 months [39]. Oral metastasis remains still a bottleneck for therapy of oral cancer. Previously, although there are a few studies showing the involvement of OLA1 in tumor metastasis, the exact role of OLA1 in oral cancer metastasis is not known yet.
Normal human oral keratinocytes (NHOKs) is normal oral cell line. UM-1 and UM-2 are oral cancer cell lines from the same patient, with the difference that UM-1 is a metastatic cell line, and UM-2 is an in-situ cell line. Cal-27 is squamous cell carcinoma with a lesion of the middle of the tongue, and poorly or undifferentiated. Our work indicated that OLA1 expression in oral cancer cells was significantly lower than that of normal oral cells, and decreased as the degree of cancer metastasis increases. When we knocked down or elevated the level of OLA1 in oral cancer cells UM-1 and UM-2, the phenotype of the cells changed significantly with a more EMT/MET (mesenchymal epithelial transition) status. It was further demonstrated by the scratch test and the Transwell experiment that OLA1 could substantially inhibit the metastasis and invasion of oral cancer.
EMT is an important mechanism of tumor metastasis. EMT is commonly found in cancer cells and provides a mechanism for transendothelial migration. In this article, we reported that OLA1 participates in the EMT process through the TGFβ/SMAD axis, which in turn affects tumor metastasis.
Blocking EMT may not directly affect cell death, but it can affect cancer cell resistance [33, 40, 41]. Although the imbalance/increase of proliferation is essential for tumor formation and growth, it is not the case for malignant tumors. For example, Snail zinc finger transcription factors trigger EMT, which confers epithelial cells migration and invasion characteristics during embryonic development and tumor progression. In addition to inducing phenotypic changes of tumor cells, Snail can also weaken the cell cycle and confer resistance to cell death induced by cell necrosis stimulating factors and pro-apoptotic signals. The resistance to cell death conferred by Snail provides a selective advantage for embryonic cells to migrate and settle in distant territories, which separates malignant cells from the primary tumor, invades and forms metastasis [42]. Elevated E-cadherin enhances the sensitivity of tumor cells to EGFR kinase inhibitors, and resistant cells are more like mesenchymal-like cells [43]. It is a well-known fact that TGFβ can induce EMT [44, 45]. There are also many reports about the involvement of TGFβ in tumor resistance [46, 47]. SMAD, as one of the important ways for TGFβ to induce EMT, has also been reported to be involved in tumor chemoresistance [48].
In addition, some chemotherapeutic drugs, while killing cancer cells, also promote the metastasis of cancer cells [34,35,36]. For example, Karagiannis et al. found that PTX could change tumor metastasis microenvironment and promote breast cancer metastasis [35]. Recently, research by Keklikoglou found that the commonly used chemotherapy drugs PTX and doxorubicin can promote the release of exosomes by tumors, change the microenvironment in the lung, and promote lung metastasis in breast cancer [36]. PTX, like cisplatin, 5-fluorouracil, and carboplatin, can be used as oral chemotherapy drugs [49]. We found that overexpression of OLA1 could increase the sensitivity of cells to PTX. Therefore, we speculate that OLA1 enhanced the sensitivity of cells to drugs by inhibiting the TGF/smad axis and EMT process. In order to verify our conjecture, we silenced SMAD2 expression in UM-2 shOLA1 cells, and the killing effect of PTX on the cells was investigated by a flow cytometry assay. Knockdown of OLA1 greatly decreased the apoptosis of UM-2 cells by PTX. However, the continued silence of SMAD2 promoted the killing of UM-2 shOLA1 cells by PTX. The experimental results show that OLA1 enhances the sensitivity of cells to PTX by inhibiting the process of TGF/SMAD and EMT. Therefore, we report that OLA1 can inhibit the EMT process induced by TGFβ/SMAD2, thereby increasing the drug sensitivity of oral cancer cells to PTX.
