
Abstract
Background
Cervical cancer is the fourth most common cancer affecting women and is caused by human Papillomavirus (HPV) infections that are sexually transmitted. There are currently commercially available prophylactic vaccines that have been shown to protect vaccinated individuals against HPV infections, however, these vaccines have no therapeutic effects for those who are previously infected with the virus. The current study’s aim was to use immunoinformatics to develop a multi-epitope vaccine with therapeutic potential against cervical cancer.
Results
In this study, T-cell epitopes from E5 and E7 proteins of HPV16/18 were predicted. These epitopes were evaluated and chosen based on their antigenicity, allergenicity, toxicity, and induction of IFN-γ production (only in helper T lymphocytes). Then, the selected epitopes were sequentially linked by appropriate linkers. In addition, a C-terminal fragment of Mycobacterium tuberculosis heat shock protein 70 (HSP70) was used as an adjuvant for the vaccine construct. The physicochemical parameters of the vaccine construct were acceptable. Furthermore, the vaccine was soluble, highly antigenic, and non-allergenic. The vaccine’s 3D model was predicted, and the structural improvement after refinement was confirmed using the Ramachandran plot and ProSA-web. The vaccine’s B-cell epitopes were predicted. Molecular docking analysis showed that the vaccine's refined 3D model had a strong interaction with the Toll-like receptor 4. The structural stability of the vaccine construct was confirmed by molecular dynamics simulation. Codon adaptation was performed in order to achieve efficient vaccine expression in Escherichia coli strain K12 (E. coli). Subsequently, in silico cloning of the multi-epitope vaccine was conducted into pET-28a ( +) expression vector.
Conclusions
According to the results of bioinformatics analyses, the multi-epitope vaccine is structurally stable, as well as a non-allergic and non-toxic antigen. However, in vitro and in vivo studies are needed to validate the vaccine’s efficacy and safety. If satisfactory results are obtained from in vitro and in vivo studies, the vaccine designed in this study may be effective as a therapeutic vaccine against cervical cancer.
Similar content being viewed by others

Background
Cervical cancer is the fourth most common cancer affecting women and is caused by human Papillomavirus (HPV) infections that are sexually transmitted [1]. HPV infection occurs in the epithelium through microscopic wounds making basal cells exposed to the virus [2]. HPV is a member of the Papillomaviridae family. HPVs are classified into five distinct major genera, known as alpha, beta, gamma, mu, and nu [3]. Approximately 200 types of HPV have been identified, which are classified into two high-risk and low-risk groups based on their carcinogenic properties. High-risk types are 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 68, and 59. It is well known that HPV16 and 18 as the most virulent types throughout the world are among the high-risk genotypes that account for 70% of all cervical cancer cases [4]. Types that are low risk include HPV 6, 11, 40, 42, 43, 44, 53, 54, 61, 72, 73, and 81 [5].
HPV genome is a circular double-stranded DNA of about 8000 base pairs that replicates in the host cell nucleus [6]. The HPV genome encompasses three regions, namely non-coding long-control region (LCR), as well as early and late regions. Early genes (E1, E2, E4, E5, E6, E7, and E8) and late genes (L1 and L2) are encoded in the two early and late regions, respectively. There are currently three commercial HPV prophylactic vaccines, Gardasil, Cervarix, and Gardasil 9, that are based on virus-like particles and can prevent high-risk HPV infections [7]. Prophylactic vaccines have been successful in preventing HPV infection and cancer. However, there is a significant population of people at high risk for HPV infection and related diseases worldwide. Therefore, the therapeutic vaccine must be designed and manufactured because prophylactic vaccines are ineffective in eradicating past infections and also do not kill infected cells [8].
The activity of p53, which stimulates the expression of genes involved in cell cycle arrest and apoptosis, is inhibited by E6 via the ubiquitin pathway and with the aid of a cellular protein known as E6-associated protein (E6AP) [9]. E7 oncoprotein inactivates the retinoblastoma (Rb) protein through ubiquitin-dependent degradation [10]. Rb prevents cell cycle progression from G1 to S through binding and inactivating transcription factors, such as members of the E2F family [11]. The majority of HPV tumorigenesis studies focus on the role of E6 and E7 proteins [12]. However, the high-risk HPV E5 protein has been demonstrated to have a significant effect on cellular pathways and signaling in human cell lines [13]. E5 protein was found to be expressed during the early stages of carcinogenesis, and its co-expression with E6 or E7 promotes transformation more than either oncoprotein alone [14]. E5 expression can increase the activity of E6 and E7 and has been shown to cause tumor progression [15]. The E5 can stimulate cancer cell proliferation by forming activating complexes with growth factor receptors, such as the Epidermal Growth Factor Receptor (EGFR), resulting in a proliferative state that lasts for a long time [16, 17]. The E5 has a function in decreasing cell death, promoting the accumulation of cells with abnormal DNA genetic mutations, and therefore accelerating the malignancy process [18]. Given all of this, it is strongly recommended that the HPV E5 protein be identified as an HPV oncoprotein and considered as a target for vaccine development [19].
The conventional approach of vaccine development is a time-consuming and expensive process requiring pathogen culture in the laboratory [20]. Reverse vaccinology uses genomic information and compute analysis for the development of vaccines without culturing microorganisms [21]. The current study aimed to use the immunoinformatics approach to develop a multi-epitope vaccine against cervical cancer.
Results
Study design
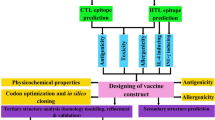
In this regard, cytotoxic T lymphocyte (CTL) and helper T lymphocyte (HTL) epitopes from the E5 and E7 proteins of HPV16 and 18, as well as the C-terminal fragment of heat shock protein 70 (HSP70) from Mycobacterium tuberculosis were used for vaccine design. Evaluation of the physicochemical properties of the vaccine candidate was performed. Subsequently, the multi-epitope vaccine's secondary and 3D structures were predicted. The vaccine’s 3D model was refined and validated after that. In the next step, molecular docking analysis of the refined three-dimensional model of the vaccine construct with toll-like receptor 4 (TLR4) was performed and then the stability of the vaccine was evaluated by molecular dynamics (MD) simulation. Finally, after testing the proposed vaccine’s expression efficacy in Escherichia coli (E. coli), the in silico cloning of the construct was conducted. The workflow followed in this study is depicted in Fig. 1.

