
Abstract
Background
Protein post-translational modification (PTM) is a key issue to investigate the mechanism of protein’s function. With the rapid development of proteomics technology, a large amount of protein sequence data has been generated, which highlights the importance of the in-depth study and analysis of PTMs in proteins.
Method
We proposed a new multi-classification machine learning pipeline MultiLyGAN to identity seven types of lysine modified sites. Using eight different sequential and five structural construction methods, 1497 valid features were remained after the filtering by Pearson correlation coefficient. To solve the data imbalance problem, Conditional Generative Adversarial Network (CGAN) and Conditional Wasserstein Generative Adversarial Network (CWGAN), two influential deep generative methods were leveraged and compared to generate new samples for the types with fewer samples. Finally, random forest algorithm was utilized to predict seven categories.
Results
In the tenfold cross-validation, accuracy (Acc) and Matthews correlation coefficient (MCC) were 0.8589 and 0.8376, respectively. In the independent test, Acc and MCC were 0.8549 and 0.8330, respectively. The results indicated that CWGAN better solved the existing data imbalance and stabilized the training error. Alternatively, an accumulated feature importance analysis reported that CKSAAP, PWM and structural features were the three most important feature-encoding schemes. MultiLyGAN can be found at https://github.com/Lab-Xu/MultiLyGAN.
Conclusions
The CWGAN greatly improved the predictive performance in all experiments. Features derived from CKSAAP, PWM and structure schemes are the most informative and had the greatest contribution to the prediction of PTM.
Similar content being viewed by others


Background
As a common occurrence in the body, protein-translational modification (PTM) plays an important role in regulating various physiological processes and functions. PTM refers to the process of covalent modification of individual amino acid residues on a protein after the mRNA has been translated into a protein [1]. However, insufficient information restricts the analysis of PTMs to delve deeper. In the past few decades, the advancement of proteomics technology and the development of “Big Data” on protein sequences shed light on the substantial study of protein nature. Although high-throughput biological technology has made tremendous achievements in protein PTM identification and analysis, the conventional approaches require expensive labor but get an unsatisfactory understanding of the relationship between structures and functions. Therefore, it is of paramount significance to develop reliable and efficient computational methods for predicting and analyzing modifications.
Alternatively, protein lysine modifications (PLMs), prevalent PTM types, which occur at active ε-amino groups of specific lysine residues in proteins and are critical for orchestrating various biological processes. So far, a series of computational prediction tools have been developed. These predictors firstly employed feature construction methods including sequences and physicochemical properties. Then, machine learning algorithms were adopted to train models. The published predictors about seven types of lysine modified sites are as follows: (1) Acetylation: NetAcet [2], PAIL [3], BRABSB-PHKA [4], PSKAcePred [5], LAceP [6], N-Ace [7], ASEB [8], ProAcePred [9] and DeepAcet [10]; (2) Glycation: GlyNN [11], PreGly [12], Gly-PseAAC [13], Glypre [14], BPB_GlySite [15], and iProtGly-SS [16]; (3) Succinylation: SucPred [17], iSuc-PseAAC [18], iSuc-PseOpt [19], SuccFind [20], SuccinSite [21], pSuc-Lys [22], SSEvol-Suc [23], and PSuccE [24]; (4) Ubiquitination: UbPred [25], CKSAAP_UbSite [26], UbiProber [27], UbiNet [28] and DeepUbi [29]; (5) SUMO: SUMOpre [30], SUMmOn [31] and seeSUMO [32]; (6) Methylation: AutoMotif Server [33], MASA [34], and PSSMe [35]; (7) Malonylation: MaloPred [36] and Mal-Lys [37]. However, these tools cannot implement classification of all potential lysine modified PTMs, only focusing on a single type, which limits the possibility of mining more information and ignores the interconnections of multiple PTMs.
The data imbalance issue was characterized by prediction bias across widely divergent categories, therefore minimizing the bias is essential for downstream exploration in the prediction of PTMs. Here, we aim to harness deep generative methodology to solve the issue. In 2014, Goodfellow et al. first proposed the Generative Adversarial Nets (GAN) [38]. GAN achieved a great success and directly inspired researchers’ interests in image generation and restoration. Later, it was widely used in various fields, especially image processing and natural language processing [39]. The common generative models based on deep learning ideas include VAE (Variational Auto Encoding), GAN, and variant models of GAN (conditional generative confrontation network (CGAN) [40]: adds the label information as well as Wasserstein Generative Adversarial Network WGAN [41]: completely solved the problem of unstable GAN training). To leverage both advantages, we integrated the CGAN and WGAN to construct the CWGAN for powerful ability of processing data-imbalance in this paper.
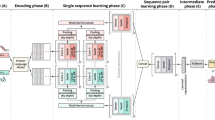
To further study the underlying mechanisms and the relationship of features and some specific modifications, Random Forest was utilized as a classifier and explain feature importance. The whole pipeline MultiLyGAN is shown in Fig. 1a.