Conclusion
We observed that OLA1 may play a negative role in the metastasis and invasion of oral cancer and this effect may be achieved through the EMT pathway, which plays a vital role in the diagnosis and treatment of clinical oral cancer. However, doubtless further investigations, including in vivo animal model studies and prospective clinical observations, are implemented. We tried to make an orthotopic oral cancer metastasis model to verify the inhibitory effect of OLA1 on oral cancer metastasis by inoculating human oral cancer metastasis cell line UM-1 in nude mice. However, we failed to make an oral cancer metastasis model in situ, which is not as simply as in subcutaneou transplantation tumor or liver metastasis model. Therefore, we plan to use OLA1(−/−) systemic knockout mice to further study the function of OLA1 in inhibiting oral cancer metastasis using a drug-induced oral carcinoma in situ in the future.
Methods
Chemicals and reagents
DMEM-F12 medium and foetal bovine serum (FBS) were purchased from the Gibco (Invitrogen, USA). Penicillin, streptomycin and BCA protein assay kits were purchased from the Solarbio (Beijing, China). Polystyrene plate for transwell was purchased from the Corning (New York, USA). The primary antibodies were diluted 1:2000 before use, including OLA1 (Cat. #ab229090, Abcam), GAPDH (Cat. #ab9485, Abcam), GAPDH (Cat. #ab8245, Abcam), N-cadherin (Cat. #ab18203, Abcam), E-cadherin (Cat. #ab76055, Abcam), Snail (Cat. #PA5–11923, Invitrogen), Snail + Slug (Cat. # ab180714, Abcam), Zeb-1 (Cat. # ab155249 Abcam) and SMAD2 (Cat. #5339, Cell Signaling Technology), p-SMAD2 (Cat. #18338, Cell Signaling Technology), SMAD4 (Cat. #9515P, Cell Signaling Technology). TGF-beta 1 antibody (Cat. # 21898–1-AP) was purchased from ProteinTech (Wuhan, China). All the chemical compounds were analytically pure reagents.
Cells and cell culture
The human OSCC cell lines UM-1 and UM-2 were donated by Prof. Hu (UCLA, USA). HN4, NHOKS and CAL-27 were purchased from the American Type Culture Collection (ATCC). Cells were cultured in a 1:1 mixture of Dulbecco’s modified Eagle medium and Nutrient Mixture F-12 medium with 10% FBS, 100 IU/mL penicillin and 100 μg/ml streptomycin in 37 °C, 5% CO2 incubator.
OSCC tissue HE staining and immunohistochemistry
We obtained five tissues diagnosed as oral squamous cell carcinoma by pathology from Dalian Stomatological Hospital. The protocols for collection and analysis of these tissues and experiments about processing and using of tissue samples was approved by the Biology and Medical Ethics Committee of Dalian University of Technology and have obtained the patients signed informed consent. Hematoxylin-eosin (HE) staining is used to judge the location of tumor tissue, and immunohistochemistry is used to evaluate the expression of OLA1 (1:200) in tumor tissue.
Small interfering RNA transfections
Small interfering RNA (siRNA) for OLA1 (siR-OLA1) marked with FAM Probe, small interfering RNA for SMAD2 (siR-SMAD2, Sense: 5′-GUCCCAUGAAAAGACUUAATT-3′, Anti-sense: 5′-UUAAGUCUUUUCAUGGGACTT-3′) and the control siRNA (Sense: 5′-UUCUCCGAACGUGUCACGUTT-3′, Anti-sense: 5′-ACGUGACACGUUCGGAGAATT-3′) were purchased from GenePharma (Shanghai, China). Cells seeded in a 6-well plate were transiently transfected with 100 pM siRNA with the Lipofectamine 2000 Transfection Reagent (Thermo Scientific) according to manufacturer’s instructions for 24 h.
Establishment of the stable OLA1 knockdown UM-2 cell lines
Small hairpin RNA (shRNA) lentiviral used for stable silencing of OLA1 (shOLA1) and the control non-targeting plasmid (shNC) were purchased from GenePharma (Shanghai, China) by inserting the following short-hairpin sequences into the pGLV3/H1/GFP/Puro vector: 5′-CCGGGAGGAAATGATTGGGCCCATTCTCGAGAATGGGCCCAATCATTTCCTCTTTTTTG-3′ for sh-OLA1 and 5′-CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTTG-3′ for Small hairpin (sh) control. Small hairpin RNA (shRNA) transfections and protocol were followed the recommendations by GenePharma (Shanghai, China). The shNC and shOLA1 vectors were transfected into UM-2 cells. The knockdown efficiency of the target gene was verified by fluorescence imaging, qRT-PCR and western blot analysis.
