
Abstract
Background
Protein-DNA interaction governs a large number of cellular processes, and it can be altered by a small fraction of interface residues, i.e., the so-called hot spots, which account for most of the interface binding free energy. Accurate prediction of hot spots is critical to understand the principle of protein-DNA interactions. There are already some computational methods that can accurately and efficiently predict a large number of hot residues. However, the insufficiency of experimentally validated hot-spot residues in protein-DNA complexes and the low diversity of the employed features limit the performance of existing methods.
Results
Here, we report a new computational method for effectively predicting hot spots in protein-DNA binding interfaces. This method, called PreHots (the abbreviation of Predicting Hotspots), adopts an ensemble stacking classifier that integrates different machine learning classifiers to generate a robust model with 19 features selected by a sequential backward feature selection algorithm. To this end, we constructed two new and reliable datasets (one benchmark for model training and one independent dataset for validation), which totally consist of 123 hot spots and 137 non-hot spots from 89 protein-DNA complexes. The data were manually collected from the literature and existing databases with a strict process of redundancy removal. Our method achieves a sensitivity of 0.813 and an AUC score of 0.868 in 10-fold cross-validation on the benchmark dataset, and a sensitivity of 0.818 and an AUC score of 0.820 on the independent test dataset. The results show that our approach outperforms the existing ones.
Conclusions
PreHots, which is based on stack ensemble of boosting algorithms, can reliably predict hot spots at the protein-DNA binding interface on a large scale. Compared with the existing methods, PreHots can achieve better prediction performance. Both the webserver of PreHots and the datasets are freely available at: http://dmb.tongji.edu.cn/tools/PreHots/.
Similar content being viewed by others


Background
With the rapid development of structural biology technologies such as X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy, a large number of tertiary structures of biological macromolecules have been generated [1]. However, the interpretation of these structures and the recognition of critical residues lie far behind the step of structure generation. Proteins and DNA are two kinds of most important biological macromolecules of life compounds. The interactions of proteins and DNA are essential for many crucial cellular processes, including gene expression and regulation, DNA replication and repair. For example, genes are regulated by the DNA-binding proteins that bind to some specific DNA sequences [2, 3]. Although DNA-protein binding interfaces contain a large number of residues, the associations between DNA and proteins are governed by a small fraction of residues with high binding affinity, which are also called hot spots. Hot spots are considered the most crucial residues for the formation and stabilization of protein complexes. Hence, accurate identification of hot spots is important to understand molecular regulation mechanisms and provide solutions to disease diagnosis and treatment [4].
At present, many experimental techniques have been used to measure protein-DNA binding free energy by site-directed mutagenesis, such as surface plasmon resonance (SPR) [5], isothermal titration calorimetry (ITC) [6] and fluorescence resonance energy transfer (FRET) [7]. However, these experimental techniques are not only inefficient and laborious, but also not suitable for dealing with the vast amounts of residues. Therefore, efficient and effective computational methods for identifying protein-DNA binding hot spots are greatly desirable and urgently needed.
Computational approaches can complement the experimental methods and make large-scale predictions efficiently. Molecular dynamics simulations and feature-based approaches are effective ways to identify hot spots. Two molecular dynamics simulation methods, SAMPDI [8] and PremPDI [9], were proposed to predict the change of protein-DNA binding free energy. SAMPDI utilizes the modified Molecular Mechanics Poisson-Boltzmann Surface Area (MM/PBSA) approach [10] along with additional knowledge-based features to predict binding affinity changes upon single mutation, while PremPDI relies on molecular mechanics force fields and fast side-chain optimization algorithms to evaluate the effects of single mutations on protein-DNA interactions. As for feature based approaches, a method called mCSN-NA [11] was developed, which uses graph-based signatures to predict the impact of a single mutation on protein-nucleic acid binding. Another feature-based approach PrPDH [12] was developed to predict protein-DNA binding hot spots. Although substantial advances have been made, there is still much space to explore for accurately identifying DNA-binding hot spots.
In this work, we develop a novel computational approach PreHots (the abbreviation of Predicting Hotspots), which is based on stack ensemble of boosting algorithms, for effectively predicting hot spots in protein-DNA binding interfaces. To this end, a dataset was constructed, which contains 260 samples from 89 protein-DNA complexes. More than half of the data are manually collected from the literature by ourselves, and the rest data are from the databases of ProNIT [13] and dbAMEPNI [14]. We totally calculated 157 features for fully representing hot spots, including not only the properties of the target residue but also target residues’ network information. From these features, a set of 19 informative features are selected by using a sequential backward selection algorithm. Extensive experiments were conducted on the benchmark dataset and the independent dataset to evaluate the proposed method, with comparison to existing methods. The experimental results show that our method can significantly boost the performance of DNA-binding hot spots prediction.
