
Abstract
Background
Availability of affordable and accessible whole genome sequencing for biomedical applications poses a number of statistical challenges and opportunities, particularly related to the analysis of rare variants and sparseness of the data. Although efforts have been devoted to address these challenges, the performance of statistical methods for rare variants analysis still needs further consideration.
Result
We introduce a new approach that applies restricted principal component analysis with convex penalization and then selects the best predictors of a phenotype by a concave penalized regression model, while estimating the impact of each genomic region on the phenotype. Using simulated data, we show that the proposed method maintains good power for association testing while keeping the false discovery rate low under a verity of genetic architectures. Illustrative data analyses reveal encouraging result of this method in comparison with other commonly applied methods for rare variants analysis.
Conclusion
By taking into account linkage disequilibrium and sparseness of the data, the proposed method improves power and controls the false discovery rate compared to other commonly applied methods for rare variant analyses.
Similar content being viewed by others


Background
Despite success in detecting associations of common variants with complex traits (www.genome.gov/gwastudies/), it has proven difficult to elucidate a comprehensive picture of the genetic architecture of risk factor and disease traits without considering the effects of both rare and common variants via whole exome or genome sequencing. Decreasing costs and increasing quality have made discovery and genotyping of rare variants, which refer to variants with minor allele frequency (MAF) less than 0.05, more accessible across a large proportion of the genome and in large sample sizes. As a result of rapid expansion of human populations, there are very large numbers of rare variants segregating and these rare variants are relatively recent in origin [1, 2]. Detecting genotype-phenotype associations and identifying novel loci having rare variant-phenotype associations are challenging since single-variant based statistical methods are inappropriate in this context due to the very large number of alleles and their low frequency. Furthermore, no or minimal effects of the majority of rare variants on a particular phenotype leads to a low signal-to-noise ratio and consequently underfitting with multiple-variant models. Hence, there is considerable interest in statistical methods that combine information across multiple variants, and thus reduce the cost of the large degrees of freedom in multivariate tests or adjustment for extensive multiple testing [3–9]. However simply combining information by pooling or collapsing does not take into account the direction of the variants’ effects on a phenotype and alternative methods have been proposed that address this limitation (see, e.g. [10–17]). Furthermore, inclusion of large numbers of correlated variants may lead to overestimation.
Transitioning from common variant analyses to rare variant analyses creates three challenges related to sparse data [18]. First, within an individual personal genome, the number of sites that differ from the reference genome is small relative to the total number of bases. Second, sequence data, unlike array-based genotype data, contain a large number of rare variants. In fact, about half of the variant alleles in a study sample are seen only one or two times [19]. And third, only a small subset of the variable sites is expected to influence a given trait of interest, and the rest is expected to be neutral. This study presents a statistical and computational method tailored for sparse data and how it can be applied to whole genome sequence data to promote novel gene and rare variant discovery. We introduce a new method called Convex-Concave Rare variant Selection (CCRS), which includes both convex and concave penalization. We leverage the fact that rare variants data have low intrinsic dimensionality and are sparse. Hence, we project the variants into a full rank space with new coordinates in order to enhance information in new predictors comparing with original variants. We obtain these new coordinates using principal component analysis that includes a convex penalty to incorporate sparsity assumption. The CCRS improves the performance of sparse principal component (SPC) based method [20] in the context of rare variants analysis by selecting the components based on their degree of association with a complex trait which is appropriate for rare variant analysis. To this end, we use a concave penalized regression model to select the most promising variants while estimating their effect simultaneously.