Schematic representation of the methodology utilized to construct a multi-epitope vaccine to combat HPV16/18
Retrieving protein sequences
Table 1 shows a list of target proteins for multi-epitope vaccine design, along with their accession numbers.
Prediction and selection of T-cell epitopes
The CTL epitopes are essential for the development of long-lasting cellular immunity and can remove the circulating viruses and cells infected with the virus [22]. The HTL epitopes regulate the adaptive immune responses by inducing T-cell cytokines [23]. In the current study, CTL and HTL epitopes were predicted by NetCTL 1.2 and NetMHCII 2.2 servers, respectively. We selected 9 and 30 high-score CTL and HTL epitopes, respectively, which were able to bind to more types of MHC alleles. Next, they were evaluated for antigenicity, allergenicity, toxicity, and IFN-γ production induction (only for HTL epitopes). Finally, a total of six CTL (Additional file 1: Table S1) and nine HTL epitopes (Additional file 1: Table S2) were selected for the four target proteins.
Molecular interaction analysis of selected CTL and HTL epitopes with HLA alleles
We used the ClusPro 2.0 server to perform molecular docking analysis to ensure that the selected T cell epitopes interacted with the HLA molecules. The results of molecular docking of CTL and HTL epitopes with HLA-A*02:01 and DRB1*01:01 molecules revealed that among CTL epitopes, the YAWVLVFVY epitope had the lowest energy score (− 1033.7 kcal/mol), while among HTL epitopes, AFTVYVFCFLLPMLL epitope with a docking score of − 1162.8 kcal/mol and FVYIPLFLIHTHARF epitope with a docking score of − 1162.6 kcal/mol had the lowest energy score (Table 2) (Additional file 1: Figs. S1 and S2).
Vaccine construct design
The selected HTL epitopes sequence was checked for overlap with the CTL epitopes sequence. It was found that the FVYIPLFLIHTHARF epitope overlapped the FLIHTHARF epitope. Consequently, the insertion of this CTL epitope in the vaccine construct was ignored. The C-terminal of Mycobacterium tuberculosis HSP70 was used as an adjuvant to achieve maximum immune response. The final structure of the multi-epitope vaccine consisted of 509 amino acids, including one adjuvant, five CTL epitopes, and nine HTL epitopes. The linkers included one EAAAK linker, four AAY linkers, and nine GPGPG linkers (Fig. 2).

Graphical map of the designed vaccine construct
Analysis of physicochemical, antigenicity, and allergenicity parameters
The ProtParam server predicted the physicochemical properties of the vaccine construct, which are shown in Table 3. The vaccine's composition included a total of 509 amino acids. Tryptophan was the least (0.2%), while alanine and glycine were the most frequent (11%) residues in the vaccine structure (Fig. 3). The solubility of the proposed vaccine was calculated to be 0.987 by the SOLpro server. The VaxiJen server computed an antigenic score of 0.5425 for the main sequence (without the adjuvant) of the vaccine construct. The final sequence antigenicity (along with the adjuvant) with the bacteria and virus models was estimated to be 0.9426 and 0.5221, respectively. The ANTIGENpro predicted the antigenicity of the main sequence of the vaccine to be 0.509057 and it increased to 0.609880 by attaching the adjuvant sequence. AllerTOP v.2.0 server identified the vaccine sequence as non-allergenic.

Composition of amino acids in the vaccine construct. The multi-epitope vaccine consists of 509 residues. Alanine and glycine are the highest and tryptophan is the lowest residues in the vaccine
Secondary structure prediction
The vaccine secondary structure was predicted by the PDBsum. The vaccine construct was composed of 12 helices, 10 helix-helix interacts, 55 beta turns, and 29 gamma turns (Fig. 4).

Graphical representation of the secondary structure of the designed vaccine
Prediction, refinement, and validation of the 3D structure
I-TASSER server generated five models for vaccine construct using the threading templates (PDB Hit: 5E85, 2KHO, 1DKX, 1U00, and 1DKZ). The C-scores of models 1–5 were − 3.71, − 2.38, − 2.31, − 2.57, and − 2.59, respectively. The C-scores range from − 5 to 2 for the predicted 3D models, and a model with the highest C-score is a high-confidence model. Therefore, model 3 with a C-score of − 2.31 was selected for the refinement (Fig. 5). The GalaxyRefine server was used to refine the vaccine structure. Model 2 was selected based on various parameters, including GDT-HA (0.9401), RMSD (0.45), MolProbity (2.293), Clash score (12.4), Poor rotamers (1.5), and Rama favored (89.9) (Fig. 5). Zlab and ProSA-web were utilized for the evaluation of the vaccine construct quality. The Ramachandran plot analysis of the unrefined showed 86.375%, 8.759%, and 4.866% of residues in the highly preferred, preferred, and questionable regions, respectively (Fig. 6A). After refinement, the highly preferred, preferred, and questionable regions had 95.134%, 3.65%, and 1.217% of residues, respectively (Fig. 6B). The overall quality score for protein structures was obtained from ProSA-web. The Z-score of the unrefined model was calculated to be − 7.32 (Fig. 6C), which reached − 7.36 after refinement (Fig. 6D).

The initial and refined models of the three-dimensional structure of the multi-epitope vaccine are shown in cyan and tan, respectively

A Ramachandran plot of the unrefined model shows that 86.375% of residues be in the highly preferred region, B while in the refined model, 95.134% of residues are in the highly preferred region. C The z-score is − 7.32 in the unrefined model and D − 7.36 in the refined model
B-cell epitopes prediction
BCPred server predicted 16 linear B-cell epitopes with a length of 20 amino acids (Table 4), which are illustrated in Fig. 7. The ElliPro server predicted 12 discontinuous B-cell epitopes (Fig. 8) with the smallest and largest predicted epitopes containing 5 and 55 amino acids, respectively (Table 5).

Linear B-cell epitopes (yellow) are highlighted in the three-dimensional structure of the multi-epitope vaccine (magenta)

The graphical representation of discontinuous B-cell epitopes on the 3D model of the multi-epitope vaccine (A–L). The vaccine construct and discontinuous B-cell epitopes are depicted in gray sticks and yellow surfaces, respectively
Molecular docking of the vaccine construct with TLR4
The ClusPro 2.0 server was used to perform molecular docking of the vaccine with TLR4, and this server generated 30 clusters. Cluster 2 had the lowest energy score and was selected as the best cluster (Fig. 9A). The energy score for the cluster was -892.5 kcal/mol. The PDBSum result showed that a total of 40 vaccine residues interacted with 46 TLR4 residues (chain A). There were 35 hydrogen bonds between the residues of TLR4 (chain A) and the vaccine construct (Fig. 9B). Table 6 shows the residues involved in the formation of these hydrogen bonds as well as the lengths of the bonds.