Methods
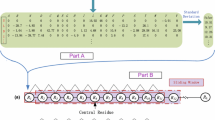
Figure 1 shows the workflow of the proposed method PreHots. First, a new reference dataset that consists of 123 hot spots and 137 non-hot spots from 89 protein-DNA complexes is constructed. The data are manually collecting from the literature and databases with a strict process of redundancy removal. Then, four types of features are encoded to characterize the target residues, including network features, exposure features, sequence features and structural features. Next, the informative features are selected by using sequential backward selection method. Following that, three boost classifiers, including categorical boosting (Catboost) [15], extreme gradient boosting (XGBoost) [16] and gradient tree boosting (GTB) [17] classifiers, are taken as the base models to form an ensemble stacking classifier (ESC), by a meta-model that adopts logistic regression (LR) [18] classifier. Finally, prediction results are output by the ESC model based on the selected feature set.

The workflow of PreHots
Datasets
We constructed an initial dataset, containing experimentally measured binding free energy changes of 660 mutations from 162 protein-DNA complexes, which were obtained by combining two databases and manually searching the literature. Among them, 79 protein-DNA crystal structures were obtained from the database of ProNIT [13] and dbAMEPNI [14], and the other 83 protein-DNA crystal structures were manually collected from the literature.
To build high quality protein-DNA binding hot spots dataset, we used two methods to determine the interface residues. Solvent accessibility area (SAS) is widely used to identify interfacial residues, which can be obtained by calculating the difference of absolute solvent accessibility (ΔASA >1Å) and the ratio of relative solvent accessibility (RASA >5%). And to make the results more accurate and stable, the ASA and RASA values of residues are calculated from protein structures by using Naccess [19]. Another method is to calculate the distance between the target residue and the DNA strand. If the distance is less than 5Å, the target residue can be considered as the interface residue. Moreover, we removed redundant homology sequences, where the similarity of protein sequences is more than 40% by using CD-HIT [20]. In this study, we define hot spots as the interface residues with the change in binding free energy (ΔΔG) ≥1.0 kcal/mol, and the others are defined as non-hot spots. Finally, the constructed dataset consists of 123 hot spots and 137 non-hot spots from 89 complexes. In order to construct a balanced dataset to reduce the potential bias of the machine learning method, 64 protein-DNA complexes were randomly selected to form the benchmark dataset, which contains 90 hot spots and 90 non-hot spots. The rest of 25 protein-DNA complexes constitute the independent dataset, including 33 hot spots and 47 non-hot spots. To the best of our knowledge, our dataset is the largest one for predicting protein-DNA binding hot spots.
Performance measures
We do performance evaluation by 10-fold cross-validation. The benchmark dataset is randomly divided into 10 subsets, each of which contains approximately the same number of samples. For each round, nine subsets are merged as the training set, while the remaining one subset is used for testing.
For comprehensively assessing the performance of our model, we adopted seven widely used evaluation metrics, including accuracy (ACC), sensitivity (SEN/Recall), specificity (SPE), precision (PRE), F1-score (F1), Matthew’s correlation coefficient (MCC) and the area under the ROC curve (AUC). ACC, SEN, SPE, PRE, F1 and MCC are defined as follows:
Above, TP is the number of true positives, FP is the number of false positives, TN is the number of true negatives and FN is the number of false negatives, respectively.
Feature description
In order to explore informative features that play important role in the prediction of protein-DNA binding hot spots, we collected a comprehensive feature set that consists of 157 features, which can be roughly divided into four groups: residue interaction network features, solvent exposure features, and traditional features based on protein sequence and structure. More details about these feature are given below.
Residue interaction network features
As a representative kind of protein structures, residue interaction networks (RINs) have been widely and successfully used for revealing the effect of residues mutation, functional region and protein folding [21]. The traditional way to build RINs is to calculate the distance between Cα atoms of two residues within a certain threshold, which ranges from 5 to 9Å [22, 23]. But in fact, the interaction of protein-DNA depends on several intermolecular factors such as hydrogen bonds, van der Waals contacts, ionic bond and several other factors [24, 25]. The stability of protein-DNA interaction is maintained by forming hydrogen bonds between amino acid side chain residues of protein and DNA bases [26]. Therefore, the construction of RINs based on whether there is an intermolecular interaction between any two nodes, including residue and DNA, in the protein-DNA complexes by using RING [27]. In this study, five intermolecular interactions are considered: hydrogen bond, Van der Waals, disulfide bond, salt bridge, π- π stacking and π-cation.