Method
The CCRS method is applicable for all variants, but in this presentation, we focus on the analysis of rare variants because they pose special opportunities (i.e. large effects sizes) and challenges (i.e. sparsity). Assume we have detected and genotyped m rare variants X=(x 1,…,x m ) in a sample of n individuals having a quantitative trait y=(y 1,…,y n ) measured on each individual. In a typical whole exome or genome sequencing scenario, m is several orders of magnitude larger than n. To combat over-determination, the typical analysis considers a subset of the variables at a time defined by physical proximity (e.g. a window) instead of functional characteristic (e.g. an annotated gene or enhancer element) because the vast majority of the rare variants are in noncoding regions in the genome. Interpretation of the results requires adjusting for multiple comparisons using accepted experiment-wise error or false discovery rate methods. Assume for the kth subset of X denoted by \(\mathbf {X}_{k}=\{\mathbf {x}_{\textit {jk}}\}_{j=1}^{p}\) where p<n, we have
where ε=(ε 1,…ε n ) is an error vector, Σ is an n×n diagonal matrix; α is the overall mean; T is an n×q covariate matrix, which includes non-genetic predictors such as age, sex and race; β k and θ are p-vector of genetic effects and q-vector of non-genetic effects, respectively. Although model (1) does not face the n≪p problem, the data lie in a lower-dimensional subspace due to dependency among rare variants [21, 22] (i.e. linkage disequilibrium, LD) and coefficients are sparse because a large proportion of variants have small or no effects on the phenotype(s) of interest. Here, we introduce a new approach for rare variants analysis to address these two issues; LD and sparsity.
The CCRS approach
In rare variant analyses, the design matrix is more likely to be singular because of the LD structure in the population [21, 22]. In addition due to low allele frequencies, there is little information about the association of each individual variant with a phenotype. Hence, applying a penalized regression model might not lead to identifying the true set of variants or genomic regions with nonzero effects. To bypass this difficulty, we project the genotype data into full rank space in order to reparameterize the regression model. Principal component analysis (PCA) is an appropriate tool for addressing collinearity and utilizes the low rank structure of the covariance matrix. One drawback of PCA is its lack of straight forward interpretability. However, in rare variant analyses each single variant is uninformative and there is a need to aggregate information in a region in order to identify association with the trait of interest. An issue of concern when applying PCA in the context of rare variants is that PCA may lead to new coordinates that include many non-influential variants due to sparseness. Accounting for such sparsity facilities identification of phenotype-influencing factors in each of the coordinates and also improves interpretability of the result because of the sparse loading matrix [23–25]. To accomplish this, we obtain a full rank approximation to the matrix X as
by imposing constraints on the columns of V and U similar to [20],
where r is the rank of X, which is smaller than min(n,p); ∥.∥ a denotes L a norm; and \(D=\{ d_{j} \}_{j=1}^{r}\) is a diagonal matrix of eigenvalues of the matrix X such that d 1≥d 2≥…≥d r ; v j and u j are the jth columns of V and U respectively. The L 1 norm penalization is equivalent to \(\sum _{r} \mid v_{\textit {ij}}\mid \), where v ij is ijth entry of V, provides sparse principal components, U D. This is an optimization problem equivalent to maximizing \(\mathbf {u}_{j}^{T} \mathbf {X} \mathbf {v}_{j}\) respect to u j and v j under constraint (2) and (3). This biconvex problem can be readily solved [26]. Therefore, we first fix u j and obtain v j when c is in the set of feasible solution \( \{ c \mid 1< c < \sqrt {p} \}\). We then obtain optimum solution of u j when \( \parallel \mathbf {u}_{j} {\parallel _{2}^{2}} \leq 1\) and for j>1, u j ⊥u 1,u 2,…,u j−1. The optimal value of c, which determines the level of sparsity, can be obtained through a cross validation approach [27, 28].
By projecting data into a lower dimensional space, we reduce the number of predictors in the model to the rank of the design matrix, which increases the degree of freedom for hypothesis test and aggregates information into fewer predictor variables which helps alleviate one aspect of the low allele frequency challenge. These two features improve the power of identifying promising genetic regions influencing a phenotype of interest (see below).
In this context, it is not appropriate to select only the first few principal components as is usual in many applications, but rather we select the PCs based on their degree of association with the phenotype. To simultaneously measure the genotype-phenotype association and carry out variable selection, we consider a linear regression model including a concave penalization with loss function
where Z=U D indicates a matrix of computed PCs with corresponding effect size γ, κ∈(0,1) and regularization parameter \(\nu \in \mathbb {R}^{+}\). Without loss of generality, hereafter, we assume the overall mean is zero.
This model is a form of Bridge regression and naturally yields sparse estimate for γ, in the sense that some of components of γ (κ,ν), may be explicitly shrunk to zero [29, 30]. The choice of κ<1 leads to nonconcave minimization problems (see, e.g., [30–32]) and provides a much sparser solution than the well-known penalized regression, lasso, with κ=1 [33].