Molecular docking of the vaccine construct with TLR4. A Docked complex of vaccine and TLR4 (chain A). The vaccine (ligand) is shown in red, and chains A, B, C, and D from TLR4 (receptor) is shown in the blue, cyan, green, and yellow, respectively. B A total of 35 hydrogen bonds (blue line) was found between the residues of TLR4 (chain A) and vaccine construct. The oval structure’s colors represent amino acid properties (positive: blue, negative: red, neutral: green, aliphatic: grey, aromatic: purple, Pro&Gly: orange, and Cys: yellow)
Molecular dynamics simulation
MD simulation of the vaccine-TLR4 docked complex was carried out using GROMACS 2019.6 software The most important method for assessing the stability of protein structures is to calculate their RMSD during MD simulation. TLR4’s RMSD increased at the beginning of the simulation and reached approximately 0.26 nm at 1000 ps. The least RMSD value was 0.19 nm at 5000 ps. Afterward, the value elevated and remained between 0.2 and 0.34 nm until the simulation period ended. The vaccine’s RMSD began with an upward trend and reached 0.58 nm after 5500 ps, and from this time to the end of the simulation period showed a slight fluctuation between 0.46 and 0.6 nm (Fig. 10A). The dynamic behavior of alpha carbon atoms in the structure contains adequate information to study important motions in proteins and reflects the general motions of the structure. Therefore, to investigate motion and structural flexibility, the RMSF of alpha carbon atoms was calculated. The vaccine construct was found to be attached to chains A of TLR4 during the molecular docking step. TLR4 has the same sequence in both chains A and B. To correctly assess the effect of the vaccine binding on chain A flexibility, the RMSF value for both chains was calculated. The flexibility of amino acids 150–450 was the same in chains A and B, while the flexibility at the beginning of chain A (amino acids 25–150) and the terminal part of chain A (amino acids 450 onwards) was lower than chain B. The vaccine’s RMSF value was very high in the 210–240 and 450–480 regions (Fig. 10B).

MD simulation of the vaccine-TLR4 complex. A The RMSD value of the vaccine shows an upward trend from the beginning of the simulation time to 5500 ps, after that, it remains between 0.46 and 0.6 nm. The RMSD score of TLR4 shows slight fluctuations indicating the stability of TLR4 in the vaccine-TLR4 complex. B The peaks indicate the regions with high flexibility
Calculating binding free energies
The MM-GBSA and MM-PBSA methods were used to calculate the binding free energies of TLR4, vaccine, and the TLR4-vaccine complex. The total MM-PBSA binding free energy was − 54,518.71 kcal/mol, − 43,712.38 kcal/mol, and − 98,299.95 kcal/mol for TLR4, vaccine, and the TLR4-vaccine complex, respectively. The total MM-GBSA binding free energy was − 55,282.62 kcal/mol for the TLR4, − 44,324.01 kcal/mol for the vaccine, and − 99,677.11 for the TLR4-vaccine complex. Based on the calculated values, the contributions of gas phase energy appear to be significant in both methods. The details contribution from each energy component are tabulated in Table 7.
Codon optimization and in silico cloning
Back translation and optimization of the codon usage of vaccine sequence in E. coli (strain K12) were conducted by JCat. The GC content and CAI value of the vaccine construct were calculated as 55% and 0.97, respectively. Finally, the DNA sequence of the vaccine was inserted between the two sites of the XhoI and EcoRI restriction enzyme in pET28a ( +) by the SnapGene tool (Fig. 11).

In silico cloning of the vaccine construct in the pET28a ( +). The vaccine's gene sequence is shown in red, and the backbone of the pET28a ( +) is shown in black
Discussion
Cervical cancer is considered to be a major public health concern [1]. There are now commercial prophylactic vaccines available that have been shown to protect vaccinated individuals against HPV infections. High-risk HPV types can cause precancerous lesions and malignant transformation in those who haven’t been vaccinated or are already infected [24]. To fight existing HPV infections and prevent disease progression in cervical cancer, the development of therapeutic HPV vaccines is crucial.
Reverse vaccinology is a novel approach that utilizes computational methods for vaccine design [25]. In recent years, this method has been widely used for designing the novel vaccine candidate against different microorganisms, including Hepatitis B Virus [26], dengue virus [27], human cytomegalovirus [28], Onchocerca volvulus [29], Klebsiella pneumoniae [30], Mycobacterium tuberculosis [31], Helicobacter pylori [32], and Candida auris [33]. Most of the efforts in reverse vaccinology to develop the therapeutic HPV vaccine have been limited to the epitopes prediction of E6 and E7 proteins.
Jabbar et al. [34] predicted the E6 and E7 antigenic peptides from HPV16 and 18 using immunoinformatics methods and analyzed their ability to bind MHC-I molecules through molecular docking and MD simulation [34]. Yao et al. [35] predicted 59 and 22 CTL epitopes from E6 and E7, respectively, using the IEDB server for the alleles that were mostly distributed in the world population [35]. In a few studies, only chimeric structures containing the epitopes of E5, E6, and E7 proteins have been designed, and no other bioinformatics evaluations have been performed on the structure. So far, only one investigation has been close to our study, in which, using bioinformatics tools for each of the E5 and E7 oncoproteins of the high-risk HPV types 16, 18, 31, and 45, a chimeric structure was designed separately and their bioinformatics evaluation was completed [36].
In the present study, for the first time, we designed the chimeric structure of the T-cell epitopes of E5 and E7 proteins from HPV16 and 18. The CTL and HTL epitopes were used as vaccine construction blocks following screening for antigenic, allergenic, and toxicity parameters. To assess the interaction of the selected epitopes with HLA alleles, molecular docking was performed between the selected epitopes and two of the most frequent alleles in the world population, including HLA-A*02:01 and DRB1*01:01 [37]. The interaction of the selected epitopes with HLA molecules indicated that they could play a role in eliciting an immune response. Feltkamp et al. pioneers in HPV epitope studies identified the HPV16-E7 sequence RAHYNIVTF (HPV16-E749-57) as an MHC-I epitope that can elicit CTL-mediated responses and eradicate established HPV l6-induced tumor cells in mice [38, 39], this epitope was also predicted and selected in our study.
