
Abstract
Background
Prediction of drug–target interaction (DTI) is an essential step for drug discovery and drug reposition. Traditional methods are mostly time-consuming and labor-intensive, and deep learning-based methods address these limitations and are applied to engineering. Most of the current deep learning methods employ representation learning of unimodal information such as SMILES sequences, molecular graphs, or molecular images of drugs. In addition, most methods focus on feature extraction from drug and target alone without fusion learning from drug–target interacting parties, which may lead to insufficient feature representation.
Motivation
In order to capture more comprehensive drug features, we utilize both molecular image and chemical features of drugs. The image of the drug mainly has the structural information and spatial features of the drug, while the chemical information includes its functions and properties, which can complement each other, making drug representation more effective and complete. Meanwhile, to enhance the interactive feature learning of drug and target, we introduce a bidirectional multi-head attention mechanism to improve the performance of DTI.
Results
To enhance feature learning between drugs and targets, we propose a novel model based on deep learning for DTI task called MCL-DTI which uses multimodal information of drug and learn the representation of drug–target interaction for drug–target prediction. In order to further explore a more comprehensive representation of drug features, this paper first exploits two multimodal information of drugs, molecular image and chemical text, to represent the drug. We also introduce to use bi-rectional multi-head corss attention (MCA) method to learn the interrelationships between drugs and targets. Thus, we build two decoders, which include an multi-head self attention (MSA) block and an MCA block, for cross-information learning. We use a decoder for the drug and target separately to obtain the interaction feature maps. Finally, we feed these feature maps generated by decoders into a fusion block for feature extraction and output the prediction results.
Conclusions
MCL-DTI achieves the best results in all the three datasets: Human, C. elegans and Davis, including the balanced datasets and an unbalanced dataset. The results on the drug–drug interaction (DDI) task show that MCL-DTI has a strong generalization capability and can be easily applied to other tasks.
Similar content being viewed by others
Introduction
Prediction of drug–target interactions (DTIs) is an essential step for drug discovery (i.e., to find new candidate drugs) and drug reposition (i.e., to find new indications for existing drugs). Drugs play an important role in the human body by interacting with multiple targets [1]. Proteins represent an important type of targets whose function can be enhanced or inhibited by drugs to achieve phenotypic effects for clinical therapeutic purposes [2]. However, traditional experiments to obtain drug candidates through bioanalysis typically take 10–15 years and cost approximately 1 billion dollars from introducing the abstract concept to release into the market [3]. Large number of computational approaches are proposed for this task aim to mitigate the costs and risks of drug development.
Over the past decades, many computational methods have been widely applied to predict DTIs [4,5,6,7,8]. These computational methods can be mainly divided into three groups: docking-based methods [9, 10], ligand-based methods [11, 12], and chemogenomic-based methods [2, 4]. Docking-based methods cannot be applied if the 3D structure information for many target proteins is unknown. Ligand-based methods will not be suitable when the number of known ligands is limited or few. The chemogenomic-based methods overcome the limitations by utilizing the chemical and genomic information of drugs and targets that are available in many online public databases. Currently, machine learning and deep learning approaches are very popular. Several studies [7, 13,14,15,16,17] have summarized the progress of ML and DL methods in DTI prediction tasks. Traditional machine learning methods include network-based methods [18,19,20,21,22], clustering-based methods [23], kernel-based methods [24,25,26,27,28], and matrix factorization-based methods [29,30,31,32,33].
Deep learning approaches generally treat the DTI task as a binary classification task by first learning the embedded representations of the drug and target separately and then connecting them for prediction. In the DTI task, according to the representation of drugs and proteins, we can categorize the mainstream deep learning methods into three groups, sequence-based methods, graph-based methods, and image-based methods.
Sequence-based approaches are more common. DeepDTA [34] uses a convolutional neural network to learn drug and protein sequence features, DrugVQA [35] uses a bi-directional long-short time memory network (BiLSTM) for feature extraction of sequence information, and TransformerCPI [36] builds a transformer architecture with a self-attention mechanism. The main idea of these methods is to construct neural networks to learn useful information from drug and target sequences for DTI task. Moltrans [37] propose an innovative FCS (Frequent Subsequence Algorithm) algorithm to decompose protein and compound sequences. By employing an augmented transformer, they successfully capture the semantic characteristics of substructures from a large volume of unlabeled biomedical texts. DeepCDA [38] combines CNN and LSTM to encode protein and compound sequences, and proposes a bidirectional attention mechanism to encode the intensity of their interaction. In order to solve the problem of sampling test and training data from different distribution domains, DeepCDA [38] also utilizes adversarial domain adaptation methods to learn the feature encoder network in the test domain.
Graph-based neural networks have become a prominent approach for extracting abstract features of drug. The RDKIT toolkit can transform the drug into graph structures, enabling the application of graph neural networks (GNN) in the CPI task [39]. GraphCPI [40] and GraphDTA [41] adopts Graph Convolutional Networks (GCN) to perform convolutional operations on compound graph structures. LGDTI [42] is based on large-scale graph representation learning to predict DTI. Compared with the existing graph based neural network methods, LGDTI adopts a unique method to extract the potential graph features of drugs and targets in complex biological network by using two different graph representation learning methods. FuHLDR [43] is a novel graph representation learning model for drug repositioning, which effectively integrates high-level and low-level biological information. It provides a new solution for constructing heterogeneous information networks for DTI tasks to improve prediction accuracy.