Result
A simulation study
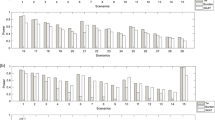
To evaluate the performance of the CCRS method, we randomly identified 1000 regions from a real whole genome sequence data set available from phs000668 study in dbGAP (http://www.ncbi.nlm.nih.gov). Each region includes 50 variants (50,000 rare variants total) sequenced for 1456 individuals. Based on our experience, we have found that 50 variants are appropriate to capture the LD structure. As an example, Fig. 1 represents this LD structure for two regions of the genome.

LD among variants with MAF <.05 in two different regions of the genome
We considered six different phenotypic effect scenarios (Table 1). We first randomly split the set of regions into two subsets to be influential regions and noninfluential regions. We then randomly selected 10 % of variants in each influential region to be causal variants with effect size +1 for Model-1 and Model-3 and with effect size ±1 for Model-2 and Model-4. In Model-5 and Model-6, the number of causal variants in a region is increased to 20 % of the total variants with different effect sizes randomly selected from U(0.5,1) and {U(−1,−0.5),U(0.5,1)}, respectively, where U denotes uniform distribution. Hence, we considered models with the same and also different effect directions.
To obtain a better understanding about the effect of LD on the result of the analysis, we selected variants based on their correlations. In Model-1 and Model-2, the causal variants are correlated with some neutral variants in their regions but in Model-3, Model-4 they are uncorrelated. For Model-5 and Model-6, both correlated and uncorrelated variants are selected (10 % of each).
In rare variants analysis, we are interested in identifying regions with significant effects on the phenotype corresponding to the following set of hypotheses for each region
To test these hypotheses, we calculated the likelihood ratio of the selected model based on CCRS to the Null model, which does not include genotype variants in the model.
We evaluated the performance of the CCRS method compared to four other commonly applied methods: Collapsing [8] denoted here as Col, CAST [3], SKAT-O [17] and sparse principal regression (SPC) [20]. The collapsing method generates a binary variable for each region to represent whether the minor allele is observed. It then tests the association between the traits level and the new binary variable \(I_{\{\sum _{j} \mathbf {x}_{j}>0\}}\) through \( \mathbf {y}=\alpha +I_{\{ \sum _{j} \mathbf {x}_{j}>0\}} {\beta } + \boldsymbol {T} \boldsymbol {\theta }+ \boldsymbol {\epsilon }\) regression model. The CAST method sums over all variants in the region and leads to \( \mathbf {y}=\alpha + \left (\sum _{j} \mathbf {x}_{j}\right) {\beta } + \boldsymbol {T} \boldsymbol {\theta }+ \boldsymbol {\epsilon }\). SKAT-O is a score based test, \((\mathbf {y}- \hat {\boldsymbol {\mu }})^{T} P_{\rho }(\mathbf {y}-\hat {\boldsymbol {\mu }})\) when β k in (1) follows an arbitrary distribution with mean 0 and variance τ and pairwise correlation ρ between different β jk s. Here, \(\hat {\boldsymbol {\mu }}\) is the predicted mean of y under H 0, \(P_{\rho } = \boldsymbol {X}_{k} R_{\rho } \boldsymbol {X}_{k}^{T}\) is an n×n kernel matrix, R ρ =(1−ρ)I+ρ 1 1 T where I is an p×p compound symmetric matrix, and 1 T=(1,…,1)T.
To examine the impact of significance level on the false and true discovery rates, we considered both α=0.01 and 0.05 and calculated false discovery rate (FDR) and true positive discovery rate (TPR) defined as
where F is the number of false positives; T is the number of true discoveries; R is the total number of significant regions; and M is the total number of regions.
To select the best model based on the CCRS method, we set ν = 0.01 and κ = 0.5 after calculating BIC of the model over for different values of ν in {0.001,0.005,0.01,0.02,0.05}. Here, BIC of the model is defined as
where d(ν) is the number of effective parameters, \(\hat {\boldsymbol {\gamma }}\) and \(\hat {\boldsymbol {\theta }}\) minimize (2.4) with a given ν [34]. A larger penalty parameter ν might be applied for problems with larger number of variants in each region.