Construction of OLA1 stable overexpression cell line
OLA1-YFP plasmid (pdEYFP-N1gen plasmid with a C-terminal YFP tag, OLA1OE) was donated by Prof. Zhengzheng Shi. UM-1 Cells in 6-well plates were transfected with the related plasmid (4 μg DNA/well) or the control plasmid using Lipofectamine 2000 (8 μl/well, Invitrogen). The overexpression efficiency of the target gene was verified by qRT-PCR and western blot analysis.
Cell viability assay
To investigate the effect of knocking down OLA1 on the proliferation ability of UM-1 and UM-2 cells, the growth curves of cells at 24 h, 48 h, 72 h and 96 h after transfection with siR-OLA1 were drawn. MTT(3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide) assay was also used to determine the viability of the treated cells. 6 × 103 cells were seeded onto 96-well plates and incubated overnight at 37 °C. PTX at different concentrations (0, 0.39, 0.78, 1.56, 3.13, 6.25, 12.5, 25.0, 50.0, 100.0 μM) were added to each well with different incubation times. The absorbance was measured at 490 nm by the microplate reader. Six replicate wells were included in each analysis, and at least three independent experiments were conducted.
qRT-PCR analysis
Total RNA was extracted from the cells using TRIzol (Cat. #15596026, Invitrogen, CA, USA), and the concentration and quality were determined by a microplate reader (DU730, Beckman, CA, USA). The nucleotides were reverse-transcribed into cDNA according to the instructions of the PrimeScript RT Reagent Kit (Cat. #RR037A, Takara, Japan). After amplification and dilution, the PCR assay was performed on the LightCycler480 II (Roche, USA). Gene primer as follows: GAPDH (glyceraldehyde 3-phosphate dehydrogenase): Forward primer: 5-CATGAGAAGTATGACAACAGCCT, Reverse primer: 5-AGTCCTTCCACGATACCAAAGT; OLA1: Forward primer: 5-TGGACAAGTATGACCCAGGT, Reverse primer: 5-GCTGCAAACCCAGCCTTAATG; CDH1: Forward primer: 5-CGAGAGCTACACGTTCACGG, Reverse primer: 5-GGGTGTCGAGGGAAAAATAGG; CDH2: Forward primer: 5-AGGCTTCTGGTGAAATCGCA, Reverse primer: 5-TGCAGTTGCTAAACTTCACATTG; SNAI1: Forward primer: 5-ACTGCAACAAGGAATACCTCAG, Reverse primer: 5-GCACTGGTACTTCTTGACATCTG;
SNAI2: Forward primer: 5-CACACGGGGGAGAAGCCTTT, Reverse primer: 5-ATTGCGTCACTCAGTGTGCT; VIM: Forward primer: 5-AGGCAAAGCAGGAGTCCACTGA, Reverse primer: 5-ATCTGGCGTTCCAGGGACTCAT; TGFB1: Forward primer: 5-GCAAGTGGACATCAACGGGT, Reverse primer: 5-TCCGTGGAGCTGAAGCAATA; TGFB2: Forward primer: 5-GGTACCTTGATGCCATCCCGCC, Reverse primer: 5-GCACTCTGGCTTTTGGGTTCTGCA; SMAD2: Forward primer: 5-CCGACACACCGAGATCCTAAC, Reverse primer: 5-GAGGTGGCGTTTCTGGAATATAA; SMAD4: Forward primer: 5-GGTTCCTTCAAGCTGCCCTA, Reverse primer: 5-ATGTGCAACCTTGCTCTCTCA;
Western blot analysis
Proteins were extracted from cells using 200 μL RIPA lysis buffer for 30 min at 4 °C, vortexed every 10 min, and then centrifuged at 12,000×g for 15 min. 20 μg sample was separated on 12% SDS-PAGE gels and blotted onto PVDF membranes. Bands were visualized by a Western Blotting Detection System (Bio-Rad Gel Doc XR+, USA).