a The pipeline of identification of multiple protein modified sites. b Sample size distributions within seven types of PTMs, including Acetylation (S1), Glycation (S2), Malonylation (S3), Methylation (S4), Succinylation (S5), Sumoylation (S6) and Ubiquitination (S7)
Results
Cross-validation results
Among the multiclassification problems, Accuracy (Acc), Confusion Entropy (CEN), Matthews Correlation Coefficient (MCC) and Cross-validation error rate (EC) and independent test error rate (EI) can be measured to evaluate statistical model performance (Details are shown in Additional file: S4). In this work, 4/5 of all samples were used as training samples (for training the model and cross-validation measurement), and the other 1/5 was utilized as an independent test set. There were 2359 features after eight kinds of sequence-encoding schemes and five structural-encoding schemes. High correlations within the features may weaken the prediction performance, resulting in low prediction accuracy, increase training difficulty and over-fitting risk. Thus, Pearson correlation coefficient (PCC) was calculated between each feature and labels, and further, we discarded features with an absolute value of the PCC greater than 0.5.
In the tenfold cross-validation of training samples, after PCC, the Acc increased by 5%; the MCC increased by 0.057; CEN and EC decreased significantly (Table 1). The remaining features after PCC played a more effective role, implying the deleted features have a negative effect on the prediction results. Moreover, the performance was clearly improved after CGAN where MCC reached 0.8114, and EC was reduced by nearly 2 times after CGAN in Table 1.
Compared with CGAN in Table 1, the indicators after the CWGAN were all improved, which demonstrated that the prediction performance of the generative network model was better after adding Wasserstein distance. Acc reached 0.8589; MCC was 0.8376; CEN and EC were the smallest compared with other schemes, which suggested the strong ability of balancing data in CWGAN. Alternatively, we analyzed there might be similar sequence characteristics or structural features among divergent lysine modification types. Table 1 in Additional file: S3 and Fig. 2 showed the confusion matrix of the tenfold cross-validation results. Samples within S1 (Acetylation) were easily predicted to be S7 (Ubiquitination); samples within S2 (Glycation) were prone to classified into S7 and S1; some samples labelled as S3 (Malonylation) were wrongly predicted as the S1, S7 and S5 (Succinylation); samples labelled as S4 (Methylation) were specifically mis-predicted as S1; samples labelled as S5 were easily incorrectly predicted as S1; samples in S6 (SUMO) were mispredicted to S1 and S7; and S7 is easily mispredicted as S1. Therefore, acetylated sequences harbor the largest similarity with other modifications, indicating its function may be interconnected with other types. Sumoylation (S6) and ubiquitination (S7) were easily confused, further demonstrating the sequence or functional correlations between the two processes.

Real-predictive label-bubble chart in tenfold cross validation. The bubble sizes depict the predicted numbers of actual samples. (Python 3.8)
Table 2 and 3 showed the prediction results in each category before and after CWGAN. Strikingly, after CWGAN, the values of Sn were significantly increased, and the false negative rate of prediction was largely reduced. For the balanced AUC value of each category, there was a substantial increase and CWGAN reflected best prediction performance. Figure 3 demonstrated the comprehensive performance of each modification using PCC, CGAN and CWGAN in the training data. To testify whether the prediction AUCs based on different methods are significant, we used DeLong test, one nonparametric test which can compare AUC of two correlated ROC curves. From Table 2 in Additional file: S3, we underscored that PCC + CWGAN + RF was significantly better than RF and PCC + RF, and for S1, S2, S4, and S6, PCC + CWGAN + RF performed significantly better than PCC + CGAN + RF. No statistically better performance in S3, S5, and S7 was shown compared PCC + CWGAN + RF with PCC + CGAN + RF.

ROC curves of seven modification types with random forest in the tenfold cross-validation after PCC, CGAN and CWGAN. True positive rate (sensitivity) as the ordinate, false positive rate as the abscissa. The performance of random forest (RF) classification (baseline), RF with PCC screening, RF with PCC screening and CGAN augmentation, RF with PCC screening and CWGAN augmentation are visualized by green, yellow, purple and red, respectively. Acetylation (S1), Glycation (S2), Malonylation (S3), Methylation (S4), Succinylation (S5), Sumoylation (S6) and Ubiquitination (S7). (Python 3.8)
Independent test results
Profiling the independent dataset which is orthogonal to training set, the results in Table 4 were consistent with the training results (Table 1), which illustrated the robustness of our predictor. Additionally, the realistic and predicted lysine modification types elucidated the similar mechanisms constant with cross-validation results, which provided an effective way to inform the possibly functional connections among different types (Fig. 4, Table 3 in Additional file: S3). The value of Acc was 0.8549, MCC was 0.8330, CEN was 0.2250 and EI was 0.1451 after CWGAN in the independent cohort. Table 5 and 6 demonstrated the better predictive performance after CWGAN for each modification type compared without CWGAN. Figure 5 enumerated ROC curves for each modification and the high AUCs suggested that MultiLyGAN harbored excellent predictive ability for the unseen data. In contract to only RF and RF without augmented data, there was a significant improvement in PCC + CWGAN + RF (Table 4 in Additional file: S3). For S1 and S3, PCC + CWGAN + RF harbored more precise prediction than PCC + CGAN + RF. The CWGAN and CGAN showed no discriminative difference in the remaining types.

