
Abstract
Background
Cancer is caused by genetic mutations, but not all somatic mutations in human DNA drive the emergence or growth of cancers. While many frequently-mutated cancer driver genes have already been identified and are being utilized for diagnostic, prognostic, or therapeutic purposes, identifying driver genes that harbor mutations occurring with low frequency in human cancers is an ongoing endeavor. Typically, mutations that do not confer growth advantage to tumors – passenger mutations – dominate the mutation landscape of tumor cell genome, making identification of low-frequency driver mutations a challenge. The leading approach for discovering new putative driver genes involves analyzing patterns of mutations in large cohorts of patients and using statistical methods to discriminate driver from passenger mutations.
Results
We propose a novel cancer driver gene detection method, QuaDMutNetEx. QuaDMutNetEx discovers cancer drivers with low mutation frequency by giving preference to genes encoding proteins that are connected in human protein-protein interaction networks, and that at the same time show low deviation from the mutual exclusivity pattern that characterizes driver mutations occurring in the same pathway or functional gene group across a cohort of cancer samples.
Conclusions
Evaluation of QuaDMutNetEx on four different tumor sample datasets show that the proposed method finds biologically-connected sets of low-frequency driver genes, including many genes that are not found if the network connectivity information is not considered. Improved quality and interpretability of the discovered putative driver gene sets compared to existing methods shows that QuaDMutNetEx is a valuable new tool for detecting driver genes. QuaDMutNetEx is available for download from https://github.com/bokhariy/QuaDMutNetExunder the GNU GPLv3 license.
Similar content being viewed by others

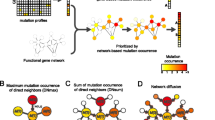
Background
Cancer driver mutations are DNA changes that are causally implicated in oncogenesis [1, 2]. Typically between two and eight mutations, targeting several cellular pathways, are needed for cancer to develop [3]. To disrupt a single pathway or a group of functionally related genes in a way that promotes cancer growth, often only one mutation is needed [4–6].
Most DNA mutations are not cancer drivers. Mutations in DNA accumulate throughout life – for example, comparing skin or gastrointestinal epithelium cells in cancer samples from patients 85 and 25 years old showed that the younger patient on average has half the number of mutation compared to the older patient. More than half of all mutations found in cancer tissue are estimated to have occurred before the start of the disease [7]. Additionally, mutation rate tends to increase in cancer cells [8], although it can differ significantly even among subclones within the tumor [9]. Most of these new random mutations do not contribute to the progression of the disease. An analysis of large number of cancer samples gathered in the Cancer Genome Atlas (TCGA) [10] shows that the total number of mutations present in a tumor tissue from a single patient can range from 10 to more than 100, and only about 2 to 6 among them are driver mutations [11]. Hence, the majority of mutations present in a cancer tissue sample are passenger mutations, with no positive impact on oncogenesis. Due to the potential of using driver genes, that is, genes that harbor driver mutations, for therapeutic, prognostic, or diagnostic purposes, assembling a comprehensive catalogue of driver genes an important ongoing endeavor [12–14]. The main challenge in this task is discovering new driver genes while avoiding false positives stemming from the abundance of passenger mutations.
Statistical and computational methods for detecting driver genes often rely on finding certain pattern of mutations in a group of driver genes across a cohort of patients. To develop cancer, multiple cellular functions must be perturbed, and in different patients, different genes with the same function may be mutated. Often, the cancer develops and is detected before a second mutation in genes with a given function occurs. Thus, for a given cancer type, for a group of patients, each patient would have at least one mutation in a functionally-related group of driver genes, but rarely would have more than one mutation – the gene set exhibits mutual exclusivity pattern of mutations. Several methods detect a set of driver genes by quantifying mutual exclusivity, including Dendrix [15] and Multi-Dendrix [16], RME [17], CoMEt [18], TiMEx [19], MEMo [20], and our own method, QuaDMutEx [21]. An alternative approach involves knowledge of networks linking genes. Frequently mutated genes and their less-frequently mutated neighbors in known human gene- or protein-level pathways or networks are detected as drivers. Methods such as HotNet2 [22, 23], MEMo [20] and DriverNet [24] adopted the network-oriented driver detection approach.
Biological network connectivity and mutual exclusivity are both important sources of information in discovering driver genes. At the same time, both types of information must be used with caution. The available biological networks are incomplete and are expected to include false positives, which might affect the true structure of the network in a way that is unknown. Deviations from mutual exclusivity pattern are expected in individual patients, especially in slow-growing tumors where random mutations have more time to accumulate before cancer is detected. Therefore, an algorithm that uses biological networks and mutual exclusivity at the same time will be able to utilize two complementary, imperfect sources of information to improve the quality of the discovered putative driver gene sets.