Transwell assay
Each Transwell chamber was filled with 1:5 diluted Matrigel 50 μL, and 37 °C stewed for 3 hours to solidify. The cells were cultured for 24 h in FBS-free medium, then were digested to make a 1 × 105 cells density suspension (1% FBS), 200 μL was added to the Transwell chamber, 500 μL of medium (10% FBS) was added to the lower compartment of the chamber. After 24 h of culture, the upper chamber cells were wiped off, washed three times with PBS, fixed with paraformaldehyde for 30 min, stained with 0.1% crystal violet for 15 min, washed three times with PBS, and photographed after air drying. Two replicate wells were included in each analysis, six pictures were captured in each well and at least three independent experiments were conducted. Metastatic cell numbers were calculated by ImageJ software (National Institutes of Health, USA).
Wound healing assay
When the cells growth in the 6-well plate covered about 80% of the bottom of the well, transfected siRNA (100pM) into the cells with Lipo2000 and MEM medium, and replace the MEM medium with DMEM-F12 medium containing 2% FBS after 5 h. When the cells filled the entire bottom of the well, a 200 μL sterile pipette tip was used to make a wound scratch on the bottom of the well, and washed with PBS for three times to remove cell debris. Take photos of wounded scratches at 0 h, 24 h or 48 h by Inverted microscope (Olympus, Japan). Two replicate wells were included in each analysis, six pictures were captured in each well and at least three independent experiments were conducted. The area of wound healing was calculated using ImageJ software (National Institutes of Health, USA).
Flow cytometry
6 × 105 cells were seeded in a 6-well plate, transfected with siRNA (100pM) for 24 h, then treated with PTX (20 μM) for 24 h. Flow cytometry was used to detect cell necrosis and apoptosis. The operation steps were carried out according to the manual introduction. The brief is as follows: cells were trypsinized and centrifuged at 300 g for 5 min at 4 °C, then washed twice with pre-cooled PBS. Staining cells with binding buffer containing Annexin V-FITC and PI for 10 min, then detected by a Flow cytometer (Attune NxT, Life, USA) and Flow cytometer (BD, USA).
Statistical analysis
All data were expressed as the Mean ± SD. All statistic results were carried by GraphPad Prism8. A two-sided tail non-paired Student’s t test or multi T-test was used to compare differences between the treated group and the control group. A value with P < 0.05 was considered significant.
Availability of data and materials
All data generated or analyzed during this study are included in this published article. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- OLA1:
-
Obg-like ATPase 1
- OSCC:
-
Oral squamous cell carcinoma
- HNSC:
-
Head and neck squamous cell cancer
- NHOKs:
-
Normal human oral keratinocytes
- EGFR:
-
Epidermal growth factor receptor
- OCC:
-
Oral cell carcinoma
- HPV:
-
Human papilloma virus
- ECS:
-
Extracapsular spread
- EMT:
-
Epithelial-mesenchymal transition
- MET:
-
Mesenchymal epithelial transition
- GEPIA :
-
Gene Expression Profiling Interactive Analysis
- GEO :
-
Gene expression omnibus
- SUMO:
-
Small ubiquitin-related modifier
- siRNA:
-
Small interfering RNA
- siR-OLA1:
-
Small interfering OLA1 RNA
- shNC:
-
Small hairpin RNA normal control
- shOLA1:
-
Small hairpin OLA1 RNA
- OLA1OE :
-
OLA1 overexpression
- siR-SMAD2:
-
Small interfering SMAD2 RNA
- PTX :
-
Paclitaxel
- FBS :
-
Foetal bovine serum
- HE staining:
-
Hematoxylin-eosin staining
- MTT :
-
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30.
Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11(1):9–22.
Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009;45(4–5):309–16.
Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6(5):963–8.
Dissanayaka WL, Pitiyage G, Kumarasiri PV, Liyanage RL, Dias KD, Tilakaratne WM. Clinical and histopathologic parameters in survival of oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113(4):518–25.