Real-predictive label bubble chart for independent test. The bubble sizes indicate the predicted numbers of actual samples. (Python 3.8)

ROC curves of seven modification types in independent test. True positive rate (sensitivity) as the ordinate, false positive rate as the abscissa. The performance of random forest (RF) classification (baseline), RF with PCC screening, RF with PCC screening and CGAN augmentation, RF with PCC screening and CWGAN augmentation are visualized by green, yellow, purple and red, respectively. Acetylation (S1), Glycation (S2), Malonylation (S3), Methylation (S4), Succinylation (S5), Sumoylation (S6) and Ubiquitination (S7). (Python 3.8)
We investigated one case study on distinguishing two confusing modifications easily to be misclassified, which was illustrated in our paper. For example, Ubiquitination and Acetylation (Fig. 2) were reported they had a direct competition [42], and the opposing role between the two PLMs was successfully supported by mass spectrometric profiling [43]. Additionally, recent papers proposed that more complicated crosstalk mechanism was revealed referring to cell cycle regulation [44]. Therefore, the signaling pathways regulated by the two PLMs might affect the function of proteins, possibly leading to difficult identification. Therefore, it is essential to detect the true label. According to Fig. 1 of Additional file: S3, we showed the detailed mis-prediction results, and the thickness of each line was proportional to the number of misclassified samples. Aided by CWGAN, there was an apparent improvement on that fragments labelled as Ubiquitination were wrongly classified into Acetylation.
Data augmentation results
In addition to the improvement of predictive performance, we evaluated CGAN and CWGAN on loss variations in neural network training. This paper adopted the average distance to evaluate simulation data after GAN training. Firstly, the mean of all real data was calculated. Secondly, the Euclidean distance was calculated between the mean value of the simulated data and the real data for each category (modification) as the distance (Fig. 6a). CGAN's distances were all above 0.1, while CWGAN's distances of 6 categories were below 0.03, which indicated that CWGAN's synthetic data were more similar to the original real data. Distances of CGAN fluctuated, while CWGAN’s was stable in different categories.

a Distance comparisons of 6 categories between CGAN and CWGAN. The Euclidean distance was calculated between the mean value of the simulated data and the real data. Distances using CGAN are all above 0.1, while distances with CWGAN of 6 categories are below 0.03, indicating that simulation data aided by CWGAN are more similar to the original real data. P value was calculated by two-sided Mann–Whitney U test. b Loss vs iterations graph of CGAN and CWGAN. The loss iteration graph reflects the stability of the iterative process of different algorithms, which proves that the CWGAN-training process is more even. (Python 3.8)
We calculated the loss of the generator (Gloss) and the loss of the discriminator (Dloss) during 50,000 iterations to compare the advantages and disadvantages of the two algorithms. The Gloss and Dloss in training CGAN and CWGAN on the acetylation modification (S1) was depicted in Fig. 6b, where the upper subgraph is an enlarged graph of CWGAN’s loss coordinate. In the early iterations, the loss of Gloss and Dloss of CGAN tended to be highly changed within 500 iterations. However, it showed long-time fluctuations in the later iterations and eventually showed no convergence. In contrast, the Gloss and Dloss of CWGAN were relatively even and showed no longer changed after 25,000 iterations. Collectively, we confirmed that more stable and convergent augmented data can be accessed in CWGAN training process. Six other modifications had similar results as acetylation.
To testify whether CWGAN had superb performance in comparison to traditional oversampling methods, we applied the Synthetic Minority Oversampling Technique (SMOTE) to conduct the same steps for comparable results. The tenfold cross-validation and independent matrices of SMOTE were lower than CWGAN, and higher than no-augmentation (Table 7, 1 and 4), implying imbalanced data types literally led to worse results and CWGAN gave the more precise predictions compared with SMOTE.
Discussion
Feature analysis
RF gave the order of importance for the 1497-dimensional features. According to the importance degree, the first nine most important features were from the PWM-encoding scheme. The frequency of different amino acids appearing in different positions of the sequence fragment was significantly different, which provided important information. The cumulative importance of different encoding schemes is summarized in Fig. 7. The importance of FoldAmyloid is 0, which provides no identification information; CKSAAP, PWM and structure features are the three most important indicators (Fig. 7a). Figure 7b shows the margin of the three most important amino acids in CKSAAP. Y**A, D**Q and V*A play key roles in the fragments, indicating that there is a significant difference. Figure 7c shows the position information of the top five amino acids in PWM. The amino acid frequency information at + 4, + 7, + 2, − 1 and − 4 positions also differs significantly during different categories. Figure 7d shows the cumulative importance of the structural information of five amino acids. CN showed no contribution, whereas angles showed the largest cumulative contribution. After analysis, it was found that the top three features in structure-encoding schemes were all from secondary structure (SS), indicating that the SS plays an important role in identification.

