
Abstract
Background
Targeted next-generation sequencing (NGS) has been widely used as a cost-effective way to identify the genetic basis of human disorders. Copy number variations (CNVs) contribute significantly to human genomic variability, some of which can lead to disease. However, effective detection of CNVs from targeted capture sequencing data remains challenging.
Results
Here we present SeqCNV, a novel CNV calling method designed to use capture NGS data. SeqCNV extracts the read depth information and utilizes the maximum penalized likelihood estimation (MPLE) model to identify the copy number ratio and CNV boundary. We applied SeqCNV to both bacterial artificial clone (BAC) and human patient NGS data to identify CNVs. These CNVs were validated by array comparative genomic hybridization (aCGH).
Conclusions
SeqCNV is able to robustly identify CNVs of different size using capture NGS data. Compared with other CNV-calling methods, SeqCNV shows a significant improvement in both sensitivity and specificity.
Similar content being viewed by others

Background
The development of Next-Generation Sequencing (NGS) technologies has enabled the generation of large-scale sequence datasets. The ability to identify and characterize genomic variants and mutations from large numbers of individuals has become feasible, driving advances in our understanding of genetic diseases. Due to the cost and the complexity of analyzing whole genome sequence data, targeted capture sequencing has become the predominant approach for genetic diagnostic purposes. Targeted capture sequencing yields significantly greater depth of coverage, providing increased quality and fidelity at a decreased cost compared with whole genome sequencing [1–4]. However, a major limitation of capture NGS is that only single nucleotide variants (SNVs) and small insertions and deletions (Indels) can be identified, while large duplication and deletions are ignored in most cases because copy number variation (CNV) identification from targeted NGS data is less reliable.
CNVs are large genomic DNA segments (≥1 kb) with variable copy number among individuals [5]. A substantial proportion of the human genome is copy number variable and more than a thousand CNV regions with a frequency of greater than 1% have been identified in the genome [6]. CNVs encompassing genes can potentially alter gene dosage, disrupt genes or perturb their expression levels [7], and are known to contribute to a number of disorders [8–15]. Additionally, CNVs have played a pivotal role in evolutionary [16–18] and population genetics analysis [19, 20].
Traditional methods for CNV identification include array comparative genomic hybridization (aCGH) [21] and SNP array technologies. In recent years, NGS has provided alternative approaches to assay CNVs [22–25]. There are two primary strategies for CNV detection using NGS data: paired-end mapping (PEM) and depth of coverage (DOC). In the PEM-based methods, both paired ends of a sequenced fragment are aligned against the reference genome, and discordantly mapped paired reads whose distances are significantly deviated from the mean insert size of fragments are predicted to possess alternations in copy number [26]. PEM-based methods are not suitable for targeted NGS as they are limited by the read length when finding large copy number gains. More importantly, they require that paired reads cross the junction. Since CNV boundaries are more likely to be located in introns or intergenic regions that are far from the targeted regions, many CNVs will be completely missed by PEM-based approaches that use targeted capture sequencing data. Another type of CNV calling methods is based on DOC windows [27]. The underlying approach is to compare the differences of DOC in particular genomic regions between case and control samples [28, 29]. Unlike the PEM-based methods that are limited by the insert size and can only detect smaller CNVs, the DOC-based methods can, in theory, detects arbitrarily large insertions. Furthermore, DOC can be effectively used with paired-end, single-end, and mixed read data. However, due to large variation of the capture NGS data, DOC-based methods usually result in significant false positives [30].
Currently, several methods have been developed to identify CNVs from capture NGS data, including CoNIFER [31], CNVnator [32], CNVer [33] and XHMM [34]. CoNIFER exploits singular value decomposition (SVD), which aims to eliminate capture biases among sample batches. XHMM is based on principal component analysis (PCA) normalization and hidden Markov model (HMM). Reliance on SVD and PCA limit the ability of CoNIFER and XHMM to perform CNV calling with a large number of samples. CNVnator and CNVer are both DOC-based methods. CNVer supplements the DOC with paired-end mapping information, where mate pairs mapping discordantly to the reference indicate the presence of variation [33]. Both of them can identify a large number of CNVs but are not effective in detecting small-size CNVs [35, 36].