The vaccine demonstrates weak immunogenicity if only epitopes are included in the vaccine structure [40]. To resolve this problem, adjuvants should be used along with these vaccines [41]. HSPs are highly conserved molecular chaperons present in both prokaryotes and eukaryotes [42]. HSPs are intracellular protective proteins that contribute to the folding of protein structure and maintenance [43]. The binding of HSP70 to the peptide leads to the further induction of peptide presentation by MHC-I and MHC-II and augments the adaptive immune response [44]. Mycobacterium tuberculosis HSP70 is of particular importance in immunity because it has been shown to activate the innate immune system and elicit specific immune responses against tumors and viral infections [45,46,47,48]. HSP70s have a high degree of homology in total proteins between different species. However, the degree of homology differs between distinct parts of the HSP70 molecule [49]. Using the C-terminal region of HSP70s with a lower degree of homology can prevent potential autoimmune reactions in vaccine design [50]. Based on the contents mentioned above, we attached the C-terminal part of Mycobacterium tuberculosis HSP70 to the vaccine as an adjuvant using an EAAAK linker. Moreover, the selected CTL and HTL epitopes were linked by AAY and GPGPG linkers in the vaccine structure, respectively. The AAY linkers promote epitope presentation and diminish junctional epitopes [51, 52]. The GPGPG linkers increase the solubility and allow adjacent domains to function more freely [53]. The vaccine candidate was analyzed for physicochemical properties. The molecular weight of the vaccine construct was calculated to be 54.34 kDa making it an effective target for vaccine development because of a molecular weight of less than 110 kDa and the ease of purification process [54]. The theoretical pI was found to be 5.66, indicating the vaccine's acidic nature. The vaccine's stability index was calculated to be 32.07, indicating that it is stable because it is less than 40 [55]. The aliphatic index was calculated to be 90.12, this high aliphatic index indicates that the vaccine is stable over a range of temperatures [56]. The GRAVY score was − 0.174, the negative value of which reveals the hydrophilic nature of the vaccine allowing it to interact better with water molecules. The vaccine sequence was antigenic both alone and with adjuvant, and the binding of an adjuvant to the N-terminal of vaccine construct improved vaccine antigenicity. In addition, our findings indicated that the proposed vaccine was non-allergenic and soluble.
The three-dimensional structure of the multi-epitope vaccine was generated by the I-TASSER server and refined by the GalaxyRefine server. According to the results of the Ramachandran plot and ProSA-web, the 3D model of the vaccine construct significantly improved after refinement. The Ramachandran plot showed that 86.375% of the residues of the unrefined model were found in the highly preferred region, while this value increased to 95.134% after refining. The z-score of the unrefined model was − 7.32, while this parameter was − 7.36 after refining. The more negative z-score confirms improvement after the refinement of the three-dimensional structure of the vaccine [57].
Several studies have revealed that TLR4 expression is upregulated in cervical cancer cells compared with other TLRs and causes apoptosis resistance [58, 59]. Therefore, TLR4 was used to conduct the vaccine's molecular docking study. From the clusters generated at the docking stage, a cluster with the lowest energy (− 892.5 kcal/mol) was selected as the input for MD simulation. The RMSD results showed that the stability of the vaccine-TLR4 complex during the simulation period (40 ns) was satisfactory and TLR4 had reached a stable state earlier than the vaccine in the complex. Evaluation of the RMSF plot related to the vaccine demonstrated that vaccine flexibility is very high in the two regions of 210–240 and 450–480, which could be due to the lack of interaction between the two regions and TLR4. The calculation of the binding free energies revealed that the TLR4-vaccine complex is more stable than its individual components. Codon adaptation was done to enhance the expression of vaccine protein in E. coli (strain K12). The GC content and CAI value are essential parameters for evaluating protein expression levels. The GC content and CAI value of the vaccine sequence were 55% and 0.97, respectively. A GC content of 30–70% is required for higher expression [60], and a CAI value of 0.8–1 is considered good for expression in the target organism [61]. As a result, our findings could be regarded as satisfactory. The results of bioinformatics analyzes of this vaccine candidate were promising. However, further in vitro and in vivo experiments are needed to confirm these findings.
Conclusion
HPV prophylactic vaccines have been successful in preventing HPV infection and cancer, but these vaccines do not have therapeutic effects for people infected with the virus. The vaccine development process is costly and time-consuming. Reverse vaccinology is one of the suggested strategies to reduce the cost and time of the vaccine development process. In this study, we used the reverse vaccinology approach to design a multi-epitope vaccine against HPV. The CTL and HTL epitopes of the HPV16/18 E5 and E7 proteins were predicted and then tested for antigenicity, allergenicity, and toxicity. Then the selected epitopes were organized into a chimeric structure along with the appropriate linkers and an adjuvant. The vaccine's physicochemical, solubility, antigenicity, and allergenicity parameters were evaluated, and the vaccine's secondary and three-dimensional structures were predicted. To evaluate the binding affinity, molecular docking was performed between the vaccine’s refined structure and the TLR4, followed by MD simulation to confirm the vaccine structure’s stability. Finally, the vaccine construct was in silico cloned into the pET28a ( +) after the reverse translation. Although the results of the bioinformatics analysis showed that the proposed vaccine can be effective against HPV, but in vitro and in vivo confirmations of these results are needed. The use of nanoparticle delivery systems may help to increase the efficacy of this vaccine.
Methods
Retrieving protein sequences
The target proteins' amino acid sequences were obtained in FASTA format from the NCBI database (https://www.ncbi.nlm.nih.gov/).