a Ranking chart of feature importance summarized by different feature-construction methods. a shows that CKSAAP, PWM and Structure coding are the three most informative methods; the importance of FoldAmyloid coding is 0, which provides no identification information. The remaining three images are the three most important amino-acid-space combination in CKSAAP (b), the position information of the top five amino acids in PWM (c), and the cumulative importance of the structural information of five signatures (d)
Comparison with other existing methods
To validate the performance of MultiLyGAN, the comparison of our models with the MusiteDeep [45] was performed. MusiteDeep, a deep-learning based predictor, provided identification for multiple PTMs, including 13 PTMs of which five are lysine-based modifications (Comparisons of number of enrolled proteins and modification sites were in Table 5 of Additional file: S3). We tested four PLMs which are also discussed in MusiteDeep. Using the same independent dataset illustrated above, we analyzed their performance in Table 8. Our method outperformed the MusiteDeep for identification of all four types of PLMs. In MusiteDeep, the Sp in each modification type was obviously higher than Sn, suggesting the lower detection ability for true positive modification types, which was improved by our method.
Conclusions
In this work, we propose a new pipeline to predict the seven types of modified sites, where the GAN was utilized to solve the data imbalance problem. We translated the multilabel prediction problem into a multiclass prediction problem. Overall, 2340 dimensional features were constructed by combining eight different sequences and five structural information-encoding schemes. Finally, 1497 dimensional features were obtained after PCC feature extraction. Through CWGAN, the generated simulation data were closer to the real data. CWGAN yielded Acc of 0.8549, MCC of 0.8330, CEN of 0.2250, and EI of 0.1451 by independent test, which were better scores than those obtained by CGAN. Meanwhile, CWGAN performed better in each of the seven modifications than CGAN.
Methods
Method overview
As illustrated in Fig. 1a, we proposed an integrated protocol including data preprocessing, feature construction, dimensionality reduction, sample augmentation and classification, which implemented stratifications of seven lysine modification types. Preparation of peptide fragments, followed by discarding homologous sequences, was finished in data preprocessing module. Subsequently, substantial sequential and structural signatures were exacted for each sample in feature construction module, after which we used Pearson correlation coefficient (PCC) to acquire principal features in a lower dimensional subspace. To minimize the influence of imbalanced problem that the minority class is prone to incorrectly classified, Conditional Generative Adversarial Network (CGAN) and Conditional Wasserstein Generative Adversarial Network (CWGAN) were carried out. Finally, we built Random Forest (RF) classifiers to identity the seven subtypes, and model performance of multiclass classification was measured by Accuracy (Acc), Confusion Entropy (CEN), Matthews Correlation Coefficient (MCC), Cross-validation error rate (EC) and independent test error rate (EI) (Details are shown in Additional file: S4). MultiLyGAN consisted of PCC, CWGAN and RF.
Data preprocessing
We collected 18 kinds of lysine modification samples from the CPLM2.0 database [46], involving a total of 284,780 modification sites from 53,501 proteins. The types of modifications were Ubiquitination, Acetylation, Succinylation, Malonylation, Sumoylation, Glycation, Methylation, Glutarylation, Propionylation, Crotonylation, Pupylation, Butyrylation, Formylation, Phosphoglycerylation, Hydroxylation, 2-hydroxyisobutyrylation, Neddylation, and Carboxylation. Peptide fragments were obtained through a sliding window technique, with length ξ = 8 in the upper and lower lysine amino acid (window size L = 17). To reduce redundancy and bias, fragments with high sequence similarity (40% or more pairwise sequence identity) were removed. After deleting homology, we obtained 46 2-hydroxyisobutyrylated, 3273 acetylated, 38 butyrylated, 16 carboxylated, 29 crotonylated, 143 formylated, 402 glutarylatd, 1454 glycated, 19 hydroxylated, 1467 malonylated, 1208 methylated, 37 neddylated, 108 phosphoglycerylated, 223 propionylated, 169 pupylated, 1855 succinylated, 1302 sumoylated and 3468 ubiquitinated lysine-centered fragments. These data totally contained 18 different modification types.
We merged the data with the same ID, site and fragments. Eighteen types of modifications contribute to theoretically 218 types of labels for each fragment. After data integration, there were a total of 58 schemes, encompassing 18 for one label, 28 for two labels, and 12 for three labels. The labels with less than 500 samples were deleted. The remaining samples are single-label data, including Ubiq (3253), Ace (3194), Succ (1692), Glyca (1416), Malon (1253), Sumo (1213) and Meth (1172). Since the feature construction consisted of the structural information of each amino acid, we discarded fragments with the length less than 17. Finally, we attained Ubiq (3185), Ace (3114), Succ (1645), Glyca (1399), Malon (1224), Sumo (1174) and Meth (1147). The detailed data of each type are shown in Fig. 1b and Table 6 in Additional file: S3.
