
Abstract
Background
Procedures for the detection of signatures of selection can be classified according to the source of information they use to reject the null hypothesis of absence of selection. Three main groups of tests can be identified that are based on: (1) the analysis of the site frequency spectrum, (2) the study of the extension of the linkage disequilibrium across the length of the haplotypes that surround the polymorphism, and (3) the differentiation among populations. The aim of this study was to compare the performance of a subset of these procedures by using a dataset on seven Spanish autochthonous beef cattle populations.
Results
Analysis of the correlations between the logarithms of the statistics that were obtained by 11 tests for detecting signatures of selection at each single nucleotide polymorphism confirmed that they can be clustered into the three main groups mentioned above. A factor analysis summarized the results of the 11 tests into three canonical axes that were each associated with one of the three groups. Moreover, the signatures of selection identified with the first and second groups of tests were shared across populations, whereas those with the third group were more breed-specific. Nevertheless, an enrichment analysis identified the metabolic pathways that were associated with each group; they coincided with canonical axes and were related to immune response, muscle development, protein biosynthesis, skin and pigmentation, glucose metabolism, fat metabolism, embryogenesis and morphology, heart and uterine metabolism, regulation of the hypothalamic–pituitary–thyroid axis, hormonal, cellular cycle, cell signaling and extracellular receptors.
Conclusions
We show that the results of the procedures used to identify signals of selection differed substantially between the three groups of tests. However, they can be classified using a factor analysis. Moreover, each canonical factor that coincided with a group of tests identified different signals of selection, which could be attributed to processes of selection that occurred at different evolutionary times. Nevertheless, the metabolic pathways that were associated with each group of tests were similar, which suggests that the selection events that occurred during the evolutionary history of the populations probably affected the same group of traits.
Similar content being viewed by others


Background
The evolutionary history of animal populations involves both natural and artificial selection. These processes not only affect the allelic frequencies at causal polymorphisms, but also the surrounding genomic regions due to the so-called “hitchhiking” effect. Thus, they may leave detectable signals on the structure of the genome that can be identified by using appropriate procedures [1, 2].
The vast majority of the procedures used to detect signatures of selection [2] is based on the null hypothesis of absence of selection, which relies on the neutral model of evolution [3]. In fact, these procedures can be classified according to the source of information they use to reject the null hypothesis. Based on the literature [2], three main groups of tests can be identified: the first group is based on the analysis of the site frequency spectrum [4–6], the second group focuses on the study of the extension of the linkage disequilibrium across the length of the haplotypes that surround a polymorphism [7, 8], and the third group is based on several measures of differentiation among populations [9–11]. In addition, the results of all these tests can be affected to some degree by demographic events and by the ascertainment bias caused by the procedure used to select the single nucleotide polymorphisms (SNPs) for the genotyping chip [12]. Thus, the results of each test may not be fully consistent with each other [13], which has led to propose strategies for summarizing results into a single statistic that either does [13] or does not [14, 15] account for the correlations between the results from different methods.
The aim of our study was to compare the performance of a subset of these procedures by using a dataset on seven autochthonous beef cattle populations (Asturiana de los Valles, Avileña-Negra Ibérica, Bruna dels Pirineus, Morucha, Pirenaica, Retinta and Rubia Gallega) which share close genetic relationships between them [16]. A second objective was to identify candidate genes and/or metabolic processes that are associated with the regions involved in the selection processes that occurred during the evolution of these populations.
Methods
Animals and sample size
A total of 171 sire/dam/offspring triplets were collected from seven Spanish beef cattle populations, including Asturiana de los Valles (AV, n = 25), Avileña-Negra Ibérica (ANI, n = 24), Bruna dels Pirineus (BP, n = 25), Morucha (Mo, n = 24), Pirenaica (Pi, n = 24), Retinta (Re, n = 24) and Rubia Gallega (RG, n = 24) breeds. The selected parents were chosen as unrelated as possible to fully represent the diversity of the populations.
SNP genotyping and phasing
Genomic DNA was extracted by standard protocols. High-density SNP genotyping was performed at a commercial laboratory (Xenética Fontao, Lugo, Spain) by using the BovineHD BeadChip (Illumina Inc, USA) according to the manufacturer’s protocol; this HD chip is designed to genotype 777,962 SNPs. The SNPs that were retained for our study were located on autosomal chromosomes at a single position. Additional requirements were a Mendelian error rate lower than 0.05, and SNP and individual call rates higher than 0.95. Quality control was performed by using PLINK software [17] and finally, 703,707 SNPs that covered 2,510,606 kb were available for the analyses with on average one SNP per 3.567 kb. Haplotypes for the parental chromosomes were derived with Beagle software [18] using the “TRIO” option.