The results of the simulation study for Model-1 and Model-2 are shown in Fig. 2. The Col method does not have sufficient power to detect the associated regions. The CAST method shows better performance at the level α=0.05, when the direction of effects are the same. At the level α=0.05, the CCRS method shows better performance than SKAT-O when the direction of effects are different. At the level α=0.01, the CCRS and SKAT-O show nearly the same performance. It is clear from the figure that the CCRS method improves performance over the SPC method.

FDR vs. TPR of Model-1 in left panel, and Model-2 in right panel, for α=0.01(∙) and α=0.05(■), compare the CCRS performance with commonly applied methods, SPC, Col, CAST, SKAT-O
Figure 3 shows the result of simulation analysis for Model-3 and Model-4. The CAST method for Model-3 and the Col method for both Model-3 and Model-4 show poor performance. In both models, the CCRS shows noticeably better performance in both α levels. The influential regions in Model-1 through Model-4 have the same effect sizes on the phenotype. Hence, comparing Figs. 2 and 3 provides insight into understanding the influence of LD between causal variants and neutral variants on the power and accuracy of selection. The FDR of the Col and CAST methods shows the largest differences between these two figures. The FDR of CCRS is robust to the correlation among causal and neutral variants in comparison to the other methods.

FDR vs. TPR of Model-3 in left panel, and Model-4 in right panel, for α=0.01(∙) and α=0.05(■), compare the CCRS performance with commonly applied methods, SPC, Col, CAST, SKAT-O
Figure 4 shows the result of analysis of Model-5 and Model-6 which include both correlated and uncorrelated effective variants. SKAT-O shows smaller FDR at the level α=0.05 in the left panel and slightly smaller TPR than CCRS, although at level α=0.01 CCRS shows better performance in terms of FDR and TPR. When the directions of effects are different (Model-6), right panel, CCRS outperforms the other methods.

FDR vs. TPR of Model-5 in left panel, and Model-6 in right panel, for α=0.01(∙) and α=0.05(■), compare the CCRS performance with commonly applied methods, SPC, Col, CAST, SKAT-O
The result of this simulation study shows that the CCRS performs better and more robust than other methods under a variety of genetic architectures, and it is much more prominent when the causal variants are not correlated with neutral variants in the region. Neglecting the presence of LD leads to overestimation of the overall effect of the regions. Although this overestimation might increase the power of detecting a region with some small effects that are correlated with some neutral variants, it increases the risk of missing more promising regions in procedure of multiple comparison of hypotheses testing.
Real data analysis
We analyzed sequencing data from the Atherosclerosis Risk in Communities (ARIC) study [35]. The data are described more fully in [19]. Briefly, 496 African-American individuals were whole genome sequenced at an average depth of 6.3-fold using an Illumine HiSeq 2000 and, after alignment, approximately 31 million high quality variants were called using SNPTools. We present here the result of an association analysis of rare and low frequency variants (MAF≤0.05) with log transformed Apolipoprotein A1 levels (ApoA1). ApoA1 is a component of high density lipoprotein (HDL), which is associated with reduced risk of coronary heart disease [36, 37]. The protein promotes lipid efflux, including cholesterol, from tissues to the liver for excretion [38].
The genotype data includes 949,986 rare variants that are mostly in noncoding regions in the genome [19]. Therefore, we used a sliding window approach to define physical proximity (window). There are approximately 38 thousand consecutive windows each including 50 rare variants and stepping 25 variants until the next window. Therefore, by design, the windows overlap and the results of consecutive windows are not independent. To detect associated regions potentially influencing plasma ApoA1 levels, we used SPC, SKAT, SKAT-O, CAST and Col methods in addition to the CCRS method introduced here.
To define the threshold for statistical significance taking into account multiple hypothesis testing, we ran 100,000 permutation test over 100 windows. Based on this threshold, 10−6, we detected one significantly associated region by the CCRS method. Figure 5 shows the p-values of 80 windows around this region. All of the approaches except CAST and Collapsing test have a peak in this region. The figure shows that the CCRS maintains power for detecting phenotype-influencing region while keeping the p-value of the null or neutral regions small. This is an important property of CCRS that controls the false discovery rate.