Prediction and selection of T-cell epitopes
CTL epitopes were predicted using the NetCTL 1.2 server (https://services.healthtech.dtu.dk/service.php?NetCTL-1.2). In this work, CTL epitopes (9-mer) were identified for 12 types of MHC-I with a threshold of 0.75 (default) [62]. The HTL epitopes of the target protein were identified by NetMHCII 2.3 server (https://services.healthtech.dtu.dk/service.php?NetMHCII-2.3). In the current study, HTL epitopes (15-mer) were predicted for the alleles of HLA-DR, HLA-DQ, and HLA-DP subtypes [63, 64]. The antigenicity, allergenicity, and toxicity of these epitopes were assessed using the VaxiJen v2.0, AllerTOP v.2.0, and ToxinPred servers, respectively. VaxiJen server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) suggests a new alignment-free method for antigen prediction to overcome the limitations of the alignment-dependent methods based on the auto cross-covariance (ACC) transformation of protein sequences into uniform equal-length vectors [65,66,67]. AllerTOP v.2.0 server (https://www.ddg-pharmfac.net/AllerTOP/) utilizes an approach based on the auto ACC conversion of amino acid sequences into regular equal-length vectors [68]. ToxinPred server (https://webs.iiitd.edu.in/raghava/toxinpred/design.php) applied a dataset of 1805 peptides to predict and develop toxic or non-toxic peptides [69]. In addition to these three parameters, the HTL epitopes were checked for IFN-γ production induction using the IFNepitope server (https://webs.iiitd.edu.in/raghava/ifnepitope/design.php). Motif and SVM hybrid was selected as the approach and IFN-γ versus non-IFN-γ as a model for prediction [70].
Molecular interaction analysis of selected CTL and HTL epitopes with HLA alleles
The 3D model of the selected epitopes was predicted by PEP-FOLD 3.5 server (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/). PEP-FOLD is a de novo peptide structure prediction method based on amino acid sequences [71]. In addition, HLA-A*02:01 (PDB ID: 1DUZ) and DRB1*01:01 (PDB ID: 1AQD) structures were collected from the Protein Data Bank (PDB) (https://www.rcsb.org/). The ClusPro 2.0 server (https://cluspro.bu.edu/publications.php) was then used to dock the selected CTL and HTL epitopes with HLA-A*02:01 and DRB1*01:01, respectively [72,73,74].
Vaccine construct design
The selected CTL and HTL epitopes were utilized to design the vaccine construct. Appropriate linkers ensure that epitopes are properly separated and that amino acid residues are flexible enough to fold into appropriate conformations [35]. AAY linkers were used to connect CTL epitopes, whereas GPGPG linkers were used to connect HTL epitopes. The C-terminal of Mycobacterium tuberculosis HSP70 (amino acids 359–625) as an adjuvant was attached at the N-terminal of the vaccine sequence using the EAAAK linker.
Analysis of physicochemical, antigenicity, and allergenicity parameters
Different physicochemical properties of the vaccine construct, such as amino acid number, molecular weight, theoretical pI, instability index, aliphatic index, and GRAVY were predicted using ExPASy ProtParam (https://web.expasy.org/protparam/) [55]. The average molecular weight of an amino acid multiplied by the total number of amino acids in the protein is a standard method for calculating the molecular weight of a protein. The average molecular weight of an amino acid is 110 daltons (Da) [75]. The isoelectric point is the pH at which a protein has no net charge because the positive and negative charges are equal, making it immobile in a direct current electric field. Knowing a protein's theoretical pI can help you choose and optimize protein purification methods like ion-exchange chromatography and isoelectric focusing electrophoresis [76]. The instability index provides an estimate of the stability of proteins in a test tube, and proteins with an instability index of less than 40 are predicted to be stable [77]. The aliphatic index of a protein is defined as the relative volume occupied by aliphatic side chains (alanine, valine, isoleucine, and leucine), and this parameter indicates the protein's heat stability [78]. The GRAVY value is calculated by dividing the sum of all amino acid hydropathy values in a protein sequence by the number of residues in the sequence [79]. The solubility of the proposed vaccine was predicted using the SOLpro server [80]. Furthermore, VaxiJen v2.0 and ANTIGENpro predicted the vaccine's antigenicity. ANTIGENpro (http://scratch.proteomics.ics.uci.edu/) predicts protein antigenicity using the data derived from protein microarray analysis [81]. Allergenicity of the vaccine construct was predicted by AllerTOP v.2.0 server.
Secondary structure prediction
PDBsum (http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=index.html) was used to predict the secondary structure of the proposed vaccine. PDBsum provides information on the protein secondary structure, protein–ligand, protein-DNA interactions, and protein structure quality [82].
Prediction, refinement, and validation of the 3D structure
I-TASSER server (https://zhanggroup.org//I-TASSER/) predicted the three-dimensional structure of the vaccine construct. I-TASSER predicts 3D models from the amino acid sequence and reports a confidence score (C-score) to assess the predicted models' accuracy [83,84,85]. The GalaxyRefine server (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) was used to increase the quality of the chosen 3D model. This server is dependent on a refining strategy that has been tested in CASP10. This method first reconstructs and repacks the side chains, and then, relaxes the overall structure through MD simulation [86]. The Zlab server and ProSA-web were used for the validation of the initial and refined models. The Zlab server (https://zlab.umassmed.edu/bu/rama/) generates the Ramachandran plot. [87]. The overall quality score for input structure is estimated by ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php). If the calculated score falls outside of a range pretty standard of native proteins, the protein structure is likely to have errors [88, 89].
B-cell epitopes prediction
BCPred server (http://ailab-projects1.ist.psu.edu:8080/bcpred/predict.html) was used to predict linear B-cell epitopes [90]. In this study, the sequence of the vaccine construct was used as input, and other parameters remained as default. Furthermore, the Ellipro server was used for the prediction of discontinuous B-cell epitopes. The ElliPro server (http://tools.iedb.org/ellipro/) uses residue clustering algorithms along with Tornton's technique to predict discontinuous B-cell epitopes [91].
Molecular docking of the vaccine construct with TLR4
Molecular docking is a computational method used to analyze the binding interactions between the ligand and receptor molecules [92]. ClusPro 2.0 server carried out the docking analysis of the vaccine construct with TLR4 (PDB ID: 4G8A). The refined vaccine model and TLR4 were introduced to the server as ligand and receptor, respectively. The interaction between vaccine residues and TLR4 residues in the docked complex was mapped using the PDBsum database (http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl) [93].
Molecular dynamics simulation
The MD simulation is a valuable model for examining the behavior of ligands and receptors in complexes formed during the docking process [94]. The selected cluster from the molecular docking stage was used as input for MD simulation. The GROMACS 2019.6 software was used to conduct MD simulation [94,95,96] and the ff99SB force field was applied to prepare the input structure. The structure's surface charge was neutralized with sodium and chlorine ions, and protein was introduced into a layer of TIP3P water molecules with a thickness of 10 angstroms using gmx solvate software. To reduce van der Waals interactions and the formation of hydrogen bonds between water and complex molecules, the energy of the structures was minimized using the steepest descent approach. The system temperature was then steadily raised from 0 to 300 K in 200 ps at constant volume, and the system was equilibrated at constant pressure. The MD simulation was conducted at a temperature of 300 K and a period of 40 ns. The calculation of non-bonded interactions with a cutoff of 10 angstroms was performed by the PME algorithms.