Image-based methods were previously underappreciated. Image-based approach to extract useful features from molecular images of drugs. PWO-CPI [44] constructs a CNN model to learn the features in molecular images as the embedding representation of the drug and uses word2vec [45] model learn the protein sequences.
These methods only considered single modal information of the drug, such as SMILES sequences, molecular graphs or molecular images. Huang et al. [46] worked out to the conclusion that the richer the variety of modalities, the more accurate the estimation of the representation space with sufficient training data. In order to obtain more comprehensive features of drugs, some researchers also use both the sequence and graph structure of drugs to achieve DTI tasks, such as SSGraphCPI [47]. This method can obtain effective information from the two modalities of drugs, which can effectively improve the effectiveness. In the field of computer vision, multimodal techniques are also widely used for various tasks, such as visual Question Answering, Image Caption, Referring Expression Comprehension and Visual Dialogue [48, 49]. In tasks such as DTI and interaction prediction, few people consider the combination of drug images and other information. Therefore, we will further discuss whether fusing and enhancing multiple modal information have improved drug and target embedding representation. In addition, TripletMultiDTI [50] is also a new multimodal DTI method, which designs a new architecture that integrates multimodal knowledge to predict affinity labels. At the same time, it also proposes a new loss function based on the triplet loss, making the model perform better. TranSynergy [51] designs an enhanced deep learning model based on knowledge and self attention machine mechanism to predict collaborative drug combinations, effectively improving the performance and interpretability of collaborative drug combination prediction.
In our previous work PWO-CPI [44], we have shown that the features of drug image can be well used for the task of DTI. In addition, the information contained in a single image is not sufficient to fully characterize the drug. We want to introduce chemical properties that are valuable for understanding compounds. Therefore, we propose that combining the images of compounds and chemical features of drugs can lead to a more comprehensive abstract characterization of drugs, which can enhance the DTI results.
Cross-attention mechanisms are often used in image description generation, visual questioning and answering, where it can cross-learn features from multiple modalities. This cross-attention mechanism enhances the expressive power of feature representation by introducing an attention mechanism to dynamically adjust the association weights between multimodal features, thus realizing effective feature fusion and interaction. Therefore, this paper proposes to introduce the cross-attention mechanism into the learning of drugs and targets features, so that the cross-learning of the above two features can be realized to extract the correlation relationship between the two, which helps to improve the performance of the DTI task.
In general, the main contributions of this paper are as follows:
-
In this study, we introduce a novel approach by integrating the multimodal information of compound images and chemical text information as input features for drugs. We can extract more comprehensive drug features from both modalities, which are effectively used for DTI tasks.
-
An innovative method of bi-directional cross-attention learning is proposed. This bi-directional cross-attention learning mechanism can learn deeper semantic relationships between drugs and targets, capturing more useful interaction features to enhance DTI prediction effects.
-
Improved predictive performance over state-of-the-art baselines on three public datasets with different scales. The DTI experimental results demonstrate the effectiveness of the method. The excellent results on the DDI task demonstrate the generalization of the method proposed in this paper.
Method
Overall workflow

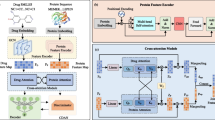
An overall architecture of MCL-DTI
DTI can be regarded as a classification problem, inputting drugs and targets into the model to predict where the two will interact with each other. If there is an interaction between the two, output 1, otherwise output 0. The method proposed in this paper inputs drug multimodal information as well as the FASTA sequence of the target into the model, and the predictive goal of the model is to output whether the two interact. The architecture of the MCL-DTI model is shown in Fig. 1. The whole model mainly consists of four modules: feature encoder module, feature decoder module, feature fusion module, and classifier. We use the Rdkit toolkit to obtain images and chemical features of drugs from SMILES sequences, used as multimodal representations of the drug. We input the multimodal representations of the drug and the sequence representation of the target into the feature extraction module, obtain the high-level abstract features of them respectively, and then feed them into the feature decoder module. The feature decoder module consists of independent drug decoder and target decoder, which are composed of MCA (Multi-head Cross Attention) Block and MSA (Multi-head Self Attention) Block. The feature decoder module can effectively decode the information of the drug and target as well as the interaction information between them. After the feature decoder, we send the two obtained feature maps to the feature fusion module, and then a classifier to get the final prediction result. We will describe each module in detail in the next few sections.
Feature encoder module
To better capture the drug features, we input the image and the chemical features text of the drug. For drug image, we construct a CNN backbone Conv similar to PWO-CPI [44]. This backbone contains convolution, batch normalization, activation and pooling layers. We first obtain the structural formula images of drugs from SMILES sequence by RDKit [52] software. These images show visual representations of molecule, as can be seen in Fig. 2a. We define the input image as \(P\in R^{h \times h}\), where h denotes the size of image. The local feature map \(x^v=Conv(P)\) of the image can be obtained by the constructed CNN backbone. Since CNN Block can only capture local information without considering global features, we build an MSA block to enhance semantic relations of features, and the specific flowchart of MSA is shown on the right side of Fig. 1. MSA block contains Layer Normalization (LN) [53] layers, multi-head self attention layer, MLP block and residual connections. Following prior works on transformers encoder in [54], we add a residual connection to the MSA computation. LN layers are applied before every block to normalize neuron nodes in the neural network. We pass the output \(x^v=Conv(P)\) of the CNN Block through the MSA block to get the image feature of the drug, \(X_{img}=MSA(x^v)\).