To make the network contain more knowledge, each edge weight is assigned with the distance between two corresponding nodes. We calculate 10 RINs features that represent the importance of the target residue in the RINs, including node degree, clustering, closeness, betweenness, eigenvector, eccentricity, average neighbor degree, flow closeness, square clustering and Katz centrality.
Solvent exposure features
Solvent exposure of amino acid is crucial for exploring and predicting protein interaction and function. Solvent exposure features consist of several types of features, including half-sphere exposure (HSE), contact number (CN), residue depth (RD), accessible surface area (ASA) and relative accessible surface area (RASA). The solvent accessible has been extensively and successfully utilized to predict protein-protein interaction hot spots [28–31]. The limitation of solvent accessible is that it cannot provide any information about completely buried residues. Compared with traditional solvent accessible, half-sphere exposure (HSE) can describe the local environment of the target residue better from another perspective [32]. RD represents the average atom depth of target residue atoms, while CN is the number of residues in the sphere within a specific distance [33].
In this study, we calculated the characteristics of half-sphere exposure, contact number and residue depth, which could complement the solvent exposure information of interface residues. Based on protein sequence, a series of computing tools have been developed for predicting HSE, CN. We choose the method of HSEpred [32] and SPOT-1D [34] to calculate these features. For protein structure, we use hsexpo [33] to calculate the above three types of features, including HSE, CN and RD.
Structure-based features
Based on the three-dimensional structures of proteins, structure-based features were calculated, including hydrogen bonds, consensus scores, secondary structures, fluctuation score and solvent accessible surface area.
1. Hydrogen bonds (Hbond).
The stability of protein-DNA interaction is maintained by forming hydrogen bonds between amino acid side chain residues of protein and DNA bases [26]. The hydrogen bond of protein-DNA complexes were calculated by using HBPLUS [35].
2. Consensus scores.
Consensus score is a linear combination of residue interface propensity score, residue energy score and residue conservation score. Here, we used ENDES [36] to calculate consensus score, while the side chain energy score and relative solvent accessibility can also be obtained.
3. Secondary structure (SS).
As an important feature, the secondary structural characteristics of residues were obtained from both sequences and structures of proteins. The definition of secondary structures of proteins (DSSP) [37] defines the secondary structure according to atomic coordinates in the protein data bank (PDB) [1]. In addition, several tools can predict the secondary structure of residue from protein sequence, including SPOT-1D [34], NetSurfp2 [38] and SPIDER3 [39].
4. Fluctuation score.
The study of protein fluctuation is helpful to understand protein structures. FlexPred was used to predict the value of residue fluctuations [40]. Meanwhile, B-factor, represents the dynamic motion of atoms in a protein, was extracted from the PDB file.
5. Solvent accessible surface area.
Solvent accessible surface area, including available surface area (ASA) and relatively accessible surface area (RASA), which has a strong correlation with hot spot prediction [12]. We applied Naccess [19] to calculate the ASA and RASA of residues from protein-DNA complexes.
Sequence-based features
Based on previous studies, we calculated many features of protein-DNA binding residues from protein sequences.
1. Position-specific scoring matrix (PSSM).
It is well known that PSSM is an essential feature for predicting hot spots [4, 28, 31]. PSSM score represents the relationship between the frequency of amino acid substitutions and that expected by chance. Negative numbers indicate less frequent substitutions than expected by chance, while positive numbers mean more frequent substitutions than expected.
2. Conservation score.
Conservative analysis of residues is extensively used to identify functionally important residues in protein sequences. The conservation score of residues can be calculated by using Jensen-Shannon divergence [41].
3. Solvent accessible surface area.
Apart from deriving solvent accessibility from protein structure, we also used SPIDER3 [39] and NetSurfp2 [38] to calculating ASA and RASA from protein sequence.
4. Physicochemical features.
Amino acid indices database (AAindex) collects various biochemical and physicochemical characteristics of amino acids [42]. In this work, protein-DNA binding hot spots are described by eight physicochemical characteristics: propensities, polarity, hydrophilicity, average accessible surface area, atom-based hydrophobic moment, flexibility parameter for no rigid neighbors, hydrophobicity and polarizability.
5. Blocks substitution matrix (BLOSUM).
BLOSUM62 [43] means that sequence similarity is more than 62% in terms of sequence alignment. We calculated BLOSUM62, the most widely used amino acid scoring matrix, whose scores indicate the similarity between two types of amino acids.
6. Local structural entropy (LSE).
Previous research found that local structural entropy is related to the stability of protein, and it was successfully used for predicting protein-protein interaction hot spots. In this work, we calculated the LSE [44] value of each residue within a protein sequence.
7. Disordered regions (DISO).
Recognizing protein disorder regions contributes to the understanding of protein function and protein fold pathway. SPOT-Disorder [45] and RaporX-Property [46] were used to predict disorder regions of protein-DNA binding residues.