In alphabetical order, S1 was set as Ace, S2 as Glyca, S3 as Malon, S4 as Meth, S5 as Succ, S6 as Sumo, and S7 as Ubiq. Thus, the total dataset S can be defined as:
Feature construction
Sequence feature
AAindex [47, 48]
Fourteen specific physical and chemical properties were selected to construct features. A 14-dimensional vector was obtained for every amino acid (L is the length of the fragment):
CKSAAP [49]
The margin K is 0, 1, and 2 between the amino acid pair. If the pair was AA, CKSAAP is “AA,” “AXA,” and “AXXA,” where X is any amino acid. The number 1, 2, 3, …, denotes amino acids according to alphabetical order A, C, D, …, Y. The sample is encoded as:
There are 400 dimensions for each margin k (0, 1, and 2) value, and a 1200-dimensional vector was obtained.
PWM [50, 51]
The position weight matrix was calculated by category to obtain the frequency information of the amino acids at each position. According to the above description, the total length of the sample fragment is L, thus, each sample can be encoded as L-dimensional vector:
\(\left(f\left(1\right), f\left(2\right),\dots \dots , f\left(L\right)\right)\). (4).
Reduced Alphabet [52, 53]
A reduced-letter code 8 is selected, and each amino acid is encoded as an 8-dimensional vector by acid, basic, aromatic, amide, small hydroxyl, sulfur, aliphatic 1 and aliphatic 2. Therefore, a sample of length L is encoded as a vector of 8 × L:
FoldAmyloid [54]
Using http://antares.protres.ru/fold-amyloid/ to predict the amyloidogenic region of the sample and finally obtain the L-dimensional vector:
\(\left(f\left(1\right), f\left(2\right),\dots \dots , f\left(L\right)\right)\). (6).
BE [5, 55]
Under BE (Binary Encoding), each amino acid is encoded into a 20-dimensional binary vector, resulting in a 20 × L-dimensional vector:
PC-PseAAC [56, 57]
Select λ = L-1 = 17–1 = 16, ω = 0.05, physicochemical properties = [‘Hydrophilic’, ‘Hydrophobic’, ‘Quality’]. Each peptide fragment ultimately obtained a (20 + L-1)-dimensional vector:
SC-PseAAC [57, 58]
Select λ = L-1 = 17–1 = 16, ω = 0.05, physicochemical properties = [‘Hydrophilic’, ‘Hydrophobic’]. Each protein fragment ultimately obtained the characteristics of a (20 + 2(L-1))-dimensional vector:
Structure feature
SPIDER3-Single [59] applies LSTM-BRNN to predict Accessible Surface Area (ASA), secondary structure (SS), backbone torsion angles (φ, ψ, θ, τ), Half Sphere Exposure (HSE) and Contact Number (CN), which had a total of 19 outputs. The first was ASA; the next 3 nodes (SS, Q3) were helix (H), strand (E) and coil (C); the next 8 (SS, Q8) were 310-helix (G), α-helix (H), \(\pi\)-helix (I), \(\beta\)-bridge (B), \(\beta\)-strand (E), \(\beta\)-turn (T), bend (S) and coil(C); the next 4 were φ, ψ, θ, and τ; the next 2 were HSEα-up and HSEα-down, and the last output code was CN. SS yields an 11-dimensional vector; ASA is 1-D; φ, ψ, θ, τ are 4-D; HSE is 2-D (HSEα-up, HSEα-down); and CN is 1-D. Therefore, we collected a 19 L-dimensional vector for each protein fragment. Combining sequence and structural features, each peptide was translated into a 2359-dimensional vector (see Table 7 in Additional file: S3).
Sample augmentation
CGAN
GAN has shown its excellent performance in training a generative model. However, there is no control on generative models in GAN and the data being generated are completely random without any information of categories, making it impossible to deal with the imbalance issue. Fortunately, the CGAN model was proposed to direct the data generation process by conditioning the model on additional information, such as class labels. An easy way to extend a GAN to a conditional model is conditioning both the generator and discriminator on some extra information y. The optimization function of CGAN as follow:
CWGAN
We used CWGAN (Conditional Wasserstein Generative Adversarial Network, CGAN under Wasserstein’s method) model, which integrates CGAN and Wasserstein’s distance. The objective of GAN is to learn best parameters for generator so as to minimize the JS divergence between the real distribution \({P}_{data}\left(x\right)\) and the simulated distribution \({P}_{G}\left(x\right)\). However, these two distributions usually have no overlap in sample space, which make their JS divergence always equal to log2 and lead to 0 gradient for parameters of generator. It is difficult for GAN to improve the performance of generator because of the 0 gradient. Therefore, a better method has been proposed to measure the divergence between distributions, which is called as Wasserstein’s distance. When Wasserstein’s distance was used in Conditional GAN, CWGAN can start training even if \({P}_{data}\left(x\right)\) and \({P}_{G}\left(x\right)\) have no intersection. The optimization function of CWGAN as follow:
For the generator, the input includes the prior noise distribution z and the categorical label y embedded as a seven-dimensional vector by one-hot encoding method. There are several main improvements in the network. CWGAN deleted the sigmoid function of the last layer of D. The loss function for G and D no longer used logarithmic transformation. Instead, it used the clip function to update the function and replace Adam with the RMSProp optimization method. Different from common WGAN that outputs the divergence of generative samples in comparison to realistic samples, the discriminator in CWGAN further adds the estimation of whether generative samples are matched to the conditional information. Therefore, CWGAN can generate samples with a specific category.