Calculating binding free energies
At this stage, the binding free energies for TLR4, vaccine, and the TLR4-vaccine complex were calculated by the MMPBSA method and using the gmx_MMPBSA tool. The calculation of binding free energies was performed using Poisson–Boltzmann (PB) and generalized Born (GB) approaches [97]. The binding energies were calculated throughout the simulation system using 1000 frames selected at regular intervals from simulation trajectories.
Codon optimization and in silico cloning
The Java Codon Adaptation Tool (JCat) (http://www.jcat.de/) performed reverse translation and codon optimization of the chimeric vaccine to be expressed in an appropriate expression vector. The protein sequence of the vaccine was used as an input, and E. coli (strain K12) was considered as the host organism. The output obtained from this tool includes the codon adaptation index (CAI) and the percentage of GC content. The XhoI and EcoRI restriction sites were added at the 5′ and the 3' end of the sequence, respectively. Finally, the SnapGene software was used for inserting the adapted gene sequence of the vaccine in the pET28a ( +) vector.
Declaration
Availability of data and materials
Datasets used in the experiments are listed as follows: (1) NCBI: https://www.ncbi.nlm.nih.gov/. (2) NetCTL 1.2 server: https://services.healthtech.dtu.dk/service.php?NetCTL-1.2. (3) NetMHCII 2.3 server: https://services.healthtech.dtu.dk/service.php?NetMHCII-2.3. (4) VaxiJen server: http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html. (5) AllerTOP v.2.0 server: https://www.ddg-pharmfac.net/AllerTOP/. (6) ToxinPred server: https://webs.iiitd.edu.in/raghava/toxinpred/design.php. (7) IFNepitope server: https://webs.iiitd.edu.in/raghava/ifnepitope/design.php. (8) ExPASy ProtParam: https://web.expasy.org/protparam/. (9) ANTIGENpro: http://scratch.proteomics.ics.uci.edu/. (10) PDBsum: http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=index.html. (11) I-TASSER server: https://zhanggroup.org//I-TASSER/. (12) GalaxyRefine server: https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE. (13) Zlab server: https://zlab.umassmed.edu/bu/rama/. (14) ProSA-web: https://prosa.services.came.sbg.ac.at/prosa.php. (15). BCPred server: http://ailab-projects1.ist.psu.edu:8080/bcpred/predict.html. ElliPro server: http://tools.iedb.org/ellipro/. ClusPro 2.0 server: https://cluspro.bu.edu/publications.php. JCat: http://www.jcat.de/.
References
Arbyn M, Weiderpass E, Bruni L, de Sanjosé S, Saraiya M, Ferlay J, et al. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. Lancet Glob Health. 2020;8:e191–203.
Fehrmann F, Laimins LA. Human papillomaviruses: targeting differentiating epithelial cells for malignant transformation. Oncogene. 2003;22:5201–7.
Bernard H-U. Taxonomy and phylogeny of papillomaviruses: an overview and recent developments. Infect Genet Evol. 2013;18:357–61.
Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–9.
Gitsch G, Kainz C, Reinthaller A, Kopp W, Tatra G, Breitenecker G. Cervical neoplasia and human papilloma virus infection in prostitutes. Sex Transm Infect. 1991;67:478–80.
Zheng Z-M, Baker CC. Papillomavirus genome structure, expression, and post-transcriptional regulation. Front Biosci. 2006;11:2286.
Schiller J, Lowy D. Explanations for the high potency of HPV prophylactic vaccines. Vaccine. 2018;36:4768–73.
Schiller JT, Castellsagué X, Garland SM. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine. 2012;30:F123–38.
Mantovani F, Banks L. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene. 2001;20:7874–87.
Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Can Res. 1996;56:4620–4.
Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene. 1999;18:7873–82.
Ganguly N, Parihar SP. Human papillomavirus E6 and E7 oncoproteins as risk factors for tumorigenesis. J Biosci. 2009;34:113–23.
Venuti A, Paolini F, Nasir L, Corteggio A, Roperto S, Campo MS, et al. Papillomavirus E5: the smallest oncoprotein with many functions. Mol Cancer. 2011;10:140.
Stöppler MC, Straight SW, Tsao G, Schlegel R, McCance DJ. The E5 gene of HPV-16 enhances keratinocyte immortalization by full-length DNA. Virology. 1996;223:251–4.
Maufort JP, Shai A, Pitot HC, Lambert PF. A role for HPV16 E5 in cervical carcinogenesis. Can Res. 2010;70:2924–31.
Pim D, Collins M, Banks L. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene. 1992;7:27–32.
Wasson CW, Morgan EL, Müller M, Ross RL, Hartley M, Roberts S, et al. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget. 2017;8:103581–600.
Oh J-M, Kim S-H, Cho E-A, Song Y-S, Kim W-H, Juhnn Y-S. Human papillomavirus type 16 E5 protein inhibits hydrogen peroxide-induced apoptosis by stimulating ubiquitin–proteasome-mediated degradation of Bax in human cervical cancer cells. Carcinogenesis. 2009;31:402–10.
Hemmat N, Baghi HB. Human papillomavirus E5 protein, the undercover culprit of tumorigenesis. Infect Agent Cancer. 2018;13:31.
Kanampalliwar AM, Soni R, Girdhar A, Tiwari A. Reverse vaccinology: basics and applications. J Vaccines Vaccin. 2013;4:2.
Flower DR, Macdonald IK, Ramakrishnan K, Davies MN, Doytchinova IA. Computer aided selection of candidate vaccine antigens. Immunome Res. 2010;6:1–16.
Doherty PC, Allan W, Eichelberger M, Carding SR. Roles of alphabeta and gammadelta T cell subsets in viral immunity. Annu Rev Immunol. 1992;10:123–51.
Kalita P, Lyngdoh DL, Padhi AK, Shukla H, Tripathi T. Development of multi-epitope driven subunit vaccine against Fasciola gigantica using immunoinformatics approach. Int J Biol Macromol. 2019;138:224–33.