Feature selection
For high-dimensional datasets, feature selection can effectively remove some irrelevant features, which contributes to lifting the efficiency of learning tasks and making the model easier to be understood. We used a sequential backward selection (SBS) algorithm to select a subset of informative features that are highly relevant to protein-DNA binding hot spots from the initial set of 157 features. Sequential backward selection (SBS), which is a heuristic search algorithm, removes one feature each time till an optimal feature subset is generated. Here, each resulting feature set is evaluated by using 10-fold cross-validation with the ESC classifier. Such 10-fold cross-validation procedure is repeated 30 times and the average performance over 30 trials is taken as the result. Besides, we combine the independent dataset and each cross-validation test dataset as the test dataset, which is used to evaluate features and obtain the evaluation score at each 10-fold cross-validation. The evaluation metric of feature selection is represented by Ec, calculated as follows:
where R is the number of cross-validation; n is the number of iterations of 10-fold cross-validation; ACCi, SENi, SPEi, MCCi, and AUCi indicate the values of accuracy, sensitivity, specificity, Matthew’s correlation coefficient and AUC score of the i-th 10-fold cross-validation, respectively.
In the SBS method, features are iteratively removed one by one from the initial feature set. In the first round, each feature is deleted once (resulting in 157 subsets of 156 features). If the ESC classifier based on a certain feature subset achieves the higher Ec, this feature subset is left for the next round of feature selection. Such a feature selection process would continues till Ec does not increase any more.
Ensemble stacking classifier
Stacking, also called super learning [47], is an ensemble machine learning method that constructs the base-level models and meta-model by combining different machine learning classifiers. The construction of base-level models is based on the benchmark dataset, and the meta-model is trained on the outputs of the base-level models. The ensemble stacking classifier (ESC) can overcome the disadvantage of single classifier and make the prediction more robust than a single model. In this study, we choose three boost classifiers as the base-level models, which are categorical boosting (Catboost) [15], extreme gradient boosting (XGBoost) [16] and gradient tree boosting (GTB) [17] classifiers, and the meta-model adopts logistic regression (LR) [18] classifier.
Results and discussion
Performance of the ensemble stacking classifier
Ensemble stacking classifier (ESC) is an ensemble technique that the output of the first-level (base) classifiers is taken as the input of the second-level classifier by constructing a two-level model. In this study, the first-level classifiers consist of categorical boosting (Catboost) [15], extreme gradient boosting (XGBoost) [16] and gradient tree boosting (GTB) [17] models, and the second-level classifier is a logistic regression (LR) [18] model. To check whether ESC is suitable for predicting hot spots in the complexes, we compared ESC with ensemble vote classifier (EVC) and some popular machine learning models, including random forests (RF) [48], GTB, support vector machine (SVM) [49], Catboost and XGBoost. Among them, the ensemble vote classifier (EVC) is another ensemble technique, which integrates different machine learning algorithms and predicts hot spots by using the average predictive probability of all algorithms. To avoid the randomness of cross-validation results, we do 10-fold cross-validation 30 times and the averaged result of all 30 cross-validation trials is taken as the final result. Table 1 shows the results of ESC and the compared methods. We can see that the ensemble techniques are generally superior to the other machine learning methods. And, ECS outperforms EVC and can significantly improves the performance of hot- spots prediction.
Performance of feature selection
Feature selection is crucial for building accurate classification models, which aims to select a small number of informative features. In this study, our initial feature set consists of 157 candidate features, which can be divided into four groups: residue contact network features (network), solvent exposure features (exposure), sequence features and structural features. We used a sequential backward selection (SBS) method to choose relevant and informative features from the initial feature set. SBS uses a stepwise feature selection scheme, which iteratively removes features one by one from the feature set. The evaluation criterion (Ec) represents the average prediction performance of ESC when selecting features. Figure 2 shows how Ec changes during the process of stepwise feature selection. Ec reaches the maximum when the number of selected features is 19. Consequently, these 19 features form our optimal feature set.

The change of Ec value in the process of stepwise feature selection
To assess the advantage of the SBS method, we compared it with four existing methods, including random forest (RF), recursive feature elimination (RFE) [50], maximum relevance minimum redundancy (mRMR) [51] and the block Hilbert-Schmidt independence criterion (HSIC) Lasso [52]. The commonly used methods are RF, RFE and mRMR, which use the mean decrease Gini index (MDGI), SVM-based recursive feature elimination and max relevance and min redundancy criteria to evaluate the importance of features, respectively. The block HSIC Lasso (HSIC Lasso) is a relatively novel method, which adopts an effective nonlinear feature selection algorithm based on HSIC Lasso to select informative biological features. To obtain reliable results, we ran 30 times of 10-fold cross-validation and took the average performance as final result. Table 2 shows the performance of the five feature selection methods on the benchmark dataset. We can see that SBS can select better features, which are helpful to predict protein-DNA binding hot spots. And the ESC classifier with SBS achieves the best prediction performance, with a 0.535 MCC and a 0.853 AUC.