We propose a tool, QuaDMutNetEx, which combines the network and exclusivity based approaches. As in our previous tool QuaDMutEx [21], the objective function that is used to select driver genes penalizes for any deviation from the mutual exclusivity pattern. Additionally, QuaDMutNetEx shows preference for genes that are connected in known human biological networks. Compared to mutual exclusivity-based tools such as QuaDMutEx or Dendrix, this additional source of information can help in finding rare driver mutations, for which neither the network connectivity and mutation frequency alone, nor exclusivity alone, are selective enough. The tool models both the network and the mutual exclusivity terms of the objective function as convex, quadratic terms, resulting in a binary quadratic problem, which is solved using our previously proposed technique of efficiently exploring the space of gene sets by using stochastic search through a series of globally optimal solutions to subproblems. Comparisons with existing state-of-the-art methods on four cancer datasets show that the approach of combining network and exclusivity approaches results in improved ability to detect highly connected, mutually exclusive rare driver genes.
Results
We evaluated QuaDMutNetEx using its default parameters that have been selected experimentally: the maximum size of the gene set is ν=50; k=1, indicating equal preference for optimizing coverage and excess coverage; C=2.5; the network parameter was set to α=0.3; the number of iterations was set to T=10,000. In the network-connectivity term of the objective function, we used combined three human protein-protein interaction networks previously used in HotNet2 [23]. The first network is the iRefIndex network, which consists of 91,872 interactions among 12,338 proteins. The second network is MultiNet network which consists of 109,597 interactions among 14,445 proteins. The last network is HINT+HI2012 which is created by considering two interactome databases: HI-2012 data in human HI2 Interactome database, HI2012, and high-quality interactomes database, HINT. The HINT+HI2012 network consists of 40,783 interactions among 10,008 proteins.
We used four datasets to evaluate the proposed algorithm: triple-negative breast cancer (TN), glioblastoma multiforme (GBM), high-grade serous ovarian cancer (HGS), and another breast cancer (METABRIC) dataset (see Table 1). These datasets were previously used in evaluating the DriverNet tool. Following standard practice, known hypermutated genes such as mucins, titin, olfactory receptors, which are unlikely to play role in cancer, were removed [25].
Quantitative and qualitative assessment of QuaDMutNetEx results
The results of the tests, presented in Table 2, show that the proposed method returns gene sets that are statistically significant at 0.05. To assess statistical significance of the results returned by QuaDMutNetEx, we used permutation test proposed in [15]. In short, we randomly permuted the patient-gene matrix in a way that preserves the number of mutations in each patient, and in each gene. This process results in a randomized dataset in which any correlations of mutations are only appearing by chance, but the gene mutation frequencies and patient mutation counts are the same as in the original dataset, which keeps the randomized dataset similar to the original. We created 256 randomized datasets and ran QuaDMutNetEx on each dataset. To obtain a p-value estimate, the final penalty score obtained from running QuaDMutNetEx on the original dataset was compared with the distribution of final penalty scores from running QuaDMutNetEx on the 256 randomized datasets.
The genes discovered by QuaDMutNetEx are presented in Table 3. To evaluate the gene’s driver status, we used COSMIC Cancer Gene Census database [26, 27] and the cancer driver gene database DriverDBv2 [28]. To check QuaDMutNetEx’s effectiveness in discovering rare cancer drivers, we focused on the genes in the solutions that are least frequently mutated in the datasets, and preformed literature review to analyze if these are true or false positives. Additionally, we visually evaluated the resulting gene networks – the largest connected component for each dataset is presented in Fig. 1. To show how inclusion of the network connectivity term in the objective function improves the solution, we have denoted genes found on the same datasets by our mutual-exclusivity-only method, QuaDMutEx. We see that most of the discovered driver genes, especially those mutated with low frequency, result from including the network connectivity term.

Known interactions between driver genes discovered by QuaDMutNetEx on the four datasets: TN: triple-negative breast cancer, GBM: glioblastoma multiforme, HGS: high-grade serous ovarian cancer, and METABRIC: breast cancer
In the triple-negative breast cancer (TN) dataset, out of thirteen identified driver genes, eight are each mutated in only two out of 94 patients, and are analyzed below. A chromatin-remodeling gene CREBBP was found to be overexpressed in breast cancers [29], and is frequently mutated in bladder cancers [30]. DAPK1 is a potential tumor suppressor gene [31, 32]. NCOA1 was found to promote angiogenesis in breast cancers [33]. SLC39A7 is a potential oncogene in colorectal cancer [34]. IDH3B is upregulated in breast cancer and is significantly involved in energy metabolism in tumor progression [35, 36]. HIST1H4A is known to play a role in cell death induction in tumor cells [37]. HIF1A functions as a tumor promoter in cancer-associated fibroblasts, and as a tumor suppressor in breast cancer cells, also it is already a vaccine target in triple-negative breast cancer [38–40]. Finally, MLL methyltransferase are known to have a haematopoietic-specific tumorigenic capability [41].