To address the limitations of current methods in detecting CNVs using target capture NGS data, we developed a robust statistical method called SeqCNV, which uses maximum penalized likelihood estimation (MPLE) to evaluate the CNV boundary and the copy number ratio. Given the variation of the sequencing depths of the case and control samples, normalization was performed using the total reads number for each chromosome in the likelihood model. A novel segmentation algorithm was developed which enabled the detection of CNVs with different lengths. We also present an assessment of its sensitivity, specificity and limitations using targeted sequencing data from both bacterial artificial clone (BAC) and human patients.
Methods
Statistical modeling and algorithm
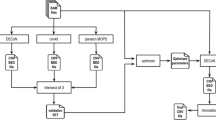
SeqCNV is a DOC-based method to identify CNVs from target capture NGS data. The workflow of SeqCNV includes four steps as shown in Fig. 1. First, with provided case and control samples, SeqCNV considers the starting point of each read as a candidate break point (CBP). Next, SeqCNV establishes the penalized log-likelihood model for all segments between case and control samples. For each CBP, SeqCNV calculates the likelihood and recursively finds the optimum starting point located upstream of it. Once the optimum starting point is determined, the region between it and the CBP is considered as a candidate CNV region. Finally, SeqCNV reports candidate regions whose likelihood values are below 0.6 (for copy loss) or above 1.4 (for copy gain). In the segmentation stage, each chromosome can be partitioned into n segments with n-1 breakpoints. When case and control reads are mixed together and given a read that is successfully mapped to the ith segment, we assume the probability that the read is from the case sample (p i ) is homogeneous across the entire targeted region. Thus, the control is diploid and the p i is normalized to the diploid genome, which is the relative probability. Therefore, the probability that the read is from the control sample is 1-p i . Thus, each short read mapped to the ith segment is an independent Bernoulli experiment with two outcomes: being a case read or a control read. t i and c i are denoted as the number of reads mapped to the ith segment in case and control samples, respectively. The log-likelihood L of the model is as follows:

Workflow of SeqCNV. A dynamic programming procedure is included in the step “Recursively find candidate regions”. It aims to quickly break iteration to find candidate regions that are most likely to be CNV events, thereby saving much time for whole algorithm running
Each segment has two parameters to be determined: the left boundary position and the copy number ratio, with the exception of the first segment, which only required the copy number ratio. Thus, the model contains 2n-1 parameters.
The optimization goal is to minimize the number of segments while keeping its fitness to the data. This task can be achieved by several criteria, such as p-value-based statistical testing, Akaike’s information criterion (AIC) [37] and Bayesian information criterion (BIC) [38]. All these criteria can be viewed as particular instances of MPLE, which attempts to maximize the following penalized likelihood (PL):
In this equation, λ is the penalization factor, as \( \lambda =\frac{1}{2}{\chi}_p^2 \) for the p-value-based criterion, λ = 1 for AIC and \( \lambda =\frac{1}{2} \ln N \) for BIC, where N is the total number of reads in the targeted genome. We recommended BIC owing to its robust statistical properties such as minimum description length [39, 40].
To find the MPLE, we proposed a dynamic programming procedure. Suppose there are M CBPs. Let s (j, i) be the log-likelihood of the segment that starts from the jth CBP and ends at the ith CBP.
where t (c) is the number of reads mapped in the segment in the case (control). Denote b(i) as the maximum penalized log-likelihood of the chromosome started at the beginning and ended at the ith CBP. Denote B (i) as the best starting CBP of the segment that ended at the ith CBP. The recursion formula is as follows:
The recursion formula has a computational complexity of O(n 2). For implementing this dynamic procedure, double loops are necessary in the coded program. The outer loop is for each i from 1 to M, traversing M CBPs. The inner loop is for j, decreasing from i to 1, searching the optimum starting point from the ending point i. To speed up, the inner loop will stop if the penalized log-likelihood at j is much lower than that at the current optimum starting point, s (j, i) + b (j-1)-2λ < b(i)j-const, where in our experiment, const = 5λ.
If the dataset contains less than 1 M shorts reads, each read can be set as a CBP and the best partition can be found within several hours with read level resolution. For larger datasets, first, CBPs can be selected instead of treating each read as a CBP as described [28]. To identify the CBP set, the dynamic programming algorithm described in formula (4) could also be utilized with lower penalty, for instance, λ = 1.92, which is equivalent to a p-value of 0.05 threshold. After CBPs being determined, the formula (4) will be run again with bigger λ to solve the optimization problem of formula (2).