Significance of selected features
By using the SBS feature selection method, we obtain an optimal feature set, which contains 19 features as shown in Table 3. The ranking of these selected features is based on F-score, which is to measure the distinguishing ability of features between hot and non-hot spots. The most important features include PSSM, hydrogen bonds, secondary structure and RINs features. Two exposure features (as novel features) are selected into the optimal feature set, which indicates that they are important features for identifying DNA-binding hot spots. Fig. 3 shows more details about the distribution of selected features in different feature categories. Six secondary structural features are selected. In previous works, secondary structural has been considered as a fundamental and essential features to improve prediction performance. In this work, we derived secondary structural features from two levels of protein structures and sequences, which can provide a more comprehensive description of secondary structural characteristics of target residues. Besides, ASA, exposure features and consensus score also contribute significantly to the prediction of hot spot residues. These results suggest that the ten categories of 19 optimal features can complement each other and accurately describe the hot spot residues, thus collectively improve the prediction performance.

The number of each class features on the optimal feature set
Performance comparison with state-of-the-art methods
Here, we further compare our method with four existing protein-DNA binding hot spots prediction methods, including PrPDH [12], PremPDI [9], mCSM-NA [11] and SAMPDI [8], on the benchmark dataset and the independent test dataset. PrPDH uses a classification model to identify hot-spots from various interface residues, while PremPDI, mCSM-NA and SAMPDI use regression models to predict the change of Protein-DNA binding free energy.
Table 4 presents the results on the benchmark dataset, where the prediction results of existing methods are from their websites. In general, our method performs better than the other methods in terms of six of the seven metrics (ACC, SEN, SPE, FRE, F1, MCC and AUC). Only our SPE is smaller than that of the mCSM-NA method.
Table 5 gives the results on the independent test dataset. Compared with the existing methods, our method significantly improves the prediction performance. Concretely, 81.8% of the true hot spots are correctly predicted (SEN = 0.818) and 76.6% of the non-hot spots are correctly predicted (SPE = 0.766). Except for SPE, our method achieves the highest values of the other metrics, especially for the comprehensive indexes MCC (0.576) and AUC (0.82). These results show that our method is superior to the existing methods in identifying protein-DNA binding hot spots.
Case study
The λ exonuclease (λexo) and DNA complex.
λexo is an ATP-independent enzyme that binds double-stranded DNA (dsDNA) to form the λexo-DNA complex (PDB ID: 3SM4, chain: A) [53]. Four mutated interfacial residues of the λexo-DNA complex have experimentally been identified and shown in Fig. 4. The hot spots residues (ΔΔG) ≥1.0 kcal/mol are K49_A and R137_A, and the rest are non-hot spots (K76_A and M53_A). Our approach successfully identified all the hot spots, while only a non-hot spot (K76_A) was wrongly identified. In addition, PremPDI, PrPDH and SAMPDI only correctly predicted two non-hot spots (K76_A and M53_A), while the two hot spots were wrongly predicted. mCSM-NA only correctly predicted one non-hot spots (M53_A). This example shows that our method can effectively identify hot spots from protein-DNA complexes than the major existing methods.

The hot spot residues of λexo-DNA complex (PDB ID: 3SM4) identified by experiments. The green surface denotes the protein chain (chain A) while the purple and yellow surfaces represent the DNA chains (purple: chain E and yellow: chain D). The red color represents experimentally identified hot spot residues and the blue color represents experimentally determined non-hot spot residues
DNA-bound SUP- 1228−121 complex.
The structure of DNA-bound SUP- 1228−121 (PDB ID:4CH1, chain: A) can provide accurate clue to the mechanism of DNA recognition [54]. The defined hot spot residues are K36_M, Y44_A, E63_K, R103_M, A110_T and G113_E, and the remaining three residues (Y78_F, N106_A and N108_A) are non-hot spots (see Fig. 5). For these nine mutated residues, PremPDI identified three of the six hot spots (K36_M, R103_M and A110_T) and one non-hot spot (Y78_F). PrPDH predicted two residues as hot spots (R103_M and A110_T) and the others as non-hot spots. SAMPDI identified one residue as hot spot (Y44_A) and the others non-hot spots, while mCSM-NA predicted all residues as non-hot spots. On the contrary, except for a hot spot (K36_M), our method predicted correctly all the other residues. This suggests that our method has the highest accuracy, which is desirable for many biological applications.