Using the sample amount of the seventh type of data as a reference, the CWGAN simulation was performed on the other six types, and the simulation data were consistent with the seventh type of data generated. The parameters tuning of CWGAN and the finial parameters of CWGAN are shown in Table 8 and Table 9 of Additional file: S3. Our training data for CWGAN was integrated samples with seven types after PCC screening, including 12,888 samples with 1497 features. After CWGAN, 71 first-class, 1786s-class, 1961 third-class, 2038 fourth-class, 1540 fifth-class, and 2011 sixth-class simulation data were generated. In total, 9407 simulated sample datasets were generated.
Random forest (RF) is a prevalent bagging approach of machine learning. The parameters of RF are shown in Table 10 of Additional file: S3.
Measurements of performance
Two-classification and Multiclassification system indicators are in Additional file: S4.
Availability of data and materials
18 kinds of lysine modification samples were retrieved from the CPLM2.0 database [46] built by authors Dr. Zexian Liu and Dr. Yu Xue (http://cplm.biocuckoo.org/).
Abbreviations
- PTM:
-
Protein post-translational modification
- GAN:
-
Generative adversarial network
- CGAN:
-
Conditional generative adversarial network
- CWGAN:
-
Conditional wasserstein generative adversarial network
- Acc:
-
Accuracy
- CEN:
-
Confusion entropy
- MCC:
-
Matthews correlation coefficient
- RF:
-
Random forest
- EI :
-
Independent test error rate
- EC :
-
Cross-validation error rate
- PCC:
-
Pearson correlation coefficient
References
Wang R, Wang G. Protein modification and autophagy activation. Adv Exp Med Biol. 2019;1206:237–59.
Kiemer L, Bendtsen JD, Blom N. NetAcet: prediction of N-terminal acetylation sites. Bioinformatics. 2005;21(7):1269–70.
Li A, Xue Y, Jin C, Wang M, Yao X. Prediction of Nepsilon-acetylation on internal lysines implemented in Bayesian Discriminant Method. Biochem Biophys Res Commun. 2006;350(4):818–24.
Shao J, Xu D, Hu L, Kwan YW, Wang Y, Kong X, Ngai SM. Systematic analysis of human lysine acetylation proteins and accurate prediction of human lysine acetylation through bi-relative adapted binomial score Bayes feature representation. Mol Biosyst. 2012;8(11):2964–73.
Suo SB, Qiu JD, Shi SP, Sun XY, Huang SY, Chen X, Liang RP. Position-specific analysis and prediction for protein lysine acetylation based on multiple features. PLoS ONE. 2012;7(11):e49108.
Hou T, Zheng G, Zhang P, Jia J, Li J, Xie L, Wei C, Li Y. LAceP: lysine acetylation site prediction using logistic regression classifiers. PLoS ONE. 2014;9(2):e89575.
Lee TY, Hsu JB, Lin FM, Chang WC, Hsu PC, Huang HD. N-Ace: using solvent accessibility and physicochemical properties to identify protein N-acetylation sites. J Comput Chem. 2010;31(15):2759–71.
Wang L, Du Y, Lu M, Li T. ASEB: a web server for KAT-specific acetylation site prediction. Nucleic Acids Res 2012;40(Web Server issue):W376–379.
Chen G, Cao M, Luo K, Wang L, Wen P, Shi S. ProAcePred: prokaryote lysine acetylation sites prediction based on elastic net feature optimization. Bioinformatics. 2018;34(23):3999–4006.
Wu M, Yang Y, Wang H, Xu Y. A deep learning method to more accurately recall known lysine acetylation sites. BMC Bioinform. 2019;20(1):49.
Johansen MB, Kiemer L, Brunak S. Analysis and prediction of mammalian protein glycation. Glycobiology. 2006;16(9):844–53.
Liu Y, Gu W, Zhang W, Wang J. Predict and analyze protein glycation Sites with the mRMR and IFS methods. Biomed Res Int. 2015;2015:561547.
Xu Y, Li L, Ding J, Wu LY, Mai G, Zhou F. Gly-PseAAC: identifying protein lysine glycation through sequences. Gene. 2017;602:1–7.
Zhao X, Zhao X, Bao L, Zhang Y, Dai J, Yin M. Glypre: in silico prediction of protein glycation sites by fusing multiple features and support vector machine. Molecules. 2017;22(11):1891.
Ju Z, Sun J, Li Y, Wang L. Predicting lysine glycation sites using bi-profile bayes feature extraction. Comput Biol Chem. 2017;71:98–103.
Islam MM, Saha S, Rahman MM, Shatabda S, Farid DM, Dehzangi A. iProtGly-SS: Identifying protein glycation sites using sequence and structure based features. Proteins. 2018;86(7):777–89.