Simulation dataset preparation
To evaluate the detection power and the false positive rate (FPR) of SeqCNV with different lengths of CNVs, we generated sequencing reads with the starting position based on the NimbleGen CCDS design file on chromosome 1, which includes 8315 targets with an average length of 168 base pairs. Detection power is defined as how many simulated one-copy gains or losses are covered by the segments that have close ratios. FPR is defined as how many segments whose ratios indicate more than one copy change actually do not overlap with the simulated ones. We assumed that the number of reads for each target followed a Poisson distribution with as the product of the affinity and length, and coordinated within the range being sampled. For the rest of the chromosome, off-target reads were assumed to be uniformly distributed and randomly sampled.
BAC spike-in experiment
DNA of nine non-overlapping BAC clones were spiked into control human genomic DNA (Additional file 1). The spike-in sample was used to mimic copy number gains in nine targeted regions.
Retinitis pigmentosa (RP) patient data analysis
RP is an inherited form of retinal degenerative disease causing progressive vision loss. Autosomal dominant RP (adRP) can be caused by loss of a single copy of PRPF31 gene on chromosome 19. To test the performance of our method, we applied SeqCNV on five adRP patients who were known to carry PRPF31 deletions. Patient DNA was extracted from peripheral blood using standard techniques.
Targeted panel design and sequencing data analysis
A custom capture panel was designed using AgilentSureSelect (Agilent Technologies, CA) targeting 18 genes (RPGR, TULP1, CABP4, RGR, MYO7A, PRPF31, ABCA4, USH2A, CNGB1, SAG, RPGRIP1, LCA5, CNGA1, MERTK, CLRN1, TTC8, CC2D2A, and PDE6A). All normal control samples, BAC spike-in samples, and adRP patient samples were sequenced using the same panel in the same batch. Resultant DNA was bar-coded, prepared, and shotgun sequenced. Reads were aligned to Human Reference Genome hg19 using Burrows Wheeler Aligner (BWA) [41]. Recalibration and realignment were performed using The Genome Analysis Toolkit (GATK) [42]. Samtools [43] was used to sort and index the resultant BAM files. Quality control analysis showed that 97% and 81% of the target area were covered with > 10× and > 40×, respectively.
aCGH validation
To validate the CNVs identified from NGS, we performed aCGH experiments on the patients with adRP. A customized aCGH platform targeting the same 18 genes including the PRPF31 (MIM: 606419) was designed using Agilent Suredesign (https://earray.chem.agilent.com/suredesign). The probes used are available upon request. The aCGH experiments were performed as per the manufacturer’s instructions and were analyzed using Agilent Genomic Workbench.
Results
Simulated results
We simulated both case and control data and applied SeqCNV to obtain the segmentations. We performed this process for 100 rounds. In each run, we randomly generated four copy changes including two gains and two losses at different sizes of 1 MB, 100 KB, 10 KB and 1 KB, containing at least one captured exon. For each of these 16 changes per sample, we mimicked a single copy number gain or loss by increasing or decreasing the number of case reads by 50% relative to the control, respectively. As shown in Fig. 2, simulated copy number changes of different sizes can be detected.

An example of simulated CNV data on chromosome 1. The data set, computationally simulated, includes two deletions and two duplications at each of four lengths. Black dots represent read density over 500 bp fixed windows along the entire chromosome. The red bands indicate the results of SeqCNV analysis. Horizontal lines mark significant points of deletion or gain
As shown in Table 1, SeqCNV is sensitive to gains of 1 KB and losses of 1 KB. Sensitivity was calculated as the ratio of correctly detected CNV regions to the total number of simulated CNV regions. A detected region was considered as a true positive only if SeqCNV determined a copy gain ratio no greater than 1.4, or loss ratio no less than 0.6 (complying with the ideal ratios of 1.5 for gain and 0.5 for loss). Additionally, the overlap of copy loss detections and simulated regions was required to exceed 50%. Due to the detection difficulty of copy number gains, any overlap of simulated regions was deemed sufficient for copy gain detections [44]. It is noted that high sensitivity is associated with 1 KB regions, which is indicative of an ability to detect a single exon copy number gain or loss.
Comparison of performance on BAC spike-in data
One of the challenges in evaluating the performance of CNV callers is the lack of a gold standard. To address this issue, we spiked in equal molar BAC DNA for nine regions into control genomic DNA to mimic copy number gains (see Additional file 1). Using this dataset, the performance of SeqCNV was compared with previous published tools in detecting CNVs from targeted NGS data.