The hot spot residues of DNA-bound SUP- 1228−121 complex (PDB ID: 4CH1) identified by experiments. The green surface denotes the protein chain (chain A) while the purple surface represents the DNA chain (chain B). The red color represents experimentally identified hot spot residues and the blue color represents experimentally determined non-hot spot residues
Webserver
A user-friendly webserver of PreHots has been implemented, which is available at: http://dmb.tongji.edu.cn/tools/PreHots/. The input to PreHots should be the PDB file, which contains at least one protein chain and one DNA strand. The user can select the chain of protein and DNA, and submit the job to the server. Then, PreHots will return a list of residues, which are predicted to be potential DNA-binding hot spots based on the ensemble classifier with optimally features. Interface residues are colored according to the predicted confidence score. For visual display, users can use the 3D viewer to display prediction results and download the results. Multiple PDB files can be submitted simultaneously, and the jobs are executed in parallel on a cluster server with multiple computing nodes to lift prediction efficiency.
Conclusion
Computational approaches can effectively and efficiently distinguish hot spots and non-hot spots from protein-DNA complexes on a large scale. In this work, we present a new computational method named PreHots for predicting hot spots in protein-DNA complexes. Compared with the existing methods, PreHots uses a high-quality dataset manually curated from literature and databases and with a strict process of redundancy removal. A large number of related features (network, exposure, sequence and structure) were calculated to characterize the residues from various aspects. To improve prediction performance, we used the SBS feature selection method to get the optimal feature set and constructed the classification model by the ESC method that integrates four well-performing models. Our method overcomes the drawbacks of single classifiers and makes the prediction more robust. We conducted extensive experiments to evaluate the proposed method, and compared it with existing methods on both a benchmark dataset and an independent test dataset. Experimental results show that our approach achieves higher overall performance than the existing methods. We believe that our method is an invaluable tool of identifying hot spot residues in protein-DNA complexes and can provide insights for the characterization of protein-DNA binding sites.
Availability of data and materials
PreHots is free available at http://dmb.tongji.edu.cn/tools/PreHots/.
Abbreviations
- PreHots:
-
The abbreviation of predicting hot spots
- SPR:
-
Surface plasmon resonance
- ITC:
-
Isothermal titration calorimetry
- MM/PBSA:
-
Molecular mechanics poisson-boltzmann surface area
- Catboost:
-
Categorical boosting
- XGBoost:
-
Extreme gradient boosting
- GTB:
-
Gradient tree boosting
- ESC:
-
Ensemble stacking classifier
- LR:
-
Logistic regression
- SAS:
-
Solvent accessibility area
- RASA:
-
Relative solvent accessibility area
- ACC:
-
Accuracy
- SEN:
-
Sensitivity
- SPE:
-
Specificity
- PRE:
-
Precision
- F1:
-
F1-score
- MCC:
-
Matthews correlation coefficient
- AUC:
-
Area under curve
- HSE:
-
Half-sphere exposure
- CN:
-
Contact number
- RD:
-
Residue depth
- Hbond:
-
Hydrogen bonds
- SS:
-
Secondary structure
- PDB:
-
Protein data bank
- PSSM:
-
Position-specific scoring matrix
- AAindex:
-
Amino acid indices database
- BLOSUM:
-
Blocks substitution matrix
- LSE:
-
Local structural entropy
- DISO:
-
Disordered regions
- SBS:
-
Sequential backward selection
- ESC:
-
Ensemble stacking classifier
- EVC:
-
Ensemble vote classifier
- RF:
-
Random forests
- SVM:
-
Support vector machine
- RFE:
-
Recursive feature elimination
- mRMR:
-
Maximum relevance minimum redundancy
- HSIC:
-
Hilbert Schmidt independence criterion
- MDGI:
-
The mean decrease Gini index
- FRET:
-
Fluorescence resonance energy transfer
References
Berman MH. The protein data bank. Nucleic Acids Res; 28(1):235–42.
Orphanides G, Reinberg D. A unified theory of gene expression. Cell. 2002; 108(4):439–51.
Roeder R. Role of general and gene-specific cofactors in the regulation of eukaryotic transcription. In: Cold Spring Harbor Symposia on Quantitative Biology, vol. 63. Cold Spring Harbor Symposia on Quantitative Biology: 1998. p. 201–18.
Pan Y, Wang Z, Zhan W, Deng L. Computational identification of binding energy hot spots in protein-RNA complexes using an ensemble approach. Bioinformatics. 2017; 34(9):1473–80.
Teh HF, Peh WY, Su X, Thomsen JS. Characterization of protein-DNA interactions using surface plasmon resonance spectroscopy with various assay schemes. Biochemistry. 2007; 46(8):2127–35.