Burd EM. Human papillomavirus and cervical cancer. Clin Microbiol Rev. 2003;16:1–17.
Rappuoli R. Reverse vaccinology. Curr Opin Microbiol. 2000;3:445–50.
Zheng J, Lin X, Wang X, Zheng L, Lan S, Jin S, et al. In silico analysis of epitope-based vaccine candidates against hepatitis B virus polymerase protein. Viruses. 2017;9:112.
Joshi GSKA, Kaushik V. T cell epitope designing for dengue peptide vaccine using docking and molecular simulation studies. Mol Simul. 2020;46:787–95.
Akhtar N, Joshi A, Singh J, Kaushik V. Design of a novel and potent multivalent epitope based human cytomegalovirus peptide vaccine: an immunoinformatics approach. J Mol Liq. 2021;335:116586.
Shey RA, Ghogomu SM, Esoh KK, Nebangwa ND, Shintouo CM, Nongley NF, et al. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci Rep. 2019;9:1–18.
Dar HA, Zaheer T, Shehroz M, Ullah N, Naz K, Muhammad SA, et al. Immunoinformatics-aided design and evaluation of a potential multi-epitope vaccine against klebsiella pneumoniae. Vaccines. 2019;7:88.
Moradi J, Tabrizi M, Izad M, Mosavari N, Feizabadi MM. Designing a novel multi-epitope DNA-based vaccine against tuberculosis: in silico approach. Jundishapur J Microbiol. 2017;10(3):1.
Urrutia-Baca VH, Gomez-Flores R, De La Garza-Ramos MA, Tamez-Guerra P, Lucio-sauceda DG, Rodríguez-padilla MC. Immunoinformatics approach to design a novel epitope-based oral vaccine against Helicobacter pylori. J Comput Biol. 2019;26:1177–90.
Akhtar N, Joshi A, Kaushik V, Kumar M, Mannan MA-u. In-silico design of a multivalent epitope-based vaccine against Candida auris. Microb Pathog. 2021;155:104879.
Jabbar B, Rafique S, Salo-Ahen OM, Ali A, Munir M, Idrees M, et al. Antigenic peptide prediction from E6 and E7 oncoproteins of HPV Types 16 and 18 for therapeutic vaccine design using immunoinformatics and MD simulation analysis. Front Immunol. 2018;9:3000.
Yao Y, Huang W, Yang X, Sun W, Liu X, Cun W, et al. HPV-16 E6 and E7 protein T cell epitopes prediction analysis based on distributions of HLA-A loci across populations: an in silico approach. Vaccine. 2013;31:2289–94.
Namvar A, Panahi HA, Agi E, Bolhassani A. Development of HPV 16, 18, 31, 45 E5 and E7 peptides-based vaccines predicted by immunoinformatics tools. Biotech Lett. 2020;42:403–18.
Singh A, Thakur M, Sharma LK, Chandra K. Designing a multi-epitope peptide based vaccine against SARS-CoV-2. Sci Rep. 2020;10:1–12.
Feltkamp MC, Smits HL, Vierboom MP, Minnaar RP, De Jongh BM, Drijfhout JW, et al. Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur J Immunol. 1993;23:2242–9.
Feltkamp MC, Vreugdenhil GR, Vierboom MP, Ras E, van der Burg SH, Schegget JT, et al. Cytotoxic T lymphocytes raised against a subdominant epitope offered as a synthetic peptide eradicate human papillomavirus type 16-induced tumors. Eur J Immunol. 1995;25:2638–42.
Suhrbier A. Multi-epitope DNA vaccines. Immunol Cell Biol. 1997;75:402–8.
Bolhassani A, Talebi S, Anvar A. Endogenous and exogenous natural adjuvants for vaccine development. Mini Rev Med Chem. 2017;17:1442–56.
Dhakal J, Brah G, Agrawal R, Pawar H, Kaur D, Verma R. Over-expression of gene encoding heat shock protein 70 from Mycobacterium tuberculosis and its evaluation as vaccine adjuvant. Indian J Med Microbiol. 2013;31:123.
Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–77.
Javid B, MacAry P, Oehlmann W, Singh M, Lehner P. Peptides complexed with the protein HSP70 generate efficient human cytolytic Tlymphocyte responses. Biochem Soc Trans. 2004;32:622–5.
Del Giudice G. Hsp70: a carrier molecule with built-in adjuvanticity. Experientia. 1994;50:1061–6.
Ebrahimi SM, Tebianian M, Toghyani H, Memarnejadian A, Attaran HR. Cloning, expression and purification of the influenza A (H9N2) virus M2e antigen and truncated Mycobacterium tuberculosis HSP70 as a fusion protein in Pichia pastoris. Protein Expr Purif. 2010;70:7–12.
Roman E, Moreno C. Synthetic peptides non-covalently bound to bacterial hsp 70 elicit peptide-specific T-cell responses in vivo. Immunology. 1996;88:487–92.
Perraut R, Lussow A, Gavoille S, Garraud O, Matile H, Tolgne C, et al. Successful primate immunization with peptides conjugated to purified protein derivative or mycobacterial heat shock proteins in the absence of adjuvants. Clin Exp Immunol. 1993;93:382–6.
Hunt C, Morimoto RI. Conserved features of eukaryotic hsp70 genes revealed by comparison with the nucleotide sequence of human hsp70. Proc Natl Acad Sci. 1985;82:6455–9.
Li X, Yang X, Li L, Liu H, Liu J. A truncated C-terminal fragment of Mycobacterium tuberculosis HSP70 gene enhanced potency of HBV DNA vaccine. Vaccine. 2006;24:3321–31.
Nezafat N, Ghasemi Y, Javadi G, Khoshnoud MJ, Omidinia E. A novel multi-epitope peptide vaccine against cancer: an in silico approach. J Theor Biol. 2014;349:121–34.
Farhadi T, Nezafat N, Ghasemi Y, Karimi Z, Hemmati S, Erfani N. Designing of complex multi-epitope peptide vaccine based on omps of Klebsiella pneumoniae: an in silico approach. Int J Pept Res Ther. 2015;21:325–41.
Kavoosi M, Creagh AL, Kilburn DG, Haynes CA. Strategy for selecting and characterizing linker peptides for CBM9-tagged fusion proteins expressed in Escherichia coli. Biotechnol Bioeng. 2007;98:599–610.