Detection of signatures of selection
The data were analysed using the following procedures for the detection of signatures of selection.
Tajima
The procedure that was developed by Tajima [4] compares two statistics to estimate the scaled mutation rate. The first statistic \(\left( {\theta_{\pi } } \right)\) is based on the number of segregating sites within a genomic region and the second \(\left( {\theta_{\kappa } } \right)\) is the average heterozygosity at segregating sites in the sample. The standardized difference between these two values, \(D = \theta_{\pi } - \theta_{\kappa } ,\) is used to infer departures from neutrality. Theoretically, if \(D < 0\) either the population has suffered expansion after a recent bottleneck or a recent selective sweep has taken place; on the contrary if \(D > 0,\) the population has either experienced a sudden population contraction or is under balancing selection. The analysis was performed over sliding windows of 100 SNPs by using own software.
Fay and Wu
This procedure [6] calculates the following statistic \(D = \theta_{\pi } - \theta_{H} ,\) where \(\theta_{H}\) depends on the number of sites at which a derived allele is present within a genomic region. In the analysis, the ancestral alleles were extracted from the study of Rocha et al. [19]. This test was computed over sliding windows of 100 SNPs by using own software.
Fu and Li
This procedure [5] is based on counting the number of singletons or alleles present in only one phase. The rationale is that a selection process will extend time to coalescence so that a larger number of mutations may take place in new or external branches of the tree and thus appear only once in the observed sample. As before, the analysis was performed over sliding windows of 100 SNPs by using own software.
iHS
This procedure [8] calculates the ratio of the integrated haplotype score (iHH) for the ancestral allele and the derived allele at a given SNP. The iHH is the integral (area) of the observed decay of the EHH (extended haplotype homozygosity) as defined by Sabeti et al. [7]. As in the previous test, ancestral alleles were extracted from Rocha et al. [19]. The iHS was calculated with the selscan software [20] using the parameters recommended by the authors. For further calculations, we used the |iHS|.
nSL
This procedure was recently presented by Ferrer-Admetlla et al. [21]. The procedure of calculation is similar to iHS, but replaces IHH by an alternative statistic (SL) that measures the length of a segment of haplotype homozygosity in terms of segregating sites. The main advantage of nSL over iHS is that it uses segregating sites as a measure of distance, while iHS needs the recombination distance. Thus, the iHS is more sensitive to recombination rate [21]. The analysis used the same parameters as in the iHS test and own software. As before, we used |nSL|.
H12
This method was recently proposed [22] with the H12 statistic being defined as:
where \(p_{j}\) is the frequency of the jth most common haplotype in the population. Here, the frequencies of the first and second most common haplotypes were combined into a single frequency. The calculation was performed over sliding windows of 100 SNPs by using own software.
Fixation index (F ST)
This procedure was described by Wright [9] and is the most classical approach to study the pattern of differentiation between populations. The fixation index \(F_{\text{ST}}\) is calculated for each SNP and for each pair of populations as \(F_{\text{ST}} = \left( {H_{O} - H_{E} } \right) /H_{E} ,\) where \(H_{O}\) and \(H_{E}\) are the observed and expected heterozygosities, respectively. Estimates for \(F_{\text{ST}}\) were averaged over sliding windows of 100 SNPs and assigned to the central SNP in each window. The procedure was computed with own software. Finally, the results for each population were computed by averaging the paired \(F_{\text{ST}}\) estimates with the other six populations.
Selestim
This procedure [10] assumes a hierarchical Bayesian model to distinguish selected polymorphisms from the background of neutral (or almost neutral) polymorphisms and also to estimate the intensity of selection in each population. The model assumes a binomial distribution of the allele counts at each locus and for each population, and the prior distribution of allelic frequencies is modeled under the assumption of a stationary density of the diffusion process [10]. The model is implemented by using a Markov chain Monte Carlo method. SelEstim software (http://www1.montpellier.inra.fr/CBGP/software/selestim/) was used for this purpose with the standard parameters that are recommended by the authors. Among the outputs provided by the Selestim approach, we extracted the \(\sigma_{ij}\) parameter [10], which represents the coefficient of selection for the ith subpopulation and the jth locus.