Multimodal information of drugs. a is the molecular image modal. b is the chemical text information modal
The chemical features are defined by a feature type and a feature family. A feature family is a general classification of features, such as hydrogen bond donors, aromaticity, etc., where pharmacophore matching is achieved based on the feature family [52]. Here we use a feature factory and choose feature family information, feature type information and feature corresponding atoms information as the chemical text information of the drug. We can obtain the chemical text information of the drug by Rdkit [52] software using the SMILES sequences, as can be seen in Fig. 2b. In order to extract features from drug text, we first use the \(k-gram\) method to segment the text sequences by words. The text sequences are divided into phrases of length k, and build a dictionary to record the order in which the phrases appear. The numerical word order of the dictionary is used to replace the original words, and these numbers are embedded for representation. Figure 3 shows \(k-gram\) method of protein sequence segmentation and embedding representation when k is 1. Similarly, we feed the embedding representation into an MSA module to obtain textual features of the compound, \(X_{text}\).

Method for protein sequence segmentation and embedding representation when k is 1
We take the sum of \(X_{img}\) and \(X_{text}\) as the drug’s features, while we assign learnable weights \(\lambda _1\) and \(\lambda _2\) to them. A higher weight indicates that the modality has a large influence on the drug feature representation. The drug is encoded as \(X_{drug}\):
For target sequence, we directly use its FASTA sequence as its text information. Similar to the chemical feature text of drug, we do the same for the FASTA sequence, first obtaining its embedding representation through k-gram, and then obtaining the abstract features of the target \(X_{tgt}\) through an MSA module.
Feature decoder module
After encoding the drug and target features, we feed the obtained \(X_{drug}\) and \(X_{tgt}\) to the feature decoder module to learn the drug–target interaction information. As shown in Fig. 1, the feature decoder module consists of two decoders and each consists of an MSA block and MCA block. MCA block have the same LN layers, MLP blocks, residual connections with MSA layers. The main difference between MSA and MCA is the calculation process of attention output. The MSA block is designed to capture the internal relationships of the features themselves, and when computing the attention output, the query, key, and value are all obtained from the same feature through a linear matrix transformation. The MCA block, on the other hand, is designed to capture the interaction information between the drug and the target. Therefore, for the MCA block of the drug decoder, not only the drug features but also the target features need to be inputted. We perform matrix linear transformation on the input target features to get the query needed to compute the attention output, and perform linear transformation on the input target features to get the key and value. Figure 4 illustrates the computational process of MCA. The target decoder is similar. With the drug decoder and the target decoder, we send their respective features to each other in both directions for two-way cross learning, and finally get two feature maps, \(Z_{drug \rightarrow target}\) and \(Z_{target \rightarrow drug}\).

Architectural elements of a cross attention block between two time-seriers form drug \(\alpha \) and target \(\beta \)
Feature fusion module
The fusion block is used to receive the input feature maps \(Z_{drug \rightarrow target}\) and \(Z_{target \rightarrow drug}\) from two decoders. We concatenate both feature maps by channel dimension and feed it into the fusion block. Fusion block contains a 2D convolution network Conv2D, a 1D convolution network Conv1D, a MLP block MLP and a fully connected layer FC. We extract the concatenated feature maps by convolution layers and finally feed them into FC layer to obtain the final prediction result P, this calculation can be expressed as:
where Z represents the feature map generated by decoder and ; denotes the concatenate operation.
Classifier
We use cross-entropy as loss function specifically as follows:
where N denotes the total number of samples, and \(y_n\) represents the true label. When model training, w choose the Adam [55] optimization algorithm as the optimizer of the model.
Experiment
In this section we present experimental comparisons of MCL-DTI with other state-of-the-art methods.
Experimental setup
Dataset
In this work, we choose three DTI public datasets for experiments including Human [56], C. elegans [56] and Davis [57]. See Table 1 for specific drug and target statistics. Human and C. elegans are both positive and negative sample balanced datasets. Their positive samples are obtained from the highest confidence biochemical databases: DrugBank [58] database and matador [59] database [56]. Davis contains 64 different drugs and 379 targets. In Davis, DTI pairs which have \(k_d\) values < 30 units are considered positive [57]. Human and C. elegans datasets are divided into 8:1:1 ratio according to train set, valid set and test set when conducting the experiments. Davis dataset division is followed by MolTrans [37]. In addition, we use the Biosnap [60] for DDI task which is to predict the interaction between drug and drug. Biosnap contains 9,648 drugs and 81,194 samples with 50.5\(\%\) of positive samples.
Metrics
In this work, we use ROC–AUC (area under the receiver operating characteristic curve), PR–AUC (area under the precision-recall curve) and recall as metrics to measure the prediction performance. The ROC–AUC is the main metric we use to evaluate all methods. The ROC–AUC curve takes into account both positive and negative examples and can effectively evaluate the overall performance of the model. The PR–AUC is more focused on positive examples, especially in data with unbalanced categories, and the value of PR–AUC is more indicative of the robustness of the model. Recall values indicate the percentage of samples predicted to be truly positive in the positive class. Recall provides good feedback on the model’s ability to learn for positive samples. The data for all results are expressed as the mean of the results and their standard deviation.