In the ovarian cancer (HGS) dataset, twenty-three out of twenty-five identified genes are low-frequency driver genes – each is mutated only in two out of 316 patients. Of these twenty-three genes, CTNNB1 is implicated in malignant ovarian transformation [42]. DAG1 and HSPA5 are already drug targets [43, 44]. ERBB2, MST1R, STAT3, VAV3, ERBB3, NTRK2 and JAK2 are known oncogenes [45–50], and FANCA is a potential oncogene [51]. GRB2 is a therapeutic target for solid tumor prevention [52]. PIK3R1 represents a critical driver of endometrial cancer pathogenesis and is a therapeutic target [53]. TSHR signaling promotes the proliferation of ovarian cancer [54]. HSP90AA1 is considered as a potential protein target in therapy of ovarian cancer. [55]. UBC is potential drug resistance-related gene in ovarian cancer [56]. Finally, WRN and CDKN2A are tumor suppressor genes [57, 58].
In the glioblastoma multiforme (GBM) dataset, out of six identified driver genes, one is mutated in five, and two in only two out of 120 patients. Of these, MAPK9 was found to be significantly upregulated in glioma stem cells [59]. MDM2 is a known oncogene [60], while RPL11 is a tumor suppressor gene that acts together with MDM2 in p53 activation pathway [61]. In the METABRIC breast cancer dataset, out of twenty five genes identified by QuaDMutNetEx, six are very rare – each mutated in only two out of 696 patients. Of these, JAK1 is known for its key role in breast cancer progression [62]. EGFR signaling pathway also has a crucial role in mammary cancers [63], and polymorphism in the EGFR ligand, EGF, was found to affect cancer progression [64]. PIK3R1 and VAV1 are known oncogenes [65, 66], SYK is a tumor suppressor gene [67], and PTPN6 has a tumor suppressor role [68]. Together, these results confirm that QuaDMutNetEx is highly effective in identifying cancer driver genes with low mutation frequency.
For comparison, we used two network-based methods, DriverNet and HotNet2. We also used a mutual exclusivity tool, Dendrix. We ran the three tools on the same four datasets: TN, GBM, HGS, and METABRIC. DriverNet was designed to utilize genomic, transcriptomic, and biological network information, HotNet2 utilizes genomic and biological network information, while Denrix used only the genomic information. In all three methods, as well as in QuaDMutNetEx, we used the default parameters. For each method, we analyzed coverage, excess coverage, conformity to the mutual exclusivity pattern as quantified by the Dendrix score \(n - \sum _{i=1}^{n} |G_{i} x - 1|\), and the number of connected components in the subgraph of the biological network consisting of the genes in the solution returned by the method.
Testing in cancer molecular subtypes dataset
Mutual exclusive pattern in tumor can be resulted from other factors [69]. Hence, methods using mutual exclusivity need to account for that. Here we are using GBM subtypes to test the effectiveness of our method [69]. Using copy number, gene expression and methylation, GBM classified into proneural, neural, classical, and mesenchymal [70, 71]. We downloaded GBM data from TCGA and divided them into four subtypes according to TCGA IDs given in [71].
The genes discovered by QuaDMutNetEx are presented in Table 4. To evaluate the gene’s driver status, we used COSMIC Cancer Gene Census database [26, 27] and the cancer driver gene database DriverDBv2 [28]. All of the resulted genes exist in both COSMIC and DriverDB2 or one of them.
Comparison with existing methods
The quality of the solutions returned by QuaDMutNetEx is higher than solutions from other methods (see Table 5). QuaDMutNetEx consistently produces more mutually exclusive gene sets than the network-based methods, and the gene sets are more highly connected – the interaction networks have fewer separate connected components. Compared to Dendrix, the tool that only considers mutual exclusivity, QuaDMutNetEx solutions show slightly lower mutual exclusivity, but at the same time are much more highly connected.