p-values of 80 windows around the selected window. The red points represent the calculated p-value by CCRS for the windows and each curves is smooth curve over calculated p-values by one approach; yellow: CAST test, red: CCRS, green: SKAT, blue: SKAT-O, black: Col, orange: SPC
The region contains the gene, FAM78B, which is expressed at high levels in myocytes, fibroblasts, endothelial cells. Little is known about the function of FAM78B. However, within the promoter for FAM78B, three binding sites for the transcription factor PPARG and two binding sites for the transcription factor HNF1A have been identified (http://www.sabiosciences.com and [39]). Pi et al. [40] have shown a significant effect of PPARG on HDL and ApoA1; the major protein component of HDL [41]. PPARs are also expressed in the cardiovascular system such as endothelial cells, vascular smooth muscle cells and monocytes/macrophages (see for e.g. [42]).
Discussion
We have introduced a new approach, CCRS, for the analysis of whole genome sequence data in order to identify regions of the genome (e.g. genes or other functional motifs) influencing a phenotype of interest. The CCRS improves the power of identifying a set of variants associated with a phenotype by taking into account the sparseness and LD structure in the data. The CCRS applies a concave penalized regression method after projecting the sequence variants in a full rank space that is more informative via sparse principal component analysis. By applying sparse PCA, the CCRS aims to enhance the information in the predictors instead of reducing dimension as typical application of sparse PCA, which might increase risk of missing important variants in rare variants analysis. While the first step of analysis (sparse PCA) is an unsupervised method, it does not increase the FDR of the method in the second step of the analysis.
Although the CCRS method can be applied to both common and rare variants, the focus of this analysis was on rare variants because of the role of these variants on phenotype variation. The CCRS method also can be easily expanded to logistic regression and applied for case control studies. However, we investigated the CCRS performance for quantitative traits while the overwhelming majority of the literature focusing on case/control studies and there is a daunting need to develop methods for quantitative traits.
Using simulated data, we show that the FDR of the CCRS method is smaller than other commonly applied genomic region-based test methods while it has higher power of identification in most of the situations. Furthermore, the FDR of CCRS is smaller and robust to the LD structure in the region in comparison to the other methods. While the statistical test for rare variants are typically region-based test, there is risk of overestimation of overall effect of regions by neglecting the LD between causal and neutral variants in the region. Consequently, the risk of missing promising regions might increase through multiple hypotheses testing.
Penalized regression and other shrinkage methods that have been introduced for sparse data applications can correctly select nonzero coefficients under specific conditions [43, 44]. Applying these approaches to large-scale genome sequence applications that include correlated variants due to LD might not lead to a true set of selected variants with nonzero coefficients. Addressing this challenge is difficult in rare variant analyses because each individual variant by itself includes little information. To resolve this problem, the CCRS reparameterizes the model via PCA restricted with L 1 norm constraints to provide a full rank design matrix. Imposing L 1 penalization in PCA generates a sparse loading matrix that renders the analysis interpretable. The CCRS method efficiently incorporates information from low frequency variants by generating new predictors that are much more informative. The CCRS uses a concave penalized regression model to simultaneously select the most important PCs regarding their association with the phenotype of interest, but also to estimate their effect sizes. The zero effect sizes can be uniquely identified due to the use of full rank approximation of the design matrix. The advantage of the concave penalty term is that the rate of shrinkage gets smaller as the effect size increases. In other words, the CCRS not only has the property of parsimony, it also avoids shrinkage over large effect sizes. Thus, the CCRS maintains power for detecting phenotype-influencing regions while keeping the p-value of the neutral regions small.
As an example real data application, we used the CCRS method and genome sequence data to analyze plasma ApoA1 levels, and one region met the experiment-wise criterion for statistical significance. The region contains the gene, FAM78B, which is expressed at high levels in myocytes, fibroblasts, endothelial cells (http://www.proteinatlas.org/ENSG00000188859-FAM78B/tissue). In a real application, annotation of the non-coding regions should be integrated into the analysis, and replication in an independent sample would be the next step to consider it as a novel discovery.