Experiment settings
The implementation of our method is based on Pytorch [62]. Each experiment is run for 100 epochs. For training, we use a server with i7 10700f, 32GB RAM and RTX 3090 GPU. For the selection of hyperparameters, we used the grid search method. The learning rate is searched in the range [1e−1, 1e−2, 1e−3, 1e−4, 1e−5], the learning rate decay coefficient is searched in the range [0.5, 0.6, 0.7, 0.8, 0.9], the batch size is searched in the range [32,64,128,256], the dropout rate is searched in the range [0.1,0.2,0.3,0.4,0.5]. We first use the grid search method to determine the learning rate and batch size, then fix the values of both, and then choose the dropout rate and learning rate decay coefficient. Through experiments, we choose the learning rate, learning rate decay coefficient, dropout rate, and batch size as 1e−3, 0.8, 0.1 and 128, respectively.
DTI experiment
Baseline.When choosing the comparative models, we mainly consider from three perspectives: Firstly, we chose representative and state-of-the-art methods, including DeepDTA [34], TransformerCPI [36], and MolTrans [37], to validate the competitiveness of our model. These methods are widely recognized and frequently used as benchmarks. Secondly, to assess the effectiveness of image-based methods, we included GNN-CPI [39] and TransformerCPI [36], which are typical examples of sequence-based and graph-based approaches. Thirdly, as the MCL-DTI model is an extension of our team’s previous work, it was essential to include our previous model, PWO-CPI [44], for comparison. At last, We compare MCL-DTI with the following methods:
-
GNN-CPI [39] uses molecular graph as drug representation and applies GNN for feature learning of embedded representation. They concatenate the outputs of the two neural networks for compound-protein interaction prediction. We follow the same hyperparameter setting described in this paper.
-
DeepDTA [34] applies CNN to two original extraction of local residual patterns using SMILES and protein sequences. The task of DeepDTA is to predict binding affinity values. We add a sigmoid activation function at the end of the model to turn it into a binary task and we set the same hyperparameters to ensure fairness.
-
DeepConv-DTI [61] uses CNN and global max pooling layers to extract local features of different lengths in protein sequences and applies the fully connected layer on drug fingerprint ECFP4. We obtain the same drug fingerprint ECFP4 and set the same hyperparameters for experimental comparison.
-
TransformerCPI [36] uses the atomic information of the drug and distance matrix as a representation of the drug and a learned representation of the protein features by wod2vec [45]. They construct a decoder with a self-attention mechanism to learn the features of compounds and proteins.
-
PWO-CPI [44] first uses drug images as molecular features. They use CNN to learn local information of drug images and apply word2vec [45] to encode protein sequences. Here, we use the same drug molecule images to represent the drugs and set the same hyperparameters for the experiments.
-
MolTrans [37] builds a large corpus and extracts the most commonly used molecular fragments. The numbers are used to replace the original characters and embedding of these numbers is used for feature learning. It conducts extensive experiment on different datasets and is the SOTA method and this is also our main method of comparison.
To ensure the fairness of the experiments, we conduct experiments for other methods on the same dataset and use the same hyperparameter settings as in the original paper. The error between the reproduced results and the original results is acceptable. We use the cross-validation strategy and conduct five experiments for each method, and the final experimental results are shown in Table 2.
For the two balanced datasets Human and C. elegans, the current deep learning methods can achieve relatively promising performance. MCL-DTI achieves the best results and exceed our previous work PWO-CPI in all metrics. PWO-CPI only uses images of drugs and does not perform the operation of feature fusion. These experimental results demonstrate that MCL-DTI can effectively conduct feature learning on balanced datasets.
In addition, the deep learning methods for experiments on Davis dataset failed to achieve satisfactory results, especially in terms of PR–AUC values. In the test set of the Davis dataset, the ratio of positive to negative samples is 1:19, which tests the model’s ability to learn the full range of sample features under the same learning environment. Compared to MolTrans as SOTA method, MCL-DTI improved by 0.014, 0.073 and 0.069 for three metrics, respectively.
In summary, these deep learning methods all utilize only single modal information about the drug molecule such as molecular graph, SMILES sequence information and molecular image. MCL-DTI utilizes multimodal information, molecular images and chemical text information, so that it can provide more comprehensive information about drug. Results on both balanced and unbalanced datasets show the competitive performance of MCL-DTI.
Our excellent prediction results can be explained from the following perspectives:
-
(1)
From the biological perspective, the structure of a molecules determines their properties. The structural characteristics of molecules can be intuitively displayed in their images, and deep learning models have excellent performance in extracting spatial structural features of images. Therefore, integrating representations from molecules images can provide a more comprehensive understanding of the biological characteristics of these molecules.
-
(2)
Chemical characteristics provides valuable information about compounds’ properties. These characteristics, such as molecular weight, polarity, or functional groups, are very relevant to the interaction between compounds and proteins. By incorporating chemical characteristics, the model can learn to recognize and exploit these properties, leading to more accurate predictions of compound-protein interactions.
-
(3)
Integrating image features with the chemical properties of compounds at an advanced semantic level can better characterize the biological characteristics of compounds. The use of a multi-head cross-attention mechanism allows the model to learn the relationship between drugs (compounds) and targets (proteins) in a more sophisticated manner. This mechanism enables the model to focus on different aspects of the compounds and proteins simultaneously, capturing their intricate interactions. By learning the complex relationships between compounds and proteins, the model can better understand the underlying biological mechanisms and predict their interactions more accurately.