Effects of parameters on QuaDMutNetEx
The behavior of proposed method can be adjusted by three parameters, based on the knowledge of the tumor under study. Parameter α quantifies the reward for gene connectivity in cellular networks – higher values indicate stronger preference for finding densely connected genes. Parameter k controls how steeply the penalty for multiple mutations in a single pathway grows – lower values of k lead to lower penalization of excess coverage in relation to coverage, and is appropriate for slower growing tumors, where additional mutations in any given pathway have more time to accumulate by chance. Finally, higher values of parameter C penalize for solutions sets with many genes.
We have analyzed how these parameters affect the solution by running QuaDMutNetEx for 100,000 iterations for parameters α=0.01,0.05,0.1,0.3,0.6,1 with C=0.25,0.5,1,1.5,2,2.5 and α=0.01,0.05,0.1,0.3,0.6,1 with k=0.25,0.5,1,1.5,2,2.5. Figure 2 shows that the parameter α achieves its design goal, that is, solution with higher α include fewer connected components and prefer connected network. The α parameter has the following effect on coverage and excess coverage: as the value of α increases, the coverage decreases and the excess coverage increases. Furthermore, as the value of α increases, it decreases the effect of C and k. Setting α to a low value, such as 0.001, makes the effect of C and k to be more dominant. Higher coverage and higher excess coverage, as expected, are observed for low k values. High values of C lead to solution sets involving only a few genes, while low values of C lead to high coverage.

Effects of parameters on QuaDMutNetEx. a, b: effect on connected components; c, d: effect on coverage; e, f: effect on excess coverage. Results shown are for the HGS dataset, the results for other datasets are similar
Discussion
The proposed method, QuaDMutNetEx, relies on two sources of information to detect cancer driver genes. It uses observed somatic mutations in a cohort of cancer patients, to detect mutual exclusivity patterns, and a biological network encoding functional relationships between genes to provide context for the observed data. Relying on two sources of information is a strength of the proposed method, since treated individually, each source is imperfect. Biological networks are know to be incomplete and contain false positives, and the functional, regulatory, and signaling relationships they capture are not all directly relevant to cancer. Mutual exclusivity patterns may not be perfectly present in the observed patient mutation data. This may be true especially for slower growing tumors, where the time from onset of the disease to its detection is long enough to allow for accumulation of additional mutations in functionally-related sets of genes that contribute to cancer. Depending on the knowledge of the analyzed type of cancer and characteristics of the studied patient cohort, the users of QuaDMutNetEx should adjust the parameters of the methods that govern the strength of preference for mutual exclusivity in relation to patient coverage, the weight assigned to the network knowledge, and the strength of preference for small driver gene sets. Users of QuaDMutNetEx should also keep in mind that it uncovers driver genes relevant to the cohort from which the mutation data comes from. That is, it detects drivers present in the particular set of patients, based on the particular type of mutation data provided.
Conclusions
Experiments on four datasets show that QuaDMutNetEx has the ability to detect driver genes mutated with low frequency, genes that may be missed by existing tools that rely on mutual exclusivity alone, or on frequency and network information alone. Improvements in the quality and interpretability of the discovered putative driver gene sets makes QuaDMutNetEx a valuable addition to the family of driver discovery tools.
Methods
Input for QuaDMutNetEx
QuaDMutNetEx input has two sources of information. The first source is the binary somatic mutation matrix as in many mutual-exclusivity tools [15, 21]. Specifically, the data for n patients, each with total of p genes explored for possible existence of somatic mutations, arrives in a form of a mutation matrix G, an n by p sparse binary matrix. We expect Gij=1 if patient i has a somatic mutation in gene j, that is, a difference between cancer tissue and matched healthy tissue from the same patient is detected; otherwise, Gij=0. The change can be a point mutation in the coding region of the gene, potentially affecting its function. It could also be a mutation in the non-coding, regulatory elements of the DNA associated with the gene, or copy number alternation of the gene in case of homozygous deletions and high-level amplifications, affecting its expression. A row of the matrix describing mutations in patient i will be referred to as a vector Gi. The second source of information is a gene connectivity matrix A, with nonzero Aij values for genes i and j that are known to be related in a biologically meaningful way, for example one gene regulates the other, or proteins encoded by the genes are known to interact in a signaling pathway. The output of the method is a column vector x of length p, with xj=1 indicating that gene j is a putative cancer driver gene, that is, its mutations can contribute to cancer growth, and zero otherwise. The non-zero elements of the solution will be referred to as the solution gene set.
Defining the quality of potential driver gene sets
For the binary solution vector x with length p genes, there are 2p−1 possible non-zero solution vectors, each encoding a different gene set. The challenge is to find a gene set that is composed of driver genes. To this end, we designed a penalty score that reflects how likely it is that genes encoded by a solution vector form a comprehensive set of driver genes impacting a single cellular function. The lower the penalty score, the more likely the solution consists of related driver genes. The penalty score has two major terms: a network term, and a mutual-exclusivity term.