XP-CLR
This approach [23] assumes that the allele frequencies of two populations that diverge from an ancestral population follow a Gaussian distribution for which the variance contains information on the history of the populations since they split. Under the assumption that the evolutionary process is reversible, the procedure defines the distribution of allelic frequencies in the first population (reference) given the allele frequencies in the second population (objective). We calculated XP-CLR by taking each pair of populations as objective and reference with the software XPCLR (http://genetics.med.harvard.edu/reich/Reich_Lab/Software.html). Then, we averaged the six available tests for each population that was treated as an objective population, to infer the signatures of selection for each breed.
XP-EHH
This approach [7] is also computed for each pair of populations. For each population, as in the iHS test, it calculates the EHH between a core SNP and a set of SNPs within a predefined genomic interval and integrates it with respect to genetic distance to calculate the integrated haplotype score (IHH) for populations A and B. Then, the statistic is computed as \(XPEHH_{AB} = \ln \left( {IHH_{A} /IHH_{B} } \right).\) As previously, we computed this statistic for each SNP and each pair of populations and the results were averaged over the six comparisons to generate a unique result for each population. We used the software selscan [20] with the parameters that are recommended by the authors.
VarLD
This procedure [24] evaluates the magnitude of the differences in linkage disequilibrium between a pair of populations. It calculates the linkage disequilibrium as the correlation coefficient between pairs of SNPs within a genomic region and creates a matrix of those correlations for each population. Then, it evaluates the differences between the matrices of both populations as the difference between its eigenvalues. The procedure was computed using the software VarLD [25] over sliding windows of 100 SNPs.
For all the above-described methods, we used the empirical distribution of the results generated along the genome as the null distribution of the test, in order to reduce the possible effects of the demographic history or the ascertainment bias. The underlying hypothesis is that, on average, both demographic events and ascertainment bias affect all the genome in a similar way, and thus, deviations or extreme values of the empirical distribution could be understood as signals of selection events.
Summary of signals of selection
In order to detect communalities and summarize the results of the 11 procedures for ease of interpretation, we normalized these results for each SNP using a logarithm transformation to make the scale of the different results comparable and, then, we calculated the correlation between the logarithms (or the negative of logarithms for the Tajima, Fu and Li and Fay and Wu procedures) for the 703,707 SNPs. In a confirmatory analysis, provided that the methods used to detect signatures of selection were classified into three groups, we performed a factor analysis restricted to a subspace of three axes using a varimax rotation [26]. The analysis was done with R [27] by using the function principal() included in the package psych.
Selection of candidate genes
First, we identified candidate genes based on the empirical distribution of the output of the three canonical axes of the factor analysis. Thus, we defined a very strict threshold by selecting the genomic 1-Mb regions with at least 25 SNPs that were in the top 0.1% of the results for each axis. Then, we used the Ensembl-Biomart database to identify the genes that were present in those genomic regions and compared our results with those in the literature to identify potential candidate genes for selection in the bovine populations.
Enrichment analysis
Finally, in order to obtain a clearer picture of the metabolic pathways that were affected by the selection processes, we identified the genomic regions that were above the top 5% of each canonical axis. The objective of the relaxation of the empirical threshold was to capture softer signals of selection. With these selected genomic regions for each canonical axis, we used the software WebGestalt [28] (http://bioinfo.vanderbilt.edu/webgestalt/) by setting the Homo sapiens genome as the reference genome. In addition, we used a hypergeometric p value to correct for multiple-testing. The results included the top 10 pathways (WikiPathways).
Results and discussion
Summarizing footprints of selection detected by 11 procedures
A large set of procedures is available for the identification of footprints of selection across the genome [2]. Most of these procedures are based on the rejection of the null hypothesis of absence of selection based on the neutral theory of evolution [3]. However, each of these methods calculates a different statistic to test this hypothesis. In addition, they are influenced to varying degrees by demographic history and ascertainment bias caused by the selection of SNPs [12]. Thus, it is expected that each test provides a different output as confirmed by the correlations between the results obtained by the 11 procedures used in this study (Fig. 1) and by the Manhattan plots generated with the results for each test and population (see Additional file 1: Figure S1, Additional file 2: Figure S2, Additional file 3: Figure S3, Additional file 4: Figure S4, Additional file 5: Figure S5, Additional file 6: Figure S6, Additional file 7: Figure S7, Additional file 8: Figure S8, Additional file 9: Figure S9, Additional file 10: Figure S10, Additional file 11: Figure S11).