Gandini S, Botteri E, Iodice S, Boniol M, Lowenfels AB, Maisonneuve P, Boyle P. Tobacco smoking and cancer: a meta-analysis. Int J Cancer. 2008;122(1):155–64.
Elwood JM, Pearson JC, Skippen DH, Jackson SM. Alcohol, smoking, social and occupational factors in the aetiology of cancer of the oral cavity, pharynx and larynx. Int J Cancer. 1984;34(5):603–12.
Reidy J, McHugh E, Stassen LF. A review of the relationship between alcohol and oral cancer. Surgeon. 2011;9(5):278–83.
Wang YX, Springer S, Mulvey CL, Silliman N, Schaefer J, Sausen M, James N, Rettig EM, Guo T, Pickering CR, et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Sci Transl Med. 2015;7(293):7.
Pignon JP, le Maitre A, Bourhis J, Group M-NC. Meta-analyses of chemotherapy in head and neck Cancer (MACH-NC): an update. Int J Radiat Oncol Biol Phys. 2007;69(2 Suppl):S112–4.
Chinn SB, Myers JN. Oral cavity carcinoma: current management, controversies, and future directions. J Clin Oncol. 2015;33(29):3269–76.
Gatta G, Botta L, Sanchez MJ, Anderson LA, Pierannunzio D, Licitra L, Grp EW. Prognoses and improvement for head and neck cancers diagnosed in Europe in early 2000s: the EUROCARE-5 population-based study. Eur J Cancer. 2015;51(15):2130–43.
Thompson L. World Health Organization Classification of Tumours: Pathology and Genetics of Head and Neck Tumours. Ear Nose Throat J. 2006;85:74.
Chi AC, Day TA, Neville BW. Oral cavity and oropharyngeal squamous cell carcinomaan update. CA-Cancer J Clin. 2015;65(5):401–21.
Thiery J. Epithelial-mesenchymal transitions in tumor progression. Nat Rev Cancer. 2002;2:442–54.
Nawshad A, LaGamba D, Hay ED. Transforming growth factor beta (TGFbeta) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Arch Oral Biol. 2004;49(9):675–89.
Eger A, Mikulits W. Models of epithelial–mesenchymal transition. Drug Discov Today Dis Model. 2005;2:57–63.
Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20(2):69–84.
Iwatsuki M, Mimori K, Yokobori T, Ishi H, Beppu T, Nakamori S, Baba H, Mori M. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010;101(2):293–9.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8.
Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415–28.
De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110.
Leipe DD, Wolf YI, Koonin EV, Aravind L. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol. 2002;317(1):41–72.
Koller-Eichhorn R, Marquardt T, Gail R, Wittinghofer A, Kostrewa D, Kutay U, Kambach C. Human OLA1 defines an ATPase subfamily in the Obg family of GTP-binding proteins. J Biol Chem. 2007;282(27):19928–37.
Czyz A, Wegrzyn G. The Obg subfamily of bacterial GTP-binding proteins: essential proteins of largely unknown functions that are evolutionarily conserved from bacteria to humans. Acta Biochim Pol. 2005;52(1):35–43.
Ding Z, Liu Y, Rubio V, He J, Minze LJ, Shi ZZ. OLA1, a translational regulator of p21, maintains optimal cell proliferation necessary for developmental progression. Mol Cell Biol. 2016;36(20):2568–82.
Chen H, Song R, Wang G, Ding Z, Yang C, Zhang J, Zeng Z, Rubio V, Wang L, Zu N, et al. OLA1 regulates protein synthesis and integrated stress response by inhibiting eIF2 ternary complex formation. Sci Rep. 2015;5:13241.
Zhang JW, Rubio V, Zheng S, Shi ZZ. Knockdown of OLA1, a regulator of oxidative stress response, inhibits motility and invasion of breast cancer cells. J Zhejiang Univ Sci B. 2009;10(11):796–804.
Jeyabal PV, Rubio V, Chen H, Zhang J, Shi ZZ. Regulation of cell-matrix adhesion by OLA1, the Obg-like ATPase 1. Biochem Biophys Res Commun. 2014;444(4):568–74.