The results are shown in Table 2. CNVnator and CNVer reported a large number of candidate CNVs, most of which were long regions whose lengths surpassed 1Mbp and turned out to be false positives. CoNIFER identified two CNVs, and both were located in the designed CNV regions, indicating high specificity but relatively low sensitivity. XHMM predicted a few CNV events, but none of them overlapped with the spiked in CNV regions. Precision is calculated as the ratio between the number of correctly detected events (the intersection between the tool calls and the known calls) and the total number of events detected by a tool [45]. The recall is calculated as the ratio between the number of correctly detected events and the total number of events in the validation set [45]. From comparative analysis of the methods, SeqCNV showed a more balanced recall and precision (Fig. 3). CNVnator is the best for recall, followed by CNVer, SeqCNV, CoNIFER and XHMM. The high recall of CNVnator is attributed to its large number of called long CNV regions, which also leads to the low precision. CoNIFER is superior in terms of precision, followed by SeqCNV, CNVer, CNVnator and XHMM. However, the recall of CoNIFER is very low since only two CNVs were called. In comparison, SeqCNV achieved high precision and moderate recall. Overall, from the results on the BAC spike-in data, SeqCNV showed the best trade-off between high precision and recall comparing with the other four methods.

Precision-Recall Contours for five CNV methods on spike-in data. Light grey contours represent F-measure levels (harmonic mean of precision and recall). SeqCNV achieved the highest F-measure value
Comparison of performance on adRP patient data
We collected targeted NGS data from five patients with adRP. Normal control samples were sequenced in the same batch. Each of these case samples contained a copy number loss in PRPF31, which was validated by aCGH (Fig. 4, Additional file 2). Considering the possibly of a non-even read distribution, we selected four samples without a copy change in PRPF31 and merged these samples with the control set. With five patient samples and the merged control samples, we ran all the CNV tools and the obtained results shown in Table 3 and Fig. 5. For CoNIFER and XHMM, we added an extra 46 samples without copy change in PRPF31 as controls due the requirement of SVD and PCA methods. The criteria of control sample selection are described in the Additional file 3. As shown in Fig. 5, both SeqCNV and CoNIFER identified deletions in PRPF31 on chromosome 19. However, CoNIFER resulted in a larger FPR. Consistent with our previous BAC spike-in experiment, both CNVnator and CNVer reported large number of CNVs, almost covering the entire chromosome. XHMM did not give any positive results. Therefore, SeqCNV outperformed the other four methods in this comparison.

aCGH validation of copy number deletion in PRPF31 gene for sample UTAD082. The shaded area indicates the CNV area with loss of one copy of the genomic segment. Validation results for other 4 samples can be found in Additional file 1 . Five samples share different deletion sizes, ranging from several exons to entire genomic region of PRPF31

CNV result for five methods on adRP patient data. a SeqCNV b CoNIFER c CNVnator d CNVer e XHMM. X-axis represents the genomic position for chromosome chr19. PRPF31 gene is located at chr19:54,618,790–54,635,150. Both SeqCNV and CoNIFER identified the PRPF31 copy number deletion
Execution time estimation
Since efficiency is an important factor to consider in evaluating the performance of the tools, we tested SeqCNV with other four methods on the BAC spike-in data, adRP human patients data and whole-exome sequencing (WES) data obtained from the 1000 Genome projects (ftp://ftp.1000genomes.ebi.ac.uk/) [46, 47]. As shown in Additional file 4, SeqCNV required 4–5 min to analyze the BAC spike-in and adRP patient data, and about 17 min to process chromosome 1 data from WES. Compared with other tools, SeqCNV was the most efficient in performing the processing and analysis of the datasets.
Discussion
SeqCNV offers many advantages over previously used methods. It does not require a large number of samples to run and is able to detect CNVs of different sizes by treating the left boundary of each reads as a CBP. As shown in both BAC spike-in data and adRP human patient data, SeqCNV exhibited the highest accuracy compared with other methods.