Heatmap of the correlations between logarithms of the results of the 11 tests applied for the detection of signatures of selection and their clustering
In order to summarize the signals of selection that were detected by the 11 tests, there are procedures to condense such results into a single statistic by using Bayes factors [14] or a combination of p values [13, 15]. However, these strategies imply that the signals of selection that are captured by the different methods are comparable. Nevertheless, as the definition of the null hypothesis varies between tests, the signals of selection identified by each procedure may correspond to different types of selection events. In fact, some authors [29] pointed out that within-population haplotype length methods [8, 21] can detect only very recent selection processes, because they become ineffective when the selected alleles reach fixation or are very close to fixation. The same authors [29] indicated that signals of selection that are based on a reduction of genetic diversity [4, 5] persist for a longer period of time and these methods can detect older signals of selection, while tests that are based on population differentiation [9, 10] occupy an intermediate position.
In this study, the correlations of the absolute logarithm of the results between the 11 methods used were low or even negative (Fig. 1). However, there are some remarkable exceptions such as the correlations between Tajima and Fu and Li, and iHS and nSL tests, that were remarkably high. It should be noted that the Tajima and Fu and Li tests are both based on the analysis of the site frequency spectrum, and that the nSL test is just a modification of iHS where the map distance is replaced by the number of segregating sites [21]. A more detailed analysis of the structure of these correlations allows to identify three main groups of tests, one group based on the site frequency spectrum (Tajima, Fu and Li, Fay and Wu); a second group based on the haplotype length (iHS and nSL), and a third group that focuses on the differentiation between populations (F ST, SelEstim and XP-CLR). The remaining tests (VarLD, H12 and XP-EHH) are in an intermediate position between the latter two, although slightly closer to tests based on population differentiation. Such a structure of the correlations between the results of these methods indicates that the implementation of a factor analysis, as suggested by Simianer et al. [30], could be appropriate to summarize the results into a few canonical axes. In addition, as described below, each axis was associated with signatures of selection of a different kind. In particular, we applied a factor analysis restricted to three canonical axes using a varimax approximation [26] that explains up to the 56% of variation.
Table 1 shows the loadings for the canonical axes that resulted from the factor analysis for each of the 11 tests used to detect selection signatures. Moreover, Table 1 presents the correlations between the canonical axes and each specific test, which are fully consistent with the results of the correlations presented in Fig. 1. The first axis explains 20% of the variation and shows a high correlation with the procedures based on the analysis of population differentiation (F ST, SelEstim, XP-EHH, XP-CLR and VarLD) and H12; the second axis explains 19% of the variation and is correlated with the methods based on the site frequency spectrum (Tajima, Fu and Li, and Fay and Wu); and, finally, the third axis is strongly correlated with methods based on the extension of linkage disequilibrium or haplotype length (iHS and nSL) and explains 17% of the total variation. For each test, the three axes explain between 32 (XP-CLR) and 90% (iHS) of the variation. The Manhattan plots of the results that relate to the three canonical axes are in Figs. 2, 3 and 4. The first two axes presented a higher level of shared signals between populations (see Figs. 2, 3) whereas the results of the third axis were, in general, breed-specific. This statement is supported by the results in Fig. 5, which shows the correlations of the results obtained for the first, second and third canonical axes between populations. An average correlation of 0.50 was found for the first axis [ranging from 0.39 (BP and Re) to 0.71 (AV and RG)]. Furthermore, the second axis also showed high correlations between populations that ranged from 0.37 (Re and Pi) to 0.60 (AV and RG) with an average of 0.49. On the contrary, the correlations for the third axis were lower with an average value of 0.08 and ranged from 0.05 (Pi and Re) to 0.16 (AV and BP). In addition, the structure of the correlations (Fig. 5) confirmed the classification of the populations into two main clusters, one composed by the ANI, Mo and Re populations and the other by the Pi, BP, RG and ANI populations, as previously reported by Cañas-Álvarez et al. [16] based on distance measures and admixture analysis.