DDI experiment
To further validate the learning ability of drug multimodality and MCA mechanism, we conduct experiments for DDI task. We use the same method as MCL-DTI for the embedding representation of drug. After obtaining the two drug embedding representations, these embedidng feature maps are fed into the same decoders as MCL-DTI to learn the interaction between different drugs respectively. Finally the prediction results are also obtained by a fusion block. We name this model for DDI tasks as MCL-DDI. Here we set ROC–AUC, PR–AUC and F1 values as indicators on Biosnap [60] dataset. Methods with which we have conducted experimental comparisons include LR [63], Nat.Prot [64], Mol2Vec [65], MoVAE, DeepDDI [66] and Caster [60].
The results of the DDI experiments are shown in Table 3. We find that MCL-DDI far exceeds the previous work in three different metrics. The performance of the model can indeed be effectively improved by multimodal and cross-attention learning of drugs. This also means that our model has strong generalization and is more suitable than previous methods for the prediction of both interactions.
Ablation study
In this section, several ablation experiments are performed on the whole model to effectively represent the influence of each module on MCL-DTI. To better represent the robustness of each module of MCL-DTI, we conduct experiments on balanced and unbalanced datasets, i.e., Human and Davis.
-
image + SMILES: we use the SMILES sequence of the drug as text information instead of the chemical text information
-
Text: we use only the chemical text modal information as the drug embedding representation
-
Image: we use only molecular image modal as the drug embedding representation.
-
MCA: we remove the MCA block from drug and target decoders, so that only the MSA block remained in the decoder.
-
MCA, image: we remove both MCA block and image modal.
-
MCA, text: we remove both MCA block and text modal.
From the results in Table 4 we can see that in MCL-DTI as a complete model achieves the best results on both datasets.
The results for image + SMILES and image are similar, and we can see that the effect is not obvious when using SMILES sequences to enhance the features. This can indicate that the image may contain the information of SMILES sequences or more. It can be inferred from MCL-DTI and image + SMILES that the chemical text information of the drug contains different information from the SMIELES sequence. In the experiments of text and image, it can be seen that images play a more important role in the features of drug molecules. In addition, it can be observed from text that the information of the chemical text improves the model. The results from these experiments further demonstrate that the multimodal and cross-attention modules have latent capabilities for feature learning.

Variation of learnable variables \(\lambda _1\) and \(\lambda _2\) on the Human and Davis datasets. The process of change of scalars during the experiment and the learning ratio correspond to the experimental results
Bias towards different modal information
It is also valuable to see that MCL-DTI introduces two learnable scalars \(\lambda _1\) and \(\lambda _2\) to combine the outputs from image modal and text modal information (Eq. 1). This leads to a by-product of MCL-DTI where \(\lambda _1\) and \(\lambda _2\) actually reflect the model’s bias towards image modal and text modal information.
We explore how different combinations of image modal and text modal affect model performance. We conduct experiments using multiple combinations of methods and summarize the results in Table 5. We perform parallel experiments on Human and Davis datasets and show the learned scalars \(\lambda _1\) and \(\lambda _2\) from different values. In addition we set a fixed \(\lambda _1\) and \(\lambda _2\) to observe whether the model has learned the scalars effectively. This observation shows a stable perference for MCL-DTI towards the different design patterns of multimodality. Again the analysis of the data results from the fixed scalars shows that the experimental results all decrease in the absence of a certain modality. We can see that the performance is promising when both scalars are working and both are learnable.
In addition, we consider separately the learning process of the two learnable scalars during the experiment. We conduct experiments to show the learnable parameters \(\lambda _1\), \(\lambda _2\) from Human and Davis datasets. From the experimental results in Fig. 5, we can see that the learning scalars stabilize in the later stages of the experiment, and the ratio between the two parameters is relatively constant. The variation of the rates in different is relatively small, especially when epoch increases. The ratio of the scalars is inversely proportional to the value of the ROC–AUC, i.e., when the difference between the two learning scalars is greater, the model is less effective. By the fact that the ratios of the final learning scalars are all relatively close, it can be seen that the design pattern in this paper is indeed useful and effective in feature learning of multimodal information.
Case study
In order to verify the practical ability of the model, we conduct a case study on two highly valuable proteins, namely 3C-like protease (3CLpro) and RNA-dependent RNA polymerase (RdRp). We select experimentally confirmed drug molecules known to interact with them, as well as unrelated drug molecules. The proposed MCL-DTI model was utilized to predict the interaction scores between them. A higher predicted score for interacting drugs and a lower predicted score for unrelated drugs would indicate the practical significance of our proposed model. The experimental results are shown in Table 6.
The 3CLpro is an enzyme found in coronaviruses. 3CLpro plays a crucial role in the replication of the virus by cleaving viral polyproteins into functional proteins necessary for viral assembly and replication. The effectiveness of 3CLpro as a target for antiviral drugs depends on its inhibition. By inhibiting 3CLpro, it is possible to disrupt the replication process of the virus, potentially reducing viral load and slowing down the progression of the disease. RdRp is an enzyme that plays a crucial role in the replication of RNA viruses. RdRp is a target for antiviral drug development, as inhibiting its activity can disrupt viral replication and potentially control viral infections. Therefore, identifying the interaction relationship between drugs and the two aforementioned targets is of great significance. We select 3CLpro and RdRp as the research subjects to determine the reliability of the MCL-DTI model in practical applications by predicting their interactions with candidate drugs such as Baritinib, Sofosbuvir, and Aspirin. Through experiments, we obtain the probability of drug binding to the target.