Zhao X, Ning Q, Chai H, Ma Z. Accurate in silico identification of protein succinylation sites using an iterative semi-supervised learning technique. J Theor Biol. 2015;374:60–5.
Xu Y, Ding YX, Ding J, Lei YH, Wu LY, Deng NY. iSuc-PseAAC: predicting lysine succinylation in proteins by incorporating peptide position-specific propensity. Sci Rep. 2015;5:10184.
Jia J, Liu Z, Xiao X, Liu B, Chou KC. iSuc-PseOpt: identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal Biochem. 2016;497:48–56.
Xu HD, Shi SP, Wen PP, Qiu JD. SuccFind: a novel succinylation sites online prediction tool via enhanced characteristic strategy. Bioinformatics. 2015;31(23):3748–50.
Hasan MM, Yang S, Zhou Y, Mollah MN. SuccinSite: a computational tool for the prediction of protein succinylation sites by exploiting the amino acid patterns and properties. Mol Biosyst. 2016;12(3):786–95.
Jia J, Liu Z, Xiao X, Liu B, Chou KC. pSuc-Lys: predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J Theor Biol. 2016;394:223–30.
Dehzangi A, Lopez Y, Lal SP, Taherzadeh G, Sattar A, Tsunoda T, Sharma A. Improving succinylation prediction accuracy by incorporating the secondary structure via helix, strand and coil, and evolutionary information from profile bigrams. PLoS ONE. 2018;13(2):e0191900.
Ning Q, Zhao X, Bao L, Ma Z, Zhao X. Detecting succinylation sites from protein sequences using ensemble support vector machine. BMC Bioinformatics. 2018;19(1):237.
Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, Goebl MG, Iakoucheva LM. Identification, analysis, and prediction of protein ubiquitination sites. Proteins. 2010;78(2):365–80.
Chen Z, Chen YZ, Wang XF, Wang C, Yan RX, Zhang Z. Prediction of ubiquitination sites by using the composition of k-spaced amino acid pairs. PLoS ONE. 2011;6(7):e22930.
Chen X, Qiu JD, Shi SP, Suo SB, Huang SY, Liang RP. Incorporating key position and amino acid residue features to identify general and species-specific Ubiquitin conjugation sites. Bioinformatics. 2013;29(13):1614–22.
Nguyen VN, Huang KY, Weng JT, Lai KR, Lee TY: UbiNet: an online resource for exploring the functional associations and regulatory networks of protein ubiquitylation. Database (Oxford) 2016.
Fu H, Yang Y, Wang X, Wang H, Xu Y. DeepUbi: a deep learning framework for prediction of ubiquitination sites in proteins. BMC Bioinform. 2019;20(1):86.
Xu J, He Y, Qiang B, Yuan J, Peng X, Pan XM. A novel method for high accuracy sumoylation site prediction from protein sequences. BMC Bioinform. 2008;9:8.
Pedrioli PG, Raught B, Zhang XD, Rogers R, Aitchison J, Matunis M, Aebersold R. Automated identification of SUMOylation sites using mass spectrometry and SUMmOn pattern recognition software. Nat Methods. 2006;3(7):533–9.
Ren J, Gao X, Jin C, Zhu M, Wang X, Shaw A, Wen L, Yao X, Xue Y. Systematic study of protein sumoylation: development of a site-specific predictor of SUMOsp 2.0. Proteomics. 2009;9(12):3409–12.
Plewczynski D, Tkacz A, Wyrwicz LS, Rychlewski L. AutoMotif server: prediction of single residue post-translational modifications in proteins. Bioinformatics. 2005;21(10):2525–7.
Shien DM, Lee TY, Chang WC, Hsu JB, Horng JT, Hsu PC, Wang TY, Huang HD. Incorporating structural characteristics for identification of protein methylation sites. J Comput Chem. 2009;30(9):1532–43.
Wen PP, Shi SP, Xu HD, Wang LN, Qiu JD. Accurate in silico prediction of species-specific methylation sites based on information gain feature optimization. Bioinformatics. 2016;32(20):3107–15.
Wang LN, Shi SP, Xu HD, Wen PP, Qiu JD. Computational prediction of species-specific malonylation sites via enhanced characteristic strategy. Bioinformatics. 2017;33(10):1457–63.
Xu Y, Ding YX, Ding J, Wu LY, Xue Y. Mal-Lys: prediction of lysine malonylation sites in proteins integrated sequence-based features with mRMR feature selection. Sci Rep. 2016;6:38318.
Goodfellow IJ, Pouget-Abadie J, Mirza M, Xu B, Warde-Farley D, Ozair S. Courville A. Bengio Y: Generat Adversarial Nets. Adv Neur In; 2014. p. 27.
Xue Y, Gao X, Cao J, Liu Z, Jin C, Wen L, Yao X, Ren J. A summary of computational resources for protein phosphorylation. Curr Protein Pept Sci. 2010;11(6):485–96.
Mirza M OS: Conditional generative adversarial nets. Comput. Sci. 2014;2672–2680.