Bai L, Yu Z, Rubio V, Chen H, Li Y, Zhang J, Yuan S, Liao C, Jeyabal PV, Shi ZZ. OLA1 contributes to epithelial-mesenchymal transition in lung cancer by modulating the GSK3β/snail/E-cadherin signaling. Oncotarget. 2016;7(9):10402–13.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102.
Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525–30.
Chang YS, Jalgaonkar SP, Middleton JD, Hai T. Stress-inducible gene Atf3 in the noncancer host cells contributes to chemotherapy-exacerbated breast cancer metastasis. Proc Natl Acad Sci U S A. 2017;114(34):E7159–E68.
Karagiannis GS, Pastoriza JM, Wang Y, Harney AS, Entenberg D, Pignatelli J, Sharma VP, Xue EA, Cheng E, D'Alfonso TM, et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci Transl Med. 2017;9(399):eaao3817.
Keklikoglou I, Cianciaruso C, Guc E, Squadrito ML, Spring LM, Tazzyman S, Lambein L, Poissonnier A, Ferraro GB, Baer C, et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat Cell Biol. 2019;21(2):190–202.
Guneri P, Epstein JB. Late stage diagnosis of oral cancer: components and possible solutions. Oral Oncol. 2014;50(12):1131–6.
Viet CT, Schmidt BL. Biologic mechanisms of oral cancer pain and implications for clinical therapy. J Dent Res. 2012;91(5):447–53.
Kowalski LP, Carvalho AL. Natural history of untreated head and neck cancer. Eur J Cancer. 2000;36(8):1032–7.
Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–51.
Liang SQ, Marti TM, Dorn P, Froment L, Hall SRR, Berezowska S, Kocher G, Schmid RA, Peng RW. Blocking the epithelial-to-mesenchymal transition pathway abrogates resistance to anti-folate chemotherapy in lung cancer. Cell Death Dis. 2015;6(7):e1824.
Vega S, Morales AV, Ocaña OH, Valdés F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18(10):1131–43.
Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita M, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66(2):944–50.
Xu J, Lamouille S, Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009;19(2):156–72.
Oshimori N, Oristian D, Fuchs E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell. 2015;160(5):963–76.
Brunen D, Willems SM, Kellner U, Midgley R, Simon I, Bernards R. TGF-β: an emerging player in drug resistance. Cell Cycle. 2013;12(18):2960–8.
Zhang H, Ren L, Ding Y, Li F, Chen X, Ouyang Y, Zhang Y, Zhang D. Hyaluronan-mediated motility receptor confers resistance to chemotherapy via TGFβ/Smad2-induced epithelial-mesenchymal transition in gastric cancer. FASEB J. 2019;33(5):6365–77.
Liu L, Liu X, Ren X, Tian Y, Chen Z, Xu X, Du Y, Jiang C, Fang Y, Liu Z, et al. Smad2 and Smad3 have differential sensitivity in relaying TGFβ signaling and inversely regulate early lineage specification. Sci Rep. 2016;6(1):21602.
Huang SH, O'Sullivan B. Oral cancer: current role of radiotherapy and chemotherapy. Med Oral Patol Oral Cir Bucal. 2013;18(2):e233–40.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81770846 and No. 81803024), Hirshberg Foundation for Pancreatic Cancer Research (GX20171003858 and GX20191005878), and Fundamental Research Funds from the Dalian Universities of Technology under Grant No. DUT17ZD308. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
GX and QY conceived and designed the experiments; QY and JL conducted the experiments and analyzed the data; JL, QY and TD wrote and GX edited the manuscript. SH, KX review the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Biology and Medical Ethics Committee of Dalian University of Technology.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
OLA1 expression profile across all tumor samples and paired normal tissues.
Additional file 2.
Original gel scan.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, J., Yang, Q., Xiao, K.C. et al. Obg-like ATPase 1 inhibited oral carcinoma cell metastasis through TGFβ/SMAD2 axis in vitro. BMC Mol and Cell Biol 21, 65 (2020). https://doi.org/10.1186/s12860-020-00311-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12860-020-00311-z