Manhattan plots for the results of the first axis (a) and genomic regions identified with at least 25 SNPs within the top 0.1% of the results (b)

Manhattan plots for the results of the second axis (a) and genomic regions identified with at least 25 SNPs within the top 0.1% of the results (b)

Manhattan plots for the results of the third axis (a) and genomic regions identified with at least 25 SNPs within the top 0.1% of the results (b)

Correlations of the results from the first, second and third canonical axes between populations
As in the study of Sabeti et al. [29], our results may indicate that old selection or adaptation processes that occurred before breed differentiation or during speciation were only detected by the site frequency spectrum methods that are associated with the second canonical axis. The signals of selection that were generated by later isolation and recent selection events within the populations are identified by the differentiation methods, which are linked to the first canonical axis. Finally, the haplotype length methods, summarized in the third canonical axis, identified more recent and, in general, less intense selection events that are mostly specific to each population. The absence of regions with strong recent signals of selection agrees with the postulate that artificial selection processes do not leave relevant signatures of selection [31]. The main reason of this absence can be due to the polygenic nature of most of the traits associated with current selection processes [32–34] or to the effect of epistasis [35].
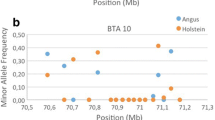
In fact, the most remarkable signal of selection from this third axis was identified on chromosome 2 around the myostatin (MTSN) gene (between 6,213,566 and 6,220,196 bp) in the AV population, where double-muscling is included as a criterion of selection in its breeding program. This specific genomic region can be used to illustrate the timing of the signatures of selection that were detected by each group of methods. In the AV population, two large signatures of selection were detected with the first and third canonical axes, respectively. The first axis is related to processes that were involved in the creation of the breed and the third axis to recent selection. In addition, a large signature of selection associated with the first canonical axis in the RG population was observed. However, in this population, there is no relevant signature in the results of the third axis. This result is consistent with several previous studies [36] that reported the presence of haplotypes associated with double-muscling also in the RG population. This may indicate that some degree of selection around this gene may have occurred during the process of breed formation, but the current breeding program does no longer put any selection pressure on double-muscling. Finally, the results from the second canonical axis are less relevant (AV) or even absent (RG), which indicates that, for this group of tests, the selection effects may be diluted because a larger number of generations without selection is considered.
Candidate genes and metabolic paths
The results of the first axis (Fig. 2) allowed us to highlight seven relevant genomic regions on Bos taurus chromosome (BTA) 2 (between 1,047,347 and 11,899,039 bp), BTA5 (between 15,920,995 and 20,321,882 bp), BTA6 (between 37,853,912 and 41,160,000 bp), BTA7 (between 47,276,124 and 47,745,164 bp), BTA11 (between 65,077,840 and 72,203,248 bp), BTA13 (between 57,430,392 and 57,754,760 bp) and BTA18 (between 12,675,262 and 16,202,289 bp). In some cases, these genomic regions were extremely large, because of strong signatures of selection such as those on BTA2 for the AV and RG populations or on BTA5, BTA6 and BTA11 for the BP population. Such huge signatures of selection imply that large genomic regions included SNPs that were associated with results above the top 0.1% of the empirical distribution along the genome. However, the localization of the strongest signals within each genomic region and for each population allowed us to narrow down the genomic regions (Fig. 2b), which are similar to those reported in a previous study on the differentiation between populations [37]. These regions included well-known genes that were previously reported as potential candidates of selection signatures in cattle [38], such as MTSN (myostatin) on BTA2, suggested in several beef cattle populations [39–41], KIT-LG (kit-ligand) on BTA5 with a very large peak in the BP population, MC1R (melanocortin 1 receptor) on BTA18, which controls the production of eumelanin (black) or pheomelanin (red) pigments [42] and appears to be relevant in populations with black (AV and Mo) or red (Re) coat color. Moreover, it should be also highlighted that the region on BTA6 that includes LAP3 (leucine aminopeptidase3), LCORL (ligand dependent nuclear receptor corepressor-like) and NCAPG (non-SMC condensing I complex, subunit G) and was identified in two meta-analyses [38, 43] as one the genomic regions that is most frequently identified with signatures of selection in the bovine genome. The genomic region identified on BTA7 includes the CAMLG (calcium modulating ligand) and TCF (transcription factor 7) genes, which are close to a strong signature of selection that was reported by Gautier [44] and is associated with the VDAC1 (voltage-dependent anion-selective channel protein 1) gene. A strong signature of selection was observed for the genomic region on BTA13, in the Re population, where is located the END3 (endothelin 3) gene that plays a role in melanocyte development [45] and was recently associated with piebald pattern [44]. Finally, there is a very strong signature of selection on BTA11 for the BP population, where the closest gene to the maximum signal is BMP10 (bone morphogenetic protein 10). Within this genomic region on BTA11, some authors [46, 47] identified signatures of selection and several genes that could be associated with fertility: PROKR1 (prokineticin receptor 1), GFPT1 (glutamine-fructose-6-phosphate transaminase 1), GMCL1 (germ cell-less spermatogenesis associated 1), PCBP1 (poly(rC) binding protein 1) and EHD3 (EH-domain containing 3).