SeqCNV can also be effectively used to analyze patient data for other regions potentially causing additional genetic diseases. Besides BAC spike-in and RP patient data, we also tested SeqCNV on WES samples from the 1000 genome project (ftp://ftp.1000genomes.ebi.ac.uk/). Similar to the methods used for analysis of the adRP patient data, we considered multiple factors such as DNA quality, DNA extraction protocol and the possibly non-even reads distribution and randomly selected three samples to pool together as a control, and randomly selected one sample as case. We validated our results with the CNVs previously reported by Conrad et al. [46, 47] in the WES samples. The results are shown in Additional file 5. Overall, SeqCNV was able to identify the known CNVs with a good recall rate (55%) and low false positive rate (~10%).
The study of CNVs in human disease is a rapidly evolving field. CNVs can result in gene dosage changes and give rise to a substantial amount of human phenotypic variability. It also has been shown that CNVs play an essential role in cancer [46, 47]. However, currently investigating CNVs for human disease is still largely overlooked due to technical issues, such as the limited accuracy of CNV detection methods from NGS data. Therefore, it is important to increase the sensitivity of detection while controlling the false positive rate with statistical tools. We recognize that although SeqCNV demonstrated the best trade-off between precision and recall compared to other approaches in our tests, it does still result in a significant number of false positives, especially when the sequencing quality is not reliable. In addition, the performance of CNV tools based on targeted capture sequencing would be limited by the capture design, although it is very efficient in detecting CNVs in known pathogenic genes. Contrarily, whole-genome sequencing may be more effective in detecting CNVs in novel regions. However, because of factors such as cost, it is not so widely used in clinical applications.
As shown in the simulation results, the length of the CNV will affect the sensitivity of detection. It is easier to detect larger CNVs while smaller size CNVs are sometimes indistinguishable from the background. One possible solution is to increase the sequencing depth, which will improve the statistical power of SeqCNV. In addition, using matched case and control files can help reduce the number of false positives resulting from sequencing bias.
Based on our comparative analysis, we observed that CNVer and CNVnator predicted a large number of CNVs. Both methods shared good recall but high FPR. Although SeqCNV requires matched control samples to perform the analysis, we can also derive a control sample by pooling other samples together, which will still serve as an effective control. As shown in the PRPF31 deletion analysis of adRP patients, we combined four normal samples that were sequenced in the same batch as the control. Furthermore, since sequencing quality can be affected by multiple factors, including DNA quality, and DNA extraction protocol, it is recommended that users select samples with good quality to pool together as controls. For example, users can calculate the evenness scores [48] representing the uniformity of sequencing and samples with highest evenness scores can be pooled as controls.
Conclusions
In this study, we devised a novel method, SeqCNV, based on the MPLE statistical model for CNV identification in targeted NGS data. Simulation analysis showed SeqCNV can detect CNVs of different sizes robustly. Additionally, we applied our method to BAC spike-in data. Compared with other methods, our method demonstrated higher sensitivity and specificity. We also tested our method on human patient datasets and causative CNVs were identified in all five samples and validated by aCGH. SeqCNV is a powerful and practical tool for CNV identification in target capture NGS data and may facilitate causative CNV discovery in genetic diseases.
Detecting CNV in targeted NGS data is a challenging area due to non-uniform distribution of read depth of target sequencing and the variation in capture efficiency. A significant difference lies in the existing tools. We think combining SeqCNV with other tools could make more reliable predictions.
A standard C++ implementation named SeqCNV can be downloaded from: http://www.iipl.fudan.edu.cn/SeqCNV.
Abbreviations
- aCGH:
-
Array comparative genomic hybridization
- adRP:
-
Autosomal dominant retinitis pigmentosa
- AIC:
-
Akaike’s information criterion
- BAC:
-
Bacterial artificial clone
- BIC:
-
Bayesian information criterion
- CBP:
-
Candidate break point
- CNV:
-
Copy number variation
- DOC:
-
Depth of coverage
- FPR:
-
False positive rate
- Indel:
-
Insertion and deletion
- MPLE:
-
Maximum penalized likelihood estimation
- NGS:
-
Next-generation sequencing
- PCA:
-
Principal Component analysis
- PEM:
-
Paired-end mapping
- PL:
-
Penalized likelihood
- RP:
-
Retinitis pigmentosa
- SNP:
-
Single nucleotide polymorphism
- SNV:
-
Single nucleotide variant
- SVD:
-
Singular value decomposition
- WES:
-
Whole exome sequencing
References
Zhao L, Wang F, Wang H, Li Y, Alexander S, Wang K, Willoughby CE, Zaneveld JE, Jiang L, Soens ZT, et al. Next-generation sequencing-based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Hum Genet. 2015;134(2):217–30.