From the experimental results, we can see that Baricitinib, Remdesivir, Lopinavir, and Ritonavir are highly likely to interact with 3CL pro and Sofosbuvir, Daclatasvir, Lopinavir, Ritonavir are highly likely to interact with RdRp. In fact, this results has been proven by many current studies and clinical trials. On the contrary, the probability of interaction between unrelated drugs aspirin and 3CL pro and RdRp is very low, which is also in line with reality. These experimental results all demonstrate the reliability of MCL-DTI, therefore, we believe that the MCL-DTI model has guiding significance in practical research and drug discovery.
Conclusion
In this work, we propose a novel model MCL-DTI for DTI task. We exploit for the first time the multimodal information of drugs which characterize them in different modal. We perform semantic learning of molecular image modal and chemical text modal as the embedding representation of the drug by a multi-head self-attentive block. Then, we propose a bi-directional cross-attention mechanism, which allows for deeper semantic learning of drug and target features. From the data results of the experiments, MCL-DTI achieves the best results in all three datasets of DTI, including the balanced datasets and unbalanced datasets. It also explained in the DDI task that MCL-DTI has a strong generalization capability and can be easily applied to other tasks. In additon, ablation experiments further demonstrate the robustness of multimodality and cross-attention block. All the results data indicate that multimodalities and cross-attention learning method can be well applied to DTI or other interaction prediction tasks. In additon, ablation experiments further demonstrate the robustness of multimodality and cross-attention block. All the results data indicate that multimodalities and cross-attention learning method can be well applied to DTI or other interaction prediction tasks. In future work, we consider incorporating other modal information to construct a more rational heterogeneous network. Besides, the effectiveness of deep learning models is still largely limited by the quality and size of the dataset. Therefore, in the next step, we hope to design useful pre-training methods to obtain useful information from large-scale unlabeled biological data in order to further improve the model’s effectiveness.
Availability of data and materials
The datasets and source codes are publicly available in the GitHub repository, https://github.com/wowowoj/MCL-DTI.git.
Abbreviations
- DTI:
-
Drug–target interaction
- MSA:
-
Multi-head self attention
- MCA:
-
Multi-head cross attention
- ML:
-
Machine learning
- GAN:
-
Generative adversarial network
- DL:
-
Deep learning
- CNN:
-
Convolution neural network
- GNN:
-
Graph neural network
- DDI:
-
Drug–drug interaction
- LN:
-
Layer normalization
- SA:
-
Self attention
- PE:
-
Positional embedding
References
Chu Y, Kaushik AC, Wang X, Wang W, Zhang Y, Shan X, Salahub DR, Xiong Y, Wei D-Q. DTI-CDF: a cascade deep forest model towards the prediction of drug–target interactions based on hybrid features. Brief Bioinform. 2021;22(1):451–62.
Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16(1):19–34.
Zhou L, Li Z, Yang J, Tian G, Liu F, Wen H, Peng L, Chen M, Xiang J, Peng L. Revealing drug–target interactions with computational models and algorithms. Molecules. 2019;24(9):1714.
Ezzat A, Wu M, Li X-L, Kwoh C-K. Computational prediction of drug–target interactions using chemogenomic approaches: an empirical survey. Brief Bioinform. 2019;20(4):1337–57.
Sachdev K, Gupta MK. A comprehensive review of feature based methods for drug target interaction prediction. J Biomed Inform. 2019;93:103159.
Wu Z, Li W, Liu G, Tang Y. Network-based methods for prediction of drug–target interactions. Front Pharmacol. 2018;1134
Zhang W, Lin W, Zhang D, Wang S, Shi J, Niu Y. Recent advances in the machine learning-based drug–target interaction prediction. Curr Drug Metab. 2019;20(3):194–202.
Nath A, Kumari P, Chaube R. Prediction of human drug targets and their interactions using machine learning methods: current and future perspectives. Comput Drug Discov Des. 2018. https://doi.org/10.1007/978-1-4939-7756-7_2.
Alonso H, Bliznyuk AA, Gready JE. Combining docking and molecular dynamic simulations in drug design. Med Res Rev. 2006;26(5):531–68.
Ma D-L, Chan DS-H, Leung C-H. Drug repositioning by structure-based virtual screening. Chem Soc Rev. 2013;42(5):2130–41.
Xu Y, Xu D, Liang J. Computational methods for protein structure prediction and modeling volume 1: basic characterization. Springer; 2007.
Lam JH, Li Y, Zhu L, Umarov R, Jiang H, Héliou A, Sheong FK, Liu T, Long Y, Li Y. A deep learning framework to predict binding preference of RNA constituents on protein surface. Nat Commun. 2019;10(1):1–13.
Chen X, Yan CC, Zhang X, Zhang X, Dai F, Yin J, Zhang Y. Drug–target interaction prediction: databases, web servers and computational models. Brief Bioinform. 2016;17(4):696–712.
Chen R, Liu X, Jin S, Lin J, Liu J. Machine learning for drug–target interaction prediction. Molecules. 2018;23(9):2208.
Anusuya S, Kesherwani M, Priya KV, Vimala A, Shanmugam G, Velmurugan D, Gromiha MM. Drug–target interactions: prediction methods and applications. Curr Prot Pept Sci. 2018;19(6):537–61.
Zhao Q, Yu H, Ji M, Zhao Y, Chen X. Computational model development of drug–target interaction prediction: a review. Curr Prot Pept Sci. 2019;20(6):492–4.
Bagherian M, Sabeti E, Wang K, Sartor MA, Nikolovska-Coleska Z, Najarian K. Machine learning approaches and databases for prediction of drug–target interaction: a survey paper. Brief Bioinform. 2021;22(1):247–69.