The results of the second canonical axis (Fig. 3) confirmed some of the signals of selection that were detected in the first axis, but also revealed several new ones that are shared by several populations and located on five chromosomes: BTA2 (between 61,684,232 and 62,199,344 and between 72,158,144 and 73,356,296 bp), BTA7 (between 20,612,988 and 21,163,812 bp), BTA13 (between 11,860,881 and 12,062,522 bp), BTA16 (between 44,612,592 and 45,846,144 bp) and BTA21 (between 32,207,264 and 32,414,316 bp). Previously, two meta-analyses [38, 43] showed that these regions were also associated with signatures of selection in other populations. Among the genes included in these regions, some of them may be good candidates for being affected by selection i.e.: (1) genes that are related to energy balance and homeostasis: R3HGM1 (R3H domain containing 1) on BTA2 [48]; CAMK1D (calcium/calmodulin-dependent protein kinase ID) on BTA13 [49]; and SLC25A33 (solute carrier family 25 (pyrimidine nucleotide carrier), member 33) and SLC2A5 (solute carrier family 2 (facilitated glucose/fructose transporter), member 5) on BTA16 [49, 50]; (2) PLIN5 (perilipin 5) on BTA7, which is involved in the regulation of lipid metabolism in rat and pigs [51, 52]; (3) SCAPER (s-phase cyclin A-associated protein in the endoplasmic reticulum) on BTA21, which regulates cell cycle progression [53]; and (4) there are two other signatures of selection that are worth noting i.e. one detected for the RG population on BTA19 (between 27,941,270 and 28,571,032 bp) where ALOX15B (arachidonate 15-lipoxygenase, type B) and ALOX12B (arachidonate 12-lipoxygenase, 12R type) are located and are involved in immune response [54], and one for the ANI population on BTA20 (between 40,854,136 and 40,996,384 bp) where the NPR3 (natriuretic peptide receptor 3) gene is located, which is related with cattle stature [55].
Although less relevant, the results of the third canonical axis (Fig. 4) confirm the signatures of selection around the MTSN gene for the AV, RG and Pi populations, and around the complex LAP–LCORL–NCAG for the BP population. There are several other interesting signatures of selection such as those located on BTA27 (between 36,466,580 and 40,862,444 bp) and BTA28 (between 41,643,416 and 45,215,488 bp) for the ANI and Mo populations, respectively. The first genomic region includes the IKBKB (inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase beta), DKK4 (dickkopf WNT signaling pathway inhibitor 4) and VDAC3 (voltage-dependent anion-selective channel protein 3) genes that are associated with immune response to trypanosoma infection in African populations [56], and the second region contains the ALOX5 (arachidonate 5-lipoxygenase) and RASSF4 (ras association (RalGDS/AF-6) domain family member 4) genes, that are related with growth [57] and feed conversion [58], respectively.
Finally, we performed a pathway enrichment analysis [59] for the identified genomic regions by applying a less restrictive criterion (top 5%). The objective of this analysis was to identify a larger number of genomic regions associated with signatures of selection although with less strong signals. The results of the top 10 pathways that were identified by the enrichment analysis are in Table 2. In general, the pathways associated with each canonical axis are coincident, which indicates that the metabolic pathways that were involved in old and recent selection events are similar, although probably with variable intensities and directions [60]. The enrichment analysis identified pathways that are related with immune response (lymphocyte TarBase), muscle development (muscle cell TarBase), protein biosynthesis (translation factors, cytoplasmic ribosomal proteins), skin and pigmentation (epithelium TarBase), glucose metabolism (insulin signaling, integrated pancreatic cancer pathway), fat metabolism (adipogenesis), embryogenesis and morphology (focal adhesion), heart (calcium regulation in the cardiac cell) and uterine metabolism (myometrial relaxation and contraction pathways), regulation of the hypothalamic–pituitary–thyroid axis (TSH signalling pathway), hormonal, cellular cycle (MAPK-signaling pathway, G1 to S cell cycle control, eukaryotic transcription initiation), cell signaling (notch signaling pathway) and extracellular receptors (GPCR, class A rhodopsin-like). Among these, 10 pathways (focal adhesion, integrated pancreatic cancer pathway, adipogenesis, myometrial relaxation and contraction pathways, adipogenesis, lymphocyte TarBase, insulin signaling, MAPK signaling pathway, focal adhesion, epithelium TarBase) had been previously identified in a meta-analysis based on a very large number of studies on selection signatures in cattle [38], which confirmed that the metabolic pathways involved in old and recent processes of selection are similar to those detected by using equivalent approaches in other cattle populations.