Fu Q, Wang F, Wang H, Xu F, Zaneveld JE, Ren H, Keser V, Lopez I, Tuan HF, Salvo JS, et al. Next-generation sequencing-based molecular diagnosis of a Chinese patient cohort with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54(6):4158–66.
Salvo J, Lyubasyuk V, Xu M, Wang H, Wang F, Nguyen D, Wang K, Luo H, Wen C, Shi C, et al. Next-generation sequencing and novel variant determination in a cohort of 92 familial exudative vitreoretinopathy patients. Invest Ophthalmol Vis Sci. 2015;56(3):1937–46.
Tajiguli A, Xu M, Fu Q, Yiming R, Wang K, Li Y, Eblimit A, Sui R, Chen R, Aisa HA. Next-generation sequencing-based molecular diagnosis of 12 inherited retinal disease probands of Uyghur ethnicity. Sci Rep. 2016;6:21384.
Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7(2):85–97.
McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, De Bakker PIW, Maller JB, Kirby A, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40(10):1166–74.
Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8(8):639–46.
Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307(5714):1434–40.
Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, Mangion J, Roberton-Lowe C, Marshall AJ, Petretto E, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439(7078):851–5.
Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, Heward JM, Gough SCL, De Smith A, Blakemore AIF, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007;39(6):721–3.
Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, Hebert M, Jones KN, Shu YL, Kitzmiller K, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): Low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. 2007;80(6):1037–54.
Fellermann K, Stange DE, Schaeffeler E, Schmalzl H, Wehkamp J, Bevins CL, Reinisch W, Teml A, Schwab M, Lichter P, et al. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am J Hum Genet. 2006;79(3):439–48.
Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39(3):319–28.
Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38(1):24–6.
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841.
Cooper GM, Nickerson DA, Eichler EE. Mutational and selective effects on copy-number variants in the human genome. Nat Genet. 2007;39:S22–9.
Perry GH, Tchinda J, McGrath SD, Zhang JJ, Picker SR, Caceres AM, Iafrate AJ, Tyler-Smith C, Scherer SW, Eichler EE, et al. Hotspots for copy number variation in chimpanzees and humans. Proc Natl Acad Sci U S A. 2006;103(21):8006–11.
Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, Pevzner PA, Eichler EE. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat Genet. 2007;39(11):1361–8.
Conrad DF, Hurles ME. The population genetics of structural variation. Nat Genet. 2007;39:S30–6.
White SJ, Vissers LELM, Van Kessel AG, De Menezes RX, Kalay E, Lehesjoki AE, Giordano PC, van de Vosse E, Breuning MH, Brunner HG, et al. Variation of CNV distribution in five different ethnic populations. Cytogenet Genome Res. 2007;118(1):19–30.
Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20(2):207–11.
Schuster SC. Next-generation sequencing transforms today’s biology. Nat Methods. 2008;5(1):16–8.
Bentley DR. Whole-genome re-sequencing. Curr Opin Genet Dev. 2006;16(6):545–52.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen ZT, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–80.
Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, Zeng K, Malek JA, Costa G, McKernan K, et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008;18(7):1051–63.
Zhao M, Wang Q, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics. 2013;14(11):S1.
Medvedev P, Stanciu M, Brudno M. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods. 2009;6(11):S13–20.
Chiang DY, Getz G, Jaffe DB, O’Kelly MJT, Zhao XJ, Carter SL, Russ C, Nusbaum C, Meyerson M, Lander ES. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009;6(1):99–103.
Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. Bmc Bioinformatics. 2009;10.
Wang K, Li MY, Hadley D, Liu R, Glessner J, Grant SFA, Hakonarson H, Bucan M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17(11):1665–74.
Krumm N, Sudmant PH, Ko A, O’Roak BJ, Malig M, Coe BP, Quinlan AR, Nickerson DA, Eichler EE, Project NES. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22(8):1525–32.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21(6):974–84.
Medvedev P, Fiume M, Dzamba M, Smith T, Brudno M. Detecting copy number variation with mated short reads. Genome Res. 2010;20(11):1613–22.
Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, Handsaker RE, McCarroll SA, O’Donovan MC, Owen MJ, et al. Discovery and Statistical Genotyping of Copy-Number Variation from Whole-Exome Sequencing Depth. Am J Hum Genet. 2012;91(4):597–607.