Conclusions
Large-scale whole genome sequencing and high-powered computing are becoming more readily available and affordable. There is an emerging shift from sequencing and computing technologies toward study design, data processing algorithms, and statistical and informatics methods for extracting usable information from the very large amount of genome sequence data that are imminent. The CCRS method presented here for the first time is a practical, powerful and efficient method for taking into account the nature of whole genome sequence variation to identify regions of the genome influencing common complex risk factor phenotypes and diseases.
References
Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012; 337(6090):64–9.
Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 2011; 147(1):32–43.
Morgenthaler S, Thilly WG. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (cast). Mutat Res Fundam Mol Mech Mutagen. 2007; 615(1):28–56.
Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008; 83(3):311–21.
Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009; 5(2):1000384.
Price AL, Kryukov GV, de Bakker PI, Purcell SM, Staples J, Wei LJ, et al. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet. 2010; 86(6):832–8.
Bansal V, Libiger O, Torkamani A, Schork NJ. Statistical analysis strategies for association studies involving rare variants. Nat Rev Genet. 2010; 11(11):773–85.
Zawistowski M, Gopalakrishnan S, Ding J, Li Y, Grimm S, Zöllner S. Extending rare-variant testing strategies: analysis of noncoding sequence and imputed genotypes. Am J Hum Genet. 2010; 87(5):604–17.
Derkach A, Lawless JF, Sun L. Pooled association tests for rare genetic variants: a review and some new results. Stat Sci. 2014; 29(2):302–21.
Han F, Pan W. Powerful multi-marker association tests: unifying genomic distance-based regression and logistic regression. Genet Epidemiol. 2010; 34(7):680–8.
Hoffmann TJ, Marini NJ, Witte JS. Comprehensive approach to analyzing rare genetic variants. PLoS One. 2010; 5(11):13584.
Bhatia G, Bansal V, Harismendy O, Schork NJ, Topol EJ, Frazer K, et al. A covering method for detecting genetic associations between rare variants and common phenotypes. PLoS Comput Biol. 2010; 6(10):1000954.
Zhang L, Pei Y-F, Li J, Papasian CJ, Deng H-W. Efficient utilization of rare variants for detection of disease-related genomic regions. PloS One. 2010; 5(12):14288.
Neale BM, Rivas MA, Voight BF, Altshuler D, Devlin B, Orho-Melander M, et al. Testing for an unusual distribution of rare variants. PLoS Genet. 2011; 7(3):1001322.
Ionita-Laza I, Buxbaum JD, Laird NM, Lange C. A new testing strategy to identify rare variants with either risk or protective effect on disease. PLoS Genet. 2011; 7(2):1001289.
Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011; 89(1):82–93.
Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012; 91(2):224–37.
Bühlmann P, Van De Geer S. Statistics for High-dimensional. Berlin: Springer Series in Statistics;2011.
Morrison AC, Voorman A, Johnson AD, Liu X, Yu J, Li A, et al. Whole genome sequence-based analysis of a model complex trait, high density lipoprotein cholesterol. Nat Genet. 2013; 45(8):899.
Witten DM, Tibshirani R, Hastie T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics. 2009; 10(3):515–534.
Talluri R, Shete S. A linkage disequilibrium–based approach to selecting disease-associated rare variants. PloS One. 2013; 8(7):69226.
Feng T, Zhu X. Whole genome sequencing data from pedigrees suggests linkage disequilibrium among rare variants created by population admixture. In: BMC proceedings. BioMed Central Ltd;2014. p. S44.
Zou H, Hastie T, Tibshirani R. Sparse principal component analysis. J Comput Graph Stat. 2006; 15(2):265–86.
Trendafilov NT, Jolliffe IT. Projected gradient approach to the numerical solution of the scotlass. Comput Stat Data Anal. 2006; 50(1):242–53.
Shen H, Huang JZ. Sparse principal component analysis via regularized low rank matrix approximation. J Multivar Anal. 2008; 99(6):1015–34.
Boyd S, Vandenberghe L. Convex Optimization. Cambridge, U.K.: Cambridge Univ. Press.; 2004.
Wold S. Cross-validatory estimation of the number of components in factor and principal components models. Technometrics. 1978; 20(4):397–405.