Conclusions
In this study, we confirm that the results of various procedures used to identify signatures of selection varied largely among groups of tests depending on the source of information they use to reject the null hypothesis of absence of selection. However, we observed some correlations between the results of each test. Accordingly, these tests could be clustered into three groups that matched with the three canonical axes of a factor analysis. Moreover, each canonical factor (or group of tests) identified different signals of selection, which were assigned to selection events that occurred at different evolutionary times. In fact, older selection events generated signatures of selection that presented communalities between populations, whereas more recent selection events were detected specifically for each population. Nevertheless, the enriched metabolic pathways associated to each group of tests showed an important degree of agreement which suggests that the traits involved in the selection events were similar during the evolutionary history of the populations.
References
Nielsen R. Molecular signatures of natural selection. Annu Rev Genet. 2005;39:197–218.
Qanbari S, Simianer H. Mapping signatures of positive selection in the genome of livestock. Livest Sci. 2014;166:133–43.
Kimura M. The neutral theory of molecular evolution. Cambridge: Cambridge University Press; 1983.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709.
Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–13.
Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–7.
Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72.
Wright S. Isolation by distance. Genetics. 1943;28:114–38.
Vitalis R, Gautier M, Dawson KJ, Beaumont MA. Detecting and measuring selection from gene frequency data. Genetics. 2014;196:799–817.
Beaumont MA, Balding DJ. Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol. 2004;13:969–80.
Nielsen R. Population genetic analysis of ascertained SNP data. Hum Genomics. 2004;1:218–24.
Ma Y, Ding X, Qanbari S, Weigend S, Zhang Q, Simianer H. Properties of different selection signature statistics and a new strategy for combining them. Heredity (Edinb). 2015;115:426–36.
Grossman SR, Shlyakhter I, Karlsson EK, Byrne EH, Morales S, Frieden G, et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science. 2006;327:883–6.
Utsunomiya YT, Perez-O’Brien AM, Sonstegard TS, Van Tassell CP, do Carmo AS, Mészáros G, et al. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS One. 2013;8:e64280.
Cañas-Álvarez JJ, González-Rodríguez A, Munilla S, Varona L, Díaz C, et al. Genetic diversity and divergence among Spanish beef cattle breeds assessed by a bovine high-density SNP chip. J Anim Sci. 2015;93:5164–74.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–23.
Rocha D, Billerey C, Samson F, Boichard D, Boussaha M. Identification of the putative ancestral allele of bovine single-nucleotide polymorphism. J Anim Breed Genet. 2014;131:483–6.
Szpiech ZA, Hernández RD. Selscan: an efficient multi-threaded program to calculate EHH-based scans for positive selection. Mol Biol Evol. 2014;31:2824–7.
Ferrer-Admetlla A, Liang M, Korneliussen T, Nielsen R. On detecting incomplete soft or hard selective sweeps using haplotype structure. Mol Biol Evol. 2014;31:1275–91.
Garud NR, Messer PW, Buzbas EO, Petrov DA. Recent selective sweeps in North American Drosophila melanogaster show signatures of soft sweeps. PLoS Genet. 2015;11:e1005004.
Chen H, Patterson N, Reich D. Population differentiation as a test for selective sweeps. Genome Res. 2010;20:393–402.
Teo YY, Fry AE, Bhattacharya K, Small KS, Kwiatkowski DP. Genome-wide comparisons of variation in linkage disequilibrium. Genome Res. 2009;19:1849–60.
Ong R, Teo YY. VarLD: a program for quantifying variation in linkage disequilibrium patterns between populations. Bioinformatics. 2010;26:1269–70.
Kaiser HF. The varimax criterion for analytic rotation in factor analysis. Psychometrika. 1958;23:187–200.