The network terms captures our preference for solution gene sets in which the products of transcribing the genes are connected in known human protein-protein interaction networks. Other sources of pairwise gene relationships could be used, for example functional similarity, or regulatory interactions. This network connectivity preference is captured by a term N(A,x) in the objective function, where A is the undirected adjacency matrix corresponding to the network, and x is the solution gene set. The new term introduces a reward for any two genes in a solution that are connected. Since the solution vector is binary, the additional term can be defined as \(N(A,x)=-x^{T}Ax=-\sum _{i,j} A_{ij} x_{i} x_{j}\). The scaled term αN(A,x) with nonnegative weight α corresponds to providing a reward of α every time two genes i and j present in the solution, that is, with xi=1 and xj=1, are connected by an edge, that is, when Aij=1. The effect of introducing the network term can be seen in Fig. 3.

Illustration of the role of the network term N(A,x). Based solely on the mutual exclusivity, potential solutions 1 and 2 are equally good, both show perfect mutual exclusivity. Inclusion of network term N(A,x) makes potential solution 2 the preferred one, since it consists of more highly connected genes
The second term in the objective function captures mutual exclusivity pattern among solution genes. We use a flexible, quadratic term previously used in our mutual exclusivity method, QuaDMutEx [21]. Briefly, the term penalizes for solutions that leave some patients not showing any mutation in the solution genes, as well as for solutions in which patients are covered by more than one mutation. The penalty for excess mutations grows quadratically with the number of mutations over one. The ration of penalty for multiple mutations to penalty for no mutation can be tweaked by parameter k. For example, for a slow growing tumor, where there is ample time for additional mutations to accumulate in a single pathway, k should be low. In addition, we define parameter C to be a penalty incurred by adding one more gene to the solution set.
For any possible solution vector x, the penalty score is a sum of the two terms described above, and is
Algorithm for finding high-quality driver gene sets
The minimization of the quadratic penalty function L(G,A,x) over binary vectors x is an example of an unconstrained binary quadratic problem (BQP). While BQPs are known to be NP-hard [72], for problems small enough the optimal solution can still be found. For example, for datasets with up to 1000 patients, if one focuses on only ν=50 genes, the solution x that is the global minimum of L(G,A,x) can be found in below a second.
As we have shown before [21], high-quality approximate solutions to BQP problems involving thousands of genes can be found efficiently by an iterative algorithm that maintains an evolving set of size ν consisting of candidate driver genes, and in each of the T iterations finds an optimal solution to a small instance of the problem in Eq. 1 involving only the current candidate genes. This allows for improving the quality of the driver gene set, while exploring a diverse set of possible genes as candidates.
A single run of QuaDMutNetEx will return a set of functionally-related driver genes with high mutual exclusivity and high network connectivity. Running QuaDMutNetEx in sequence, removing discovered genes from input matrices G and A after each iteration, will allow to uncover genes from multiple pathways needed for oncogenesis, although the joint solution is no longer expected to have high connectivity, nor to conform to the mutual exclusivity pattern.
Abbreviations
- TCGA:
-
The Cancer Genome Atlas
- TN:
-
Triple negative breast cancer
- GBM:
-
Glioblastoma multiforme
- HGS:
-
High-grade serous ovarian cancer
- COSMIC:
-
Catalogue of somatic mutations in cancer
- DDBv2:
-
DriverDBv2
References
Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, et al.Signatures of mutation and selection in the cancer genome. Nature. 2010; 463(7283):893–8.
Knudson AG. Cancer genetics. Am J Med Genet. 2002; 111(1):96–102.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer Genome Landsc. Science. 2013; 339(6127):1546–58.
McCormick F. Signalling networks that cause cancer. Trends Biochem Sci. 1999; 24(12):53–6.
Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004; 10(8):789–99.
Yeang C-H, McCormick F, Levine A. Combinatorial patterns of somatic gene mutations in cancer. FASEB J. 2008; 22(8):2605–22.
Tomasetti C, Vogelstein B, Parmigiani G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci. 2013; 110(6):1999–2004.
Loeb LA. Human cancers express mutator phenotypes: origin, consequences and targeting. Nat Rev Cancer. 2011; 11(6):450–457.
Kennedy SR, Schultz EM, Chappell TM, Kohrn B, Knowels GM, Herr AJ. Volatility of mutator phenotypes at single cell resolution. PLoS Genet. 2015; 11(4):1005151.
Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM, Network CGAR, et al.The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013; 45(10):1113–20.