Owen AB, Perry PO. Bi-cross-validation of the svd and the nonnegative matrix factorization. Ann Appl Stat. 2009; 3(2):564–94.
Frank LE, Friedman JH. A statistical view of some chemometrics regression tools. Technometrics. 1993; 35(2):109–35.
Fu WJ. Penalized regressions: the bridge versus the lasso. J Comput Graph Stat. 1998; 7(3):397–416.
Zou H, Li R. One-step sparse estimates in nonconcave penalized likelihood models. Ann Stat. 2008; 36(4):1509–33.
Armagan A. Variational bridge regression. Journal of Machine Learning Research, Workshop and Conference Proceedings. 2009; 5:17–24.
Tibshirani R. Regression shrinkage and selection via the lasso. J R Stat Soc Ser B. 1996; 58:267–88.
Huang J, Ma S, Xie H, Zhang CH. A group bridge approach for variable selection. Biometrika. 2009; 96(2):339–55.
Aric Investigators. The atherosclerosis risk in communit (aric) stui) y: Design and objectwes. American journal of epidemiology. 1989; 129(4):687–702.
Sing CF, Boerwinkle E, Moll P, Davignon J. Apolipoproteins and cardiovascular risk: genetics and epidemiology. Ann Biol Clin. 1985; 43:407–417.
Virani SS, Brautbar A, Davis BC, Nambi V, Hoogeveen RC, Sharrett AR, et al. Associations between lipoprotein (a) levels and cardiovascular outcomes in black and white subjects the atherosclerosis risk in communities (aric) study. Circulation. 2012; 125(2):241–9.
Glomset JA. The plasma lecithin: cholesterol acyltransferase reaction. J Lipid Res. 1968; 9(2):155–67.
Rosenbloom KR, Dreszer TR, Pheasant M, Barber GP, Meyer LR, Pohl A, et al. Encode whole-genome data in the ucsc genome browser. Nucleic Acids Res. 2010; 38(suppl 1):620–5.
Pei W, Baron H, Müller-Myhsok B, Knoblauch H, Ali Al-Yahyaee S, Hui R, et al. Support for linkage of familial combined hyperlipidemia to chromosome 1q21–q23 in chinese and german families. Clin Genet. 2000; 57(1):29–34.
DiDonato JA, Huang Y, Aulak KS, Even-Or O, Gerstenecker G, Gogonea V, et al. Function and distribution of apolipoprotein a1 in the artery wall are markedly distinct from those in plasma. Circulation. 2013; 128(15):1644–55.
Das SK, Chakrabarti R. Role of ppar in cardiovascular diseases. Recent Pat Cardiovasc Drug Discov. 2006; 1(2):193–209.
Knight K, Fu W. Asymptotics for lasso-type estimators. Ann Stat. 2000; 28(5):1356–78.
Huang J, Horowitz JL, Ma S. Asymptotic properties of bridge estimators in sparse high-dimensional regression models. Ann Stat. 2008; 36:587–613.
Acknowledgements
Atherosclerosis Risk in Communities (ARIC) Study: This ARIC study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (H H S N 268201100005C, H H S N 2682011000 06C, H H S N 268201100007C, H H S N 268201100008C, H H S N 268201100009C, H H S N 268201100010C, H H S N 268201100011C,and H H S N 268201100012C).
The authors thank the staff and participants of the ARIC study for their important contributions. Funding support for “Building on GWAS for NHLBI-diseases: the U.S. CHARGE consortium” was provided by the NIH through the American Recovery and Reinvestment Act of 2009 (ARRA) (5RC2HL102419). Sequencing was carried out at the Baylor Genome Center (U54 HG003273).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AY (first author) developed method, carried out simulated and real data analysis, interpreted results and made initial drafting of the manuscript. AY (second author) provided valuable comments and advice on discussions and interpretation. EB conceived of project and study question, provided funding, generation of the real data, aided in interpreting of results and made critical edits to the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License(http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yazdani, A., Yazdani, A. & Boerwinkle, E. Rare variants analysis using penalization methods for whole genome sequence data. BMC Bioinformatics 16, 405 (2015). https://doi.org/10.1186/s12859-015-0825-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-015-0825-4