Quan TM, Nguyen-Duc T, Jeong WK. Compressed sensing MRI reconstruction using a generative adversarial network with a cyclic loss. IEEE Trans Med Imaging. 2018;37(6):1488–97.
Tang X, Wen S, Zheng D, Tucker L, Cao L, Pantazatos D, Moss SF, Ramratnam B. Acetylation of drosha on the N-terminus inhibits its degradation by ubiquitination. PLoS ONE. 2013;8(8):e72503.
Danielsen JM, Sylvestersen KB, Bekker-Jensen S, Szklarczyk D, Poulsen JW, Horn H, Jensen LJ, Mailand N, Nielsen ML. Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol Cell Proteom. 2011;10(3):3590.
Liu X, Xiao W, Wang XD, Li YF, Han J, Li Y. The p38-interacting protein (p38IP) regulates G2/M progression by promoting alpha-tubulin acetylation via inhibiting ubiquitination-induced degradation of the acetyltransferase GCN5. J Biol Chem. 2013;288(51):36648–61.
Wang D, Liu D, Yuchi J, et al. MusiteDeep: a deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res. 2020;48:W140–6.
Liu ZX, Wang YB, Gao TS, Pan ZC, Cheng H, Yang Q, Cheng ZY, Guo AY, Ren J, Xue Y. CPLM: a database of protein lysine modifications. Nucleic Acids Res. 2014;42(D1):D531–6.
Saethang T, Payne DM, Avihingsanon Y, Pisitkun T. A machine learning strategy for predicting localization of post-translational modification sites in protein-protein interacting regions. BMC Bioinform. 2016;17(1):307.
Su MG, Huang KY, Lu CT, Kao HJ, Chang YH, Lee TY. topPTM: a new module of dbPTM for identifying functional post-translational modifications in transmembrane proteins. Nucleic Acids Res. 2014;42((Database issue)):537–45.
Wuyun QQG, Zheng W, Zhang YP, Ruan JS, Hu G. Improved species-specific lysine acetylation site prediction based on a large variety of features set. Plos ONE 2016;11(5).
Kao HJ, Weng SL, Huang KY, Kaunang FJ, Hsu JBK, Huang CH, Lee TY: MDD-carb: a combinatorial model for the identification of protein carbonylation sites with substrate motifs. Bmc Syst Biol 2017;11.
Chang WC, Lee TY, Shien DM, Hsu JB, Horng JT, Hsu PC, Wang TY, Huang HD, Pan RL. Incorporating support vector machine for identifying protein tyrosine sulfation sites. J Comput Chem. 2009;30(15):2526–37.
Wong YH, Lee TY, Liang HK, Huang CM, Wang TY, Yang YH, Chu CH, Huang HD, Ko MT, Hwang JK: KinasePhos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic acids research 2007;35(Web Server issue):W588–594.
Yu CS, Chen YC, Lu CH, Hwang JK. Prediction of protein subcellular localization. Proteins. 2006;64(3):643–51.
Garbuzynskiy SO, Lobanov MY, Galzitskaya OV. FoldAmyloid: a method of prediction of amyloidogenic regions from protein sequence. Bioinformatics. 2010;26(3):326–32.
Li TT, Du PF, Xu NF: Identifying Human Kinase-Specific Protein Phosphorylation Sites by Integrating Heterogeneous Information from Various Sources. Plos One 2010, 5(11).
Chou KC. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins-Structure Function And Genetics. 2001;43(3):246–55.
Liu B, Liu F, Wang X, Chen J, Fang L, Chou KC. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015;43(W1):W65-71.
Chou KC. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics. 2005;21(1):10–9.
Heffernan R, Paliwal K, Lyons J, Singh J, Yang Y, Zhou Y. Single-sequence-based prediction of protein secondary structures and solvent accessibility by deep whole-sequence learning. J Comput Chem. 2018;39(26):2210–6.
Acknowledgements
We thanked the anonymous reviewers which gave us very valuable suggestions.
Funding
This work was supported by grants from the Natural Science Foundation of China (12071024) and the Ministry of Science and Technology of China (2019AAA0105103). The funders had no role in the design of the study, the collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
Y.X. and Y.Y. conceived and designed the experiments. Y.Y. and H.W. performed the experiments and data analysis. W.L., H.W. and Y.X. wrote the paper. S.W., Y.L. and X.W. revised the manuscript. All the authors read and agreed on the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent to publication
Not applicable.
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. S1
: Sequence preprocessing. The supplementary material introduces amino acid window sliding technology and feature construction that convert amino acid sequences into numerical vectors.
Additional file 2. S2
: CWGAN generative model.
Additional file 3. S3
: Supplementary tables.
Additional file 4. S4
: Classification system indicators.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, Y., Wang, H., Li, W. et al. Prediction and analysis of multiple protein lysine modified sites based on conditional wasserstein generative adversarial networks. BMC Bioinformatics 22, 171 (2021). https://doi.org/10.1186/s12859-021-04101-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-021-04101-y