Cheng F, Liu C, Jiang J, Lu W, Li W, Liu G, Zhou W, Huang J, Tang Y. Prediction of drug–target interactions and drug repositioning via network-based inference. PLoS Comput Biol. 2012;8(5):1002503.
Chen X, Liu M-X, Yan G-Y. Drug–target interaction prediction by random walk on the heterogeneous network. Mol BioSyst. 2012;8(7):1970–8.
Fu G, Ding Y, Seal A, Chen B, Sun Y, Bolton E. Predicting drug target interactions using meta-path-based semantic network analysis. BMC Bioinform. 2016;17(1):1–10.
Luo Y, Zhao X, Zhou J, Yang J, Zhang Y, Kuang W, Peng J, Chen L, Zeng J. A network integration approach for drug–target interaction prediction and computational drug repositioning from heterogeneous information. Nat Commun. 2017;8(1):1–13.
Wu Z, Cheng F, Li J, Li W, Liu G, Tang Y. Sdtnbi: an integrated network and chemoinformatics tool for systematic prediction of drug–target interactions and drug repositioning. Brief Bioinform. 2017;18(2):333–47.
Zhang X, Li L, Ng MK, Zhang S. Drug–target interaction prediction by integrating multiview network data. Comput Biol Chem. 2017;69:185–93.
Jacob L, Vert J-P. Protein–ligand interaction prediction: an improved chemogenomics approach. Bioinformatics. 2008;24(19):2149–56.
Xia Z, Wu L-Y, Zhou X, Wong ST. Semi-supervised drug-protein interaction prediction from heterogeneous biological spaces. In BMC systems biology; 2010. vol. 4, pp. 1–16. BioMed Central
Van Laarhoven T, Nabuurs SB, Marchiori E. Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics. 2011;27(21):3036–43.
Shang F, Jiao L, Liu Y. Integrating spectral kernel learning and constraints in semi-supervised classification. Neural Process Lett. 2012;36(2):101–15.
Nascimento AC, Prudêncio RB, Costa IG. A multiple kernel learning algorithm for drug–target interaction prediction. BMC Bioinform. 2016;17(1):1–16.
Gönen M. Predicting drug–target interactions from chemical and genomic kernels using Bayesian matrix factorization. Bioinformatics. 2012;28(18):2304–10.
Liu Y, Wu M, Miao C, Zhao P, Li X-L. Neighborhood regularized logistic matrix factorization for drug–target interaction prediction. PLoS Comput Biol. 2016;12(2):1004760.
Hao M, Bryant SH, Wang Y. Predicting drug–target interactions by dual-network integrated logistic matrix factorization. Sci Rep. 2017;7(1):1–11.
Bolgár B, Antal P. VB-MK-LMF: fusion of drugs, targets and interactions using variational bayesian multiple kernel logistic matrix factorization. BMC Bioinform. 2017;18(1):1–18.
Bagherian M, Kim RB, Jiang C, Sartor MA, Derksen H, Najarian K. Coupled matrix–matrix and coupled tensor-matrix completion methods for predicting drug–target interactions. Brief Bioinform. 2021;22(2):2161–71.
Öztürk H, Özgür A, Ozkirimli E. Deepdta: deep drug–target binding affinity prediction. Bioinformatics. 2018;34(17):821–9.
Zheng S, Li Y, Chen S, Xu J, Yang Y. Predicting drug–protein interaction using quasi-visual question answering system. Nat Mach Intell. 2020;2(2):134–40.
Chen L, Tan X, Wang D, Zhong F, Liu X, Yang T, Luo X, Chen K, Jiang H, Zheng M. Transformercpi: improving compound-protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics. 2020;36(16):4406–14.
Huang Kexin, Xiao Cao, Glass Lucas M, Sun Jimeng. MolTrans: molecular interaction transformer for drug–target interaction prediction. Bioinformatics. 2021;37(6):830–6.
Abbasi K, Razzaghi P, Poso A, et al. DeepCDA: deep cross-domain compound–protein affinity prediction through LSTM and convolutional neural networks. Bioinformatics. 2020;36(17):4633–42.
Tsubaki M, Tomii K, Sese J. Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics. 2019;35(2):309–18.
Quan Z, Guo Y, Lin X, Wang Z-J, Zeng X. Graphcpi: graph neural representation learning for compound-protein interaction. In: 2019 IEEE international conference on bioinformatics and biomedicine (BIBM), IEEE; 2019. pp. 717–722
Nguyen Thin, Le Hang, Quinn Thomas P, Nguyen Tri, Le Thuc Duy, Venkatesh Svetha. GraphDTA: predicting drug–target binding affinity with graph neural networks. Bioinformatics. 2021;37(8):1140–7.
Zhao Bo-Wei, You Zhu-Hong, Hu Lun, Guo Zhen-Hao, Wang Lei, Chen Zhan-Heng, Wong Leon. A novel method to predict drug–target interactions based on large-scale graph representation learning. Cancers. 2021;13(9):2111.
Zhao B-W, Wang L, Hu P-W, Wong L, Su X-R, Wang B-Q, You Z-H, Hu L. Fusing Higher and Lower-order biological information for drug repositioning via graph representation learning. IEEE Trans Emerg Topics Comput. 2023. https://doi.org/10.1109/TETC.2023.3239949.