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al.Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502(7471):333–9.
Dimitrakopoulos CM, Beerenwinkel N. Computational approaches for the identification of cancer genes and pathways. Wiley Interdiscip Rev Syst Biol Med. 2017; 9(1). https://doi.org/10.1002/wsbm.1364.
Ding L, Wendl MC, McMichael JF, Raphael BJ. Expanding the computational toolbox for mining cancer genomes. Nat Rev Genet. 2014; 15(8):556.
Chen J, Sun M, Shen B. Deciphering oncogenic drivers: from single genes to integrated pathways. Brief Bioinforma. 2014; 16(3):413–28.
Vandin F, Upfal E, Raphael BJ. De novo discovery of mutated driver pathways in cancer. Genome Res. 2012; 22(2):375–85.
Leiserson MD, Blokh D, Sharan R, Raphael BJ. Simultaneous identification of multiple driver pathways in cancer. PLoS Comput Biol. 2013; 9(5):1003054.
Miller CA, Settle SH, Sulman EP, Aldape KD, Milosavljevic A. Discovering functional modules by identifying recurrent and mutually exclusive mutational patterns in tumors. BMC Med Genom. 2011; 4(1):1.
Leiserson MD, Wu H-T, Vandin F, Raphael BJ. CoMEt: a statistical approach to identify combinations of mutually exclusive alterations in cancer. Genome Biol. 2015; 16(1):1.
Constantinescu S, Szczurek E, Mohammadi P, Rahnenführer J, Beerenwinkel N. TiMEx: a waiting time model for mutually exclusive cancer alterations. Bioinformatics. 2015; 32(7):968–75.
Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012; 22(2):398–406.
Bokhari Y, Arodz T. QuaDMutEx: quadratic driver mutation explorer. BMC Bioinformatics. 2017; 18(1):458.
Vandin F, Upfal E, Raphael BJ. Algorithms for detecting significantly mutated pathways in cancer. J Comput Biol. 2011; 18(3):507–22.
Leiserson MD, Vandin F, Wu H-T, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, et al.Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet. 2015; 47(2):106–14.
Bashashati A, Haffari G, Ding J, Ha G, Lui K, Rosner J, Huntsman DG, Caldas C, Aparicio SA, Shah SP. DriverNet: uncovering the impact of somatic driver mutations on transcriptional networks in cancer. Genome Biol. 2012; 13(12):124.
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al.Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499(7457):214–8.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al.Cosmic: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2018; 47(D1):941–7.
cancer.sanger.ac.uk. Accessed 14 Dec 2019.
Chung I-F, Chen C-Y, Su S-C, Li C-Y, Wu K-J, Wang H-W, Cheng W-C. DriverDBv2: a database for human cancer driver gene research. Nucleic Acids Res. 2016; 44(D1):975–9.
Chin SF, Teschendorff AE, Marioni JC, Wang Y, Barbosa-Morais NL, Thorne NP, Costa JL, Pinder SE, van De Wiel MA, Green AR, et al.High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer. Genome Biol. 2007; 8(10):215.
Duex JE, Swain KE, Dancik GM, Paucek RD, Owens C, Churchill ME, Theodorescu D. Functional impact of chromatin remodeling gene mutations and predictive signature for therapeutic response in bladder cancer. Mol Cancer Res. 2018; 16(1):69–77.
Yadav P, Masroor M, Nandi K, Kaza R, Jain S, Khurana N, Saxena A. Promoter methylation of BRCA1, DAPK1 and RASSF1A is associated with increased mortality among indian women with breast cancer. Asian Pac J Cancer Prev APJCP. 2018; 19(2):443.
Qin R, Wolfenson H, Saxena M, Sheetz M. Tumor suppressor DAPK1 catalyzes adhesion assembly on rigid but anoikis on soft matrices. bioRxiv. 2018:320739. https://doi.org/10.1101/320739.
Qin L, Xu Y, Xu Y, Ma G, Liao L, Wu Y, Li Y, Wang X, Wang X, Jiang J, et al.NCOA1 promotes angiogenesis in breast tumors by simultaneously enhancing both HIF1 α- and AP-1-mediated VEGFa transcription. Oncotarget. 2015; 6(27):23890.
Ventura-Bixenshpaner H, Asraf H, Chakraborty M, Elkabets M, Sekler I, Taylor KM, Hershfinkel M. Enhanced ZnR/GPR39 activity in breast cancer, an alternative trigger of signaling leading to cell growth. Sci Rep. 2018; 8(1):8119.
Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N, Howell A, Martinez-Outschoorn UE, et al.Ketones and lactate "fuel" tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010; 9(17):3506–514.