Freire E, Mayorga OL, Straume M. Isothermal titration calorimetry. Anal Chem. 1990; 62(18):950–9.
Hillisch A, Lorenz M, Diekmann S. Recent advances in fret: distance determination in protein-DNA complexes. Curr Opin Struct Biol. 2001; 11(2):201–7.
Peng Y, Sun L, Jia Z, Li L, Alexov E. Predicting protein-DNA binding free energy change upon missense mutations using modified MM/PBSA approach: SAMPDI webserver. Bioinformatics. 2017; 34(5):779–86.
Zhang N, Chen Y, Zhao F, Yang Q, Simonetti FL, Li M. PremPDI estimates and interprets the effects of missense mutations on protein-DNA interactions. PLoS Comput Biol. 2018; 14(12):1006615.
Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. the accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2010; 51(1):69–82.
Pires DE, Ascher DB. mCSM-NA: predicting the effects of mutations on protein–nucleic acids interactions. Nucleic Acids Res. 2017; 45(W1):241–6.
Zhang S, Zhao L, Zheng C-H, Xia J. A feature-based approach to predict hot spots in protein-DNA binding interfaces. Brief Bioinform. 2019; 21(3):1038–46.
Kumar MS, Bava KA, Gromiha MM, Prabakaran P, Kitajima K, Uedaira H, Sarai A. Protherm and pronit: thermodynamic databases for proteins and protein–nucleic acid interactions. Nucleic Acids Res. 2006; 34(suppl_1):204–6.
Liu L, Xiong Y, Gao H, Wei D-Q, Mitchell JC, Zhu X. dbAMEPNI: a database of alanine mutagenic effects for protein–nucleic acid interactions. Database. 2018; 2018. https://doi.org/10.1093/database/bay034.
Dorogush AV, Ershov V, Gulin A. Catboost: gradient boosting with categorical features support. 2018. arXiv preprint arXiv:1810.11363.
Chen T, Guestrin C. Xgboost: A scalable tree boosting system. In: In 22nd SIGKDD Conference on Knowledge Discovery and Data Mining. ACM: 2016. p. 785–94.
Friedman JH. Stochastic gradient boosting. Comput Stat Data Anal. 2002; 38(4):367–78.
Wright RE. Logistic regression. Reading & Understanding Multivariate Stats. 1995; 68(3):497–07.
Hubbard SJ, Thornton JM. Naccess. Computer Program, Department of Biochemistry and Molecular Biology, University College London. 1993; 2(1).
Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006; 22(13):1658–9.
Yan W, Zhou J, Sun M, Chen J, Hu G, Shen B. The construction of an amino acid network for understanding protein structure and function. Amino Acids. 2014; 46(6):1419–39.
Chakrabarty B, Parekh N. NAPS: Network analysis of protein structures. Nucleic Acids Res. 2016; 44(W1):375–82.
Pan Y, Liu D, Deng L. Accurate prediction of functional effects for variants by combining gradient tree boosting with optimal neighborhood properties. PLoS ONE. 2017; 12(6):0179314.
Hogan M, Austin RH. Importance of DNA stiffness in protein-DNA binding specificity. Nature. 1987; 329(6136):263.
Luscombe NM, Laskowski RA, Thornton JM. Amino acid–base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001; 29(13):2860–74.
Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. The role of dna shape in protein-DNA recognition. Nature. 2009; 461(7268):1248.
Piovesan D, Minervini G, Tosatto SC. The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Res. 2016; 44(W1):367–74.
Deng L, Guan J, Wei X, Yi Y, Zhang QC, Zhou S. Boosting prediction performance of protein-protein interaction hot spots by using structural neighborhood properties. J Comput Biol. 2013; 20(11):878–91.
Deng L, Zhang QC, Chen Z, Meng Y, Guan J, Zhou S. PredHS: a web server for predicting protein–protein interaction hot spots by using structural neighborhood properties. Nucleic Acids Res. 2014; 42(Webserver-Issue):290–5.
Tuncbag N, Gursoy A, Keskin O. Identification of computational hot spots in protein interfaces: combining solvent accessibility and inter-residue potentials improves the accuracy. Bioinformatics. 2009; 25(12):1513–20.
Deng L, Guan J, Dong Q, Zhou S. Prediction of protein-protein interaction sites using an ensemble method. BMC Bioinformatics. 2009; 10(1):426.
Song J, Tan H, Takemoto K, Akutsu T. HSEpred: predict half-sphere exposure from protein sequences. Bioinformatics. 2008; 24(13):1489–97.
Hamelryck T. An amino acid has two sides: a new 2D measure provides a different view of solvent exposure. Proteins Struct Funct Bioinforma. 2005; 59(1):38–48.