Shen Y, Gu Y, Pe’er I. A hidden Markov model for copy number variant prediction from whole genome resequencing data. BMC Bioinformatics. 2011;12(6):S4.
Suzuki S, Yasuda T, Shiraishi Y, Miyano S, Nagasaki M. ClipCrop: a tool for detecting structural variations with single-base resolution using soft-clipping information. BMC Bioinformatics. 2011;12(14):S7.
Akaike H. Information theory and an extension of the maximum likelihood principle. In: Selected Papers of Hirotugu Akaike. New York: Springer; 1998. p. 199–213.
Schwarz G. Estimating the dimension of a model. Ann Stat. 1978;6(2):461–4.
Lam W, Bacchus F. Learning Bayesian belief networks: An approach based on the MDL principle. Comput Intell. 1994;10(3):269–93.
Cruz-Ramírez N, Acosta-Mesa H-G, Barrientos-Martínez R-E, Nava-Fernández L-A. How good are the Bayesian information criterion and the minimum description length principle for model selection? A Bayesian network analysis. In: MICAI 2006: Advances in Artificial Intelligence. New York: Springer; 2006. p. 494–504.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Daines B, Wang H, Li YM, Han Y, Gibbs R, Chen R. High-Throughput Multiplex Sequencing to Discover Copy Number Variants in Drosophila. Genetics. 2009;182(4):935–41.
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang QY, Locke DP, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6(9):677–U676.
Magi A, Tattini L, Cifola I, D’Aurizio R, Benelli M, Mangano E, Battaglia C, Bonora E, Kurg A, Seri M, et al. EXCAVATOR: detecting copy number variants from whole-exome sequencing data. Genome Biol. 2013;14:10.
Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini I, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97(9):643–55.
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905.
Mokry M, Feitsma H, Nijman IJ, De Bruijn E, van der Zaag PJ, Guryev V, Cuppen E. Accurate SNP and mutation detection by targeted custom microarray-based genomic enrichment of short-fragment sequencing libraries. Nucleic Acids Res. 2010;38(10), e116.
Acknowledgement
Not applicable.
Funding
FW was supported by National Natural Science Foundation of China (61472086) and National Key Research and Development Program of China (2016YFC0902100). RC was supported by the Retinal Research Foundation, Foundation Fighting Blindness (BR-GE-0613-0618-BCM), and the National Eye Institute (R01EY022356, R01EY018571, R01EY020540).
Availability of data and materials
A standard C++ implementation named SeqCNV can be downloaded from: http://www.iipl.fudan.edu.cn/SeqCNV.
Authors’ contributions
YC, LZ, YW, and MC performed code development and implementation, and drafted the manuscript. VG, MX, SAA, YL and SPD performed aCGH analysis and CNV validation. RG, FW, and RC conceived the project and participated in the software design. All authors critically revised the manuscript. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All the patients provided consent to participate in this study and publish the results. All the data collection and usage were approved by the Baylor College of Medicine Institutional Review Board.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1:
BAC spike-in regions. CNVs for 9 genes performed in BAC spike-in experiment. (PDF 86 kb)
Additional file 2:
Additional aCGH validation. aCGH validations for copy number loss in PRPF31 gene for other samples. (A), UTAD034; (B), UTAD069. (C), UTAD411; (D), UTAD611. (PDF 1564 kb)
Additional file 3:
The criteria of control sample selection. This file describes the criteria for control sample selection. (PDF 8 kb)
Additional file 4:
Execution time comparison. Estimation of the execution time for SeqCNV compared with other tools on BAC spike-in data, retinitis pigmentosa data and whole-exome sequencing data (chr1 only) in our study. (PDF 11 kb)
Additional file 5:
SepCNV results on WES data. As we did on retinitis pigmentosa data, considering multiple factors such as DNA quality, DNA extraction protocol and the possibly non-even reads distribution, we randomly selected three samples to pool together as control (NA19152, NA18973, and NA19206), and randomly select one sample as case (NA10847). (PDF 7 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, Y., Zhao, L., Wang, Y. et al. SeqCNV: a novel method for identification of copy number variations in targeted next-generation sequencing data. BMC Bioinformatics 18, 147 (2017). https://doi.org/10.1186/s12859-017-1566-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-017-1566-3