Naz A, Awan FM, Obaid A, Muhammad SA, Paracha RZ, Ahmad J, et al. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: a reverse vaccinology based approach. Infect Genet Evol. 2015;32:280–91.
Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: The proteomics protocols handbook. Springer; 2005. pp. 571–607.
Foroutan M, Ghaffarifar F, Sharifi Z, Dalimi A, Pirestani M. Bioinformatics analysis of ROP8 protein to improve vaccine design against Toxoplasma gondii. Infect Genet Evol. 2018;62:193–204.
Yakubu A, De Donato M, Imumorin I. Modelling functional and structural impact of non-synonymous single nucleotide polymorphisms of the DQA1 gene of three Nigerian goat breeds. South Afr J Anim Sci. 2017;47:146–56.
Wang Y, Weng Y, Shi Y, Xia X, Wang S, Duan H. Expression and functional analysis of Toll-like receptor 4 in human cervical carcinoma. J Membr Biol. 2014;247:591–9.
Yang X, Cheng Y, Li C. The role of TLRs in cervical cancer with HPV infection: a review. Sign Transduct Target Ther. 2017;2:17055.
Ali M, Pandey RK, Khatoon N, Narula A, Mishra A, Prajapati VK. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci Rep. 2017;7:1–13.
Morla S, Makhija A, Kumar S. Synonymous codon usage pattern in glycoprotein gene of rabies virus. Gene. 2016;584:1–6.
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007;8:424.
Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinform. 2009;10:296.
Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinform. 2007;8:238.
Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8:4.
Doytchinova IA, Flower DR. Identifying candidate subunit vaccines using an alignment-independent method based on principal amino acid properties. Vaccine. 2007;25:856–66.
Doytchinova IA, Flower DR. Bioinformatic approach for identifying parasite and fungal candidate subunit vaccines. Open Vaccine J. 2008;1:4.
Dimitrov I, Flower DR, Doytchinova I: AllerTOP-a server for in silico prediction of allergens. In: BMC bioinformatics. vol. 14: BioMed Central; 2013: pp. 1–9.
Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP, et al. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE. 2013;8:e73957.
Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013;8:30.
Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tufféry P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucl Acids Res. 2012;40:W288–93.
Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, et al. The ClusPro web server for protein–protein docking. Nat Protoc. 2017;12:255.
Vajda S, Yueh C, Beglov D, Bohnuud T, Mottarella SE, Xia B, et al. New additions to the C lus P ro server motivated by CAPRI. Proteins: Struct, Funct Bioinform. 2017;85:435–44.
Kozakov D, Beglov D, Bohnuud T, Mottarella SE, Xia B, Hall DR, et al. How good is automated protein docking? Proteins: Struct Funct Bioinform. 2013;81:2159–66.
Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook. 2005:571–607.
Pergande MR, Cologna SM. Isoelectric point separations of peptides and proteins. Proteomes. 2017;5(1):4.
Guruprasad K, Reddy BV, Pandit MW. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. 1990;4:155–61.
Ikai A. Thermostability and aliphatic index of globular proteins. J Biochem. 1980;88:1895–8.
Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–32.
Magnan CN, Randall A, Baldi P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics. 2009;25:2200–7.
Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, Felgner PL, et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26:2936–43.
Laskowski RA, Jabłońska J, Pravda L, Vařeková RS, Thornton JM. PDBsum: structural summaries of PDB entries. Protein Sci. 2018;27:129–34.
Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–38.
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8.
Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008;9:40.
Ko J, Park H, Heo L, Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucl Acids Res. 2012;40:W294–7.
Anderson RJ, Weng Z, Campbell RK, Jiang X. Main-chain conformational tendencies of amino acids. Proteins. 2005;60:679–89.
Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucl Acids Res. 2007;35:W407–10.
Sippl MJ. Recognition of errors in three-dimensional structures of proteins. Proteins: Struct Funct Bioinform. 1993;17:355–62.
El-Manzalawy Y, Dobbs D, Honavar V. Predicting linear B-cell epitopes using string kernels. J Mol Recognit: Interdiscip J. 2008;21:243–55.
Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008;9:514.
Pandey RK, Kumbhar BV, Srivastava S, Malik R, Sundar S, Kunwar A, et al. Febrifugine analogues as Leishmania donovani trypanothione reductase inhibitors: binding energy analysis assisted by molecular docking, ADMET and molecular dynamics simulation. J Biomol Struct Dyn. 2017;35:141–58.
Laskowski RA. PDBsum new things. Nucl Acids Res. 2009;37:D355–9.
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25.
Páll S, Abraham MJ, Kutzner C, Hess B, Lindahl E. Tackling exascale software challenges in molecular dynamics simulations with GROMACS. In: International conference on exascale applications and software. Springer; 2014: p. 3–27.
Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–54.
Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10:449–61.
Acknowledgements
The authors would like to thank the Cellular and Molecular Research Center, Basic Health Sciences Institute, Shahrekord University of Medical Sciences, Shahrekord, Iran.
Funding
This work was supported by a grant from Shahrekord University of Medical Sciences with Grant Number 3429.
Author information
Authors and Affiliations
Contributions
SS and NB conceptualized the study and wrote the original draft. KAD, HPT, FAD, and GRM performed bioinformatics analyzes. MA, MSN, MRK, and MGD reviewed and edited the manuscript and rewrote it. All authors contributed to the article and approved the submitted manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The ethical committee of Shahrekord University of Medical Sciences approved this study with the number: IR.SKUMS.REC.1399.210.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file1
Table S1: Results of CTL epitope prediction of E5 and E7 proteins from HPV16/18. Table S2: Results of HTL epitope prediction of E5 and E7 proteins from HPV16/18. Figure S1: Molecular docking of the selected CTL epitopes with HLA-A*02:01 molecule. Figure (A–F) represents the CTL epitopes E1-E6 respectively. The HLA-A*02:01 molecule is depicted in tan, and the CTL epitopes are depicted in blue. Figure S2: Molecular docking of the selected HTL epitopes with DRB1*01:01 molecule. Figure (A–I) represents the HTL epitopes E7-E15 respectively. The DRB1*01:01 molecule is shown in gray, and the HTL epitopes are shown in red.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sanami, S., Rafieian-Kopaei, M., Dehkordi, K.A. et al. In silico design of a multi-epitope vaccine against HPV16/18. BMC Bioinformatics 23, 311 (2022). https://doi.org/10.1186/s12859-022-04784-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-022-04784-x