Hanson J, Paliwal K, Litfin T, Yang Y, Zhou Y. Improving prediction of protein secondary structure, backbone angles, solvent accessibility and contact numbers by using predicted contact maps and an ensemble of recurrent and residual convolutional neural networks. Bioinformatics. 2018; 10:2403–10.
McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol. 1994; 238(5):777–93.
Liang S, Meroueh SO, Wang G, Qiu C, Zhou Y. Consensus scoring for enriching near-native structures from protein–protein docking decoys. Proteins Struct Funct Bioinforma. 2009; 75(2):397–403.
Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers Orig Res Biomol. 1983; 22(12):2577–637.
Klausen MS, Jespersen MC, Nielsen H, Jensen KK, Jurtz VI, Soenderby CK, Sommer MOA, Winther O, Nielsen M, Petersen B, et al. Netsurfp-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins Struct Funct Bioinforma. 2019; 87(6):520–7.
Heffernan R, Yang Y, Paliwal K, Zhou Y. Capturing non-local interactions by long short-term memory bidirectional recurrent neural networks for improving prediction of protein secondary structure, backbone angles, contact numbers and solvent accessibility. Bioinformatics. 2017; 33(18):2842–9.
Jamroz M, Kolinski A, Kihara D. Structural features that predict real-value fluctuations of globular proteins. Proteins Struct Funct Bioinforma. 2012; 80(5):1425–35.
Capra JA, Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007; 23(15):1875–82.
Kawashima S, Kanehisa M. AAindex: amino acid index database. Nucleic Acids Res. 2000; 28(1):374.
Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci. 1992; 89(22):10915–9.
Chan C-H, Liang H-K, Hsiao N-W, Ko M-T, Lyu P-C, Hwang J-K. Relationship between local structural entropy and protein thermostabilty. Proteins Struct Funct Bioinforma. 2004; 57(4):684–91.
Hanson J, Yang Y, Paliwal K, Zhou Y. Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinformatics. 2016; 33(5):685–692.
Wang S, Li W, Liu S, Xu J. Raptorx-property: a web server for protein structure property prediction. Nucleic Acids Res. 2016; 44(W1):430–5.
Van der Laan MJ, Polley EC, Hubbard AE. Super learner. Stat Appl Genet Mol Biol. 2007; 6(1).
Breiman L. Random forests. Mach Learn. 2001; 45(1):5–32.
Chang C-C, Lin C-J. Libsvm: A library for support vector machines. ACM Trans Intell Syst Technol (TIST). 2011; 2(3):27.
Guyon I, Weston J, Barnhill S, Vapnik V. Gene selection for cancer classification using support vector machines. Mach Learn. 2002; 46(1-3):389–422.
Peng H, Long F, Ding C. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell. 2005; 8:1226–38. https://doi.org/10.1109/tpami.2005.159.
Climente-González H, Azencott C-A, Kaski S, Yamada M. Block hsic lasso: model-free biomarker detection for ultra-high dimensional data. bioRxiv. 2019:532192. https://doi.org/10.1093/bioinformatics/btz333.
Pan X, Smith CE, Zhang J, McCabe KA, Fu J, Bell CE. A structure–activity analysis for probing the mechanism of processive double-stranded DNA digestion by λ exonuclease trimers. Biochemistry. 2015; 54(39):6139–48.
Amrane S, Rebora K, Zniber I, Dupuy D, Mackereth CD. Backbone-independent nucleic acid binding by splicing factor sup-12 reveals key aspects of molecular recognition. Nat Commun. 2014; 5:4595.
Acknowledgements
Not applicable.
About this supplement
This article has been published as part of BMC Bioinformatics Volume 21 Supplement 13, 2020: Selected articles from the 18th Asia Pacific Bioinformatics Conference (APBC 2020): bioinformatics. The full contents of the supplement are available online at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-21-supplement-13.
Funding
This work was supported by the National Natural Science Foundation of China (61772367, 61972100) and the National Key Research and Development Program of China (grant No. 2016YFC0901704). Publication costs were funded by the National Natural Science Foundation of China (61772367, 61972100).
Author information
Authors and Affiliations
Contributions
YL and JH conceived this work and designed the experiments. YL carried out the experiments, drafted the manuscript and developed the webserver. YL and JH collected the data and analyzed the results. SG participated technical discussions and revised the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pan, Y., Zhou, S. & Guan, J. Computationally identifying hot spots in protein-DNA binding interfaces using an ensemble approach. BMC Bioinformatics 21 (Suppl 13), 384 (2020). https://doi.org/10.1186/s12859-020-03675-3
Published:
DOI: https://doi.org/10.1186/s12859-020-03675-3