Cai Y, Nogales-Cadenas R, Zhang Q, Lin J-R, Zhang W, O’Brien K, Montagna C, Zhang ZD. Transcriptomic dynamics of breast cancer progression in the MMTV-PyMT mouse model. BMC Genomics. 2017; 18(1):185.
Yan-Fang T, Zhi-Heng L, Li-Xiao X, Fang F, Jun L, Gang L, Lan C, Na-Na W, Xiao-Juan D, Li-Chao S, et al.Molecular mechanism of the cell death induced by the histone deacetylase pan inhibitor LBH589 (panobinostat) in wilms tumor cells. PloS ONE. 2015; 10(7):0126566.
Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, Tanowitz HB, Casimiro MC, Wang C, Pestell RG, et al.HIF1-alpha functions as a tumor promoter in cancer-associated fibroblasts, and as a tumor suppressor in breast cancer cells: autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010; 9(17):3534–51.
Ponente M, Campanini L, Cuttano R, Piunti A, Delledonne GA, Coltella N, Valsecchi R, Villa A, Cavallaro U, Pattini L, et al.PML promotes metastasis of triple-negative breast cancer through transcriptional regulation of HIF1A target genes. JCI Insight. 2017; 2(4). https://doi.org/10.1172/jci.insight.87380.
Ssempala A. Vaccine targeting HIF1A in triple negative breast cancer. Eur J Cancer. 2017; 72:26.
Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015; 15(6):334.
Palacios J, Gamallo C. Mutations in the β-catenin gene (CTNNB1) in endometrioid ovarian carcinomas. Cancer Res. 1998; 58(7):1344–7.
Pathak HB, Zhou Y, Sethi G, Hirst J, Schilder RJ, Golemis EA, Godwin AK. A synthetic lethality screen using a focused siRNA library to identify sensitizers to dasatinib therapy for the treatment of epithelial ovarian cancer. PloS ONE. 2015; 10(12):0144126.
Sethi G, Pathak HB, Zhang H, Zhou Y, Einarson MB, Vathipadiekal V, Gunewardena S, Birrer MJ, Godwin AK. An RNA interference lethality screen of the human druggable genome to identify molecular vulnerabilities in epithelial ovarian cancer. PLoS ONE. 2012; 7(10):47086.
Revillion F, Bonneterre J, Peyrat J. ERBB2 oncogene in human breast cancer and its clinical significance. Eur J Cancer. 1998; 34(6):791–808.
Bonomi S, Gallo S, Catillo M, Pignataro D, Biamonti G, Ghigna C. Oncogenic alternative splicing switches: role in cancer progression and prospects for therapy. Int J Cell Biol. 2013; 2013. https://doi.org/10.1155/2013/962038.
Lee K, Liu Y, Mo JQ, Zhang J, Dong Z, Lu S. Vav3 oncogene activates estrogen receptor and its overexpression may be involved in human breast cancer. BMC Cancer. 2008; 8(1):158.
Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA, et al.Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013; 23(5):603–17.
Jones DT, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz H-J, Zichner T, Lambert SR, Ryzhova M, Quang DAK, et al.Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013; 45(8):927.
Sakai K, Ukita M, Schmidt J, Wu L, De Velasco MA, Roter A, Jevons L, Nishio K, Mandai M. Clonal composition of human ovarian cancer based on copy number analysis reveals a reciprocal relation with oncogenic mutation status. Cancer Lett. 2017; 405:22–8. https://doi.org/10.1016/j.canlet.2017.07.013.
Naderi A, Teschendorff A, Barbosa-Morais N, Pinder S, Green A, Powe D, Robertson J, Aparicio S, Ellis I, Brenton J, et al.A gene-expression signature to predict survival in breast cancer across independent data sets. Oncogene. 2007; 26(10):1507.
Giubellino A, Burke TR, Bottaro DP. Grb2 signaling in cell motility and cancer. Expert Opin Ther Targets. 2008; 12(8):1021–33.
Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, et al.High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011; 11. https://doi.org/10.1158/2159-8290.cd-11-0039.
Huang W-L, Li Z, Lin T-Y, Wang S-W, Wu F-J, Luo C-W. Thyrostimulin-TSHR signaling promotes the proliferation of NIH: OVCAR-3 ovarian cancer cells via trans-regulation of the EGFR pathway. Sci Rep. 2016; 6:27471.
Chu S. -h., Liu Y. -w., Zhang L, Liu B, Li L, Shi J. -z.Regulation of survival and chemoresistance by HSP90AA1 in ovarian cancer SKOV3 cells. Mol Biol Rep. 2013; 40(1):1–6.