Qian Y, Li X, Wu J, Zhou A, Xu Z, Zhang Q. Picture-word order compound protein interaction: predicting compound-protein interaction using structural images of compounds. J Comput Chem. 2022;43(4):255–64.
Mikolov T, Chen K, Corrado G, Dean J. Efficient estimation of word representations in vector space; 2013. arXiv preprint arXiv:1301.3781
Huang Y, Du C, Xue Z, Chen X, Zhao H, Huang L. What makes multi-modal learning better than single (provably). Adv Neural Inf Process Syst. 2021;34:10944–56.
Wang X, Liu J, Zhang C, Wang S. SSGraphCPI: a novel model for predicting compound–protein interactions based on deep learning. Int J Mol Sci. 2022;23(7):3780.
Han K, Wang Y, Chen H, Chen X, Tao D. A survey on visual transformer 2020
Liu Y, Zhang Y, Wang Y, Hou F, Yuan J, Tian J, Zhang Y, Shi Z, Fan J, He Z. A survey of visual transformers; 2021. arXiv e-prints
Dehghan A, Razzaghi P, Abbasi K, et al. TripletMultiDTI: multimodal representation learning in drug-target interaction prediction with triplet loss function. Expert Syst Appl. 2023;232:120754.
Liu Q, Xie L. TranSynergy: mechanism-driven interpretable deep neural network for the synergistic prediction and pathway deconvolution of drug combinations. PLoS Comput Biol. 2021;17(2):e1008653.
Landrum G. Rdkit documentation. Release. 2013;1(1–79):4.
Ba JL, Kiros JR, Hinton GE. Layer normalization; 2016. arXiv preprint arXiv:1607.06450
Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, Kaiser Ł, Polosukhin I Attention is all you need. Adv Neural Inf Process Systems 2017;30
Kingma DP, Ba J Adam: a method for stochastic optimization; 2014. arXiv preprint arXiv:1412.6980
Liu H, Sun J, Guan J, Zheng J, Zhou S. Improving compound-protein interaction prediction by building up highly credible negative samples. Bioinformatics. 2015;31(12):221–9.
Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Natu Biotechnol. 2011;29(11):1046–51.
Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. Drugbank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36(suppl–1):901–6.
Günther S, Kuhn M, Dunkel M, Campillos M, Senger C, Petsalaki E, Ahmed J, Urdiales EG, Gewiess A, Jensen LJ. Supertarget and matador: resources for exploring drug–target relationships. Nucleic Acids Res. 2007;36(suppl-1):919–22.
Huang K, Xiao C, Hoang T, Glass L, Sun J. Caster: predicting drug interactions with chemical substructure representation. In: Proceedings of the AAAI conference on artificial intelligence 2020; Vol. 34, pp. 702–709
Lee I, Keum J, Nam H. Deepconv-DTI: Prediction of drug–target interactions via deep learning with convolution on protein sequences. PLoS Comput Biol. 2019;15(6):1007129.
Paszke A, Gross S, Massa F, Lerer A, Bradbury J, Chanan G, Killeen T, Lin Z, Gimelshein N, Antiga L, et al. Pytorch: An imperative style, high-performance deep learning library. Adv Neural Inf Process Syst. 2019;32.
Wright RE. Logistic regression (1995).
Gómez-Bombarelli R, Wei JN, Duvenaud D, Hernández-Lobato JM, Sánchez-Lengeling B, Sheberla D, Aguilera-Iparraguirre J, Hirzel TD, Adams RP, Aspuru-Guzik A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent Sci. 2018;4(2):268–76.
Vilar S, Uriarte E, Santana L, Lorberbaum T, Hripcsak G, Friedman C, Tatonetti NP. Similarity-based modeling in large-scale prediction of drug–drug interactions. Nat Protoc. 2014;9(9):2147–63.
Ryu JY, Kim HU, Lee SY. Deep learning improves prediction of drug–drug and drug–food interactions. Proc Natl Acad Sci. 2018;115(18):4304–11.
Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V, Marconi VC, Ruiz-Palacios GM, Hsieh L, Kline S, et al. Baricitinib plus remdesivir for hospitalized adults with Covid-19. New Engl J Med. 2021;384(9):795–807.
Elfiky Abdo A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci. 2020;253:117592.
Stower H. Lopinavir–ritonavir in severe COVID-19. Nat Med. 2020;26(4):465–465.
Sadeghi A, Ali Asgari A, Norouzi A, Kheiri Z, Anushirvani A, Montazeri M, Hosamirudsai H, Afhami S, Akbarpour E, Aliannejad R, Radmard AR. Sofosbuvir and daclatasvir compared with standard of care in the treatment of patients admitted to hospital with moderate or severe coronavirus infection (COVID-19): a randomized controlled trial. J Antimicrob Chemother. 2020;75(11):3379–85.
Acknowledgements
We are grateful to the anonymous reviewers for their constructive comments on the original manuscript.
Funding
None.
Author information
Authors and Affiliations
Contributions
YQ, XYL, JW, and QZ conceived of the presented idea and designed the study. The experimental part is done for XYL and JW. The manuscript was drafted by JW and revised by XYL with the support from YQ and QZ. All authors have discussed the results and contributed to the final manuscript. All authors have read and approved to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qian, Y., Li, X., Wu, J. et al. MCL-DTI: using drug multimodal information and bi-directional cross-attention learning method for predicting drug–target interaction. BMC Bioinformatics 24, 323 (2023). https://doi.org/10.1186/s12859-023-05447-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-023-05447-1