Yin F, Liu X, Li D, Wang Q, Zhang W, Li L. Tumor suppressor genes associated with drug resistance in ovarian cancer. Oncol Rep. 2013; 30(1):3–10.
Ayyildiz D, Gov E, Sinha R, Arga KY. Ovarian cancer differential interactome and network entropy analysis reveal new candidate biomarkers. Omics: J Integr Biol. 2017; 21(5):285–94.
Wrzeszczynski KO, Varadan V, Byrnes J, Lum E, Kamalakaran S, Levine DA, Dimitrova N, Zhang MQ, Lucito R. Identification of tumor suppressors and oncogenes from genomic and epigenetic features in ovarian cancer. PloS ONE. 2011; 6(12):28503.
Kim S-H, Ezhilarasan R, Phillips E, Gallego-Perez D, Sparks A, Taylor D, Ladner K, Furuta T, Sabit H, Chhipa R, et al.Serine/threonine kinase MLK4 determines mesenchymal identity in glioma stem cells in an NF- κB-dependent manner. Cancer Cell. 2016; 29(2):201–13.
Schiebe M, Ohneseit P, Hoffmann W, Meyermann R, Rodemann H-P, Bamberg M. Analysis of mdm2 and p53 gene alterations in glioblastomas and its correlation with clinical factors. J Neuro-Oncol. 2000; 49(3):197–203.
Zheng J, Lang Y, Zhang Q, Cui D, Sun H, Jiang L, Chen Z, Zhang R, Gao Y, Tian W, et al.Structure of human MDM2 complexed with RPL11 reveals the molecular basis of p53 activation. Genes Dev. 2015; 29(14):1524–34.
Wehde BL, Rädler PD, Shrestha H, Johnson SJ, Triplett AA, Wagner K-U. Janus kinase 1 plays a critical role in mammary cancer progression. Cell Rep. 2018; 25(8):2192–207.
Hardy KM, Booth BW, Hendrix MJ, Salomon DS, Strizzi L. ErbB/EGF signaling and EMT in mammary development and breast cancer. J Mammary Gland Biol Neoplasia. 2010; 15(2):191–9.
Bhowmick DA, Zhuang Z, Wait SD, Weil RJ. A functional polymorphism in the EGF gene is found with increased frequency in glioblastoma multiforme patients and is associated with more aggressive disease. Cancer Res. 2004; 64(4):1220–3.
Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, Thomas RJ, Phillips WA. The phosphatidylinositol 3’-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001; 61(20):7426–9.
Shalom B, Farago M, Pikarsky E, Katzav S. Vav1 mutations identified in human cancers give rise to different oncogenic phenotypes. Oncogenesis. 2018; 7(10):80.
Coopman PJ, Do MT, Barth M, Bowden ET, Hayes AJ, Basyuk E, Blancato JK, Vezza PR, McLeskey SW, Mangeat PH, et al.The Syk tyrosine kinase suppresses malignant growth of human breast cancer cells. Nature. 2000; 406(6797):742.
Lazo JS, McQueeney KE, Burnett JC, Wipf P, Sharlow ER. Small molecule targeting of PTPs in cancer. Int J Biochem Cell Biol. 2018; 96:171–81.
van de Haar J, Canisius S, Michael KY, Voest EE, Wessels LF, Ideker T. Identifying epistasis in cancer genomes: A delicate affair. Cell. 2019; 177(6):1375–83.
Cancer Genome Atlas Research Network and others, et al.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455(7216):1061.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al.The somatic genomic landscape of glioblastoma. Cell. 2013; 155(2):462–77.
Kochenberger G, Hao J-K, Glover F, Lewis M, Lü Z, Wang H, Wang Y. The unconstrained binary quadratic programming problem: a survey. J Comb Optim. 2014; 28(1):58–81.
Acknowledgments
The authors thank the development team of Gurobi for providing free, academic license for their optimization software.
Funding
TA is supported by NSF grant IIS-1453658. NSF provided funds for the article processing fee and for the corresponding author’s work on the research presented in this manuscript. The funding agency had no role in study design, in data collection, analysis and interpretation, or in manuscript preparation.
Author information
Authors and Affiliations
Contributions
YB and TA conceived the method. YB, AA and TA drafted the manuscript. YB implemented the method. YB and TA performed and analyzed the experiments. YB, AA and TA read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bokhari, Y., Alhareeri, A. & Arodz, T. QuaDMutNetEx: a method for detecting cancer driver genes with low mutation frequency. BMC Bioinformatics 21, 122 (2020). https://doi.org/10.1186/s12859-020-3449-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-020-3449-2