R Development Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2008. ISBN 3-900051-07-0. http://www.R-project.org.
Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt). Nucleic Acids Res. 2013;41:W77–83.
Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, et al. Positive natural selection in human lineage. Science. 2006;312:1614–20.
Simianer H, Ma Y, Qanbari S. Statistical problems in livestock population genomics. In: Proceedings, 10th World Congress of genetics applied to livestock production. Vancouver; 2014. https://asas.org/docs/default-source/wcgalp-proceedings-oral/202_paper_10373_manuscript_1346_0.pdf?sfvrsn=2.
Kemper KE, Saxton SJ, Bolormaa S, Hayes BJ, Goddard ME. Selection for complex traits leaves little or no classic signatures of selection. BMC Genomics. 2014;15:246.
Cole JB, VanRaden PM, O’Connell JR, Van Tassell CP, Sonstegard TS, Schnabel RD, et al. Distribution and location of genetic effects for dairy traits. J Dairy Sci. 2009;92:2931–46.
Hayes BJ, Pryce J, Chamberlain AJ, Bowman PJ, Goddard ME. Genetic architecture of complex traits and accuracy of genomic prediction: coat colour, milk-fat percentage and type in Holstein cattle as contrasting model traits. PLoS Genet. 2010;23:e1001139.
Pimentel EG, Erbe M, König S, Simianer H. Genome partitioning of genetic variation for milk production and composition traits in Holstein cattle. Front Genet. 2011;2:19.
Shao H, Burrage LC, Sinasac DS, Hill AE, Ernest SR, O’Brien W, et al. Genetic architecture of complex traits: large phenotypic effects and pervasive epistasis. Proc Natl Acad Sci USA. 2008;105:19910–4.
Dunner S, Miranda ME, Amigues Y, Cañon J, Georges M, Hanset R, et al. Haplotype diversity of the myostatin gene among beef cattle breeds. Genet Sel Evol. 2003;35:103–18.
González-Rodríguez A, Toro MA, Varona L, Carabaño MJ, Cañas-Álvarez JJ, Altarriba J, et al. Genome-wide analysis of genetic diversity in Autochthonous Spanish populations of beef cattle. In: Proceedings of the 10th World Congress of genetics applied to livestock production. Vancouver; 2014. https://asas.org/docs/default-source/wcgalp-proceedings-oral/255_paper_9607_manuscript_1524_0.pdf?sfvrsn=2.
Gutierrez-Gil B, Arranz JJ, Wiener P. An interpretive review of selective sweep studies in Bos taurus cattle populations: identification of unique and shared selection signals across breeds. Front Genet. 2015;6:167.
Wiener P, Gutierrez-Gil B. Assessment of selection mapping near the myostatin gene (GDF-8) in cattle. Anim Genet. 2009;40:598–608.
Boitard S, Rocha D. Detection of signatures of selective sweeps in the Blonde d’Aquitaine cattle breed. Anim Genet. 2013;44:579–83.
Druet T, Pérez-Pardal L, Charlier C, Gautier M. Identification of large selective sweeps associated with major genes in cattle. Anim Genet. 2013;44:758–62.
Werth LA, Hawkins GA, Eggen A, Petit E, Elduque C, Kreigesmann B, et al. Rapid communication: melanocyte stimulating hormone receptor (MC1R) maps to bovine chromosome 18. J Anim Sci. 1996;74:262.
Randhawa IAS, Khatkar MS, Thomson PC, Raadsma HW. A meta-assembly of selection signatures in cCattle. PLoS One. 2016;11:e0152013.
Gautier M. Genome-wide scan for adaptive divergence and association with population-specific covariates. Genetics. 2015;201:1555–79.
Saldana-Caboverde A, Kos L. Roles of endothelin signaling in melanocyte development and melanoma. Pigment Cell Melanoma Res. 2010;23:160–70.
Stella A, Ajmone-Marsan P, Lazzari B, Boettcher P. Identification of selection signatures in cattle breeds selected for dairy production. Genetics. 2010;185:1451–61.
Rothammer S, Seichter D, Förster M, Medugorac I. A genomic scan for signatures of differential artificial selection in ten cattle breeds. BMC Genomics. 2013;14:908.
Burt DW. The cattle genome reveals its secrets. J Biol. 2009;8:36.
Randhawa IAS, Khatkar MS, Thomson PC, Raadsma HW. Composite selection signals can localize the trait specific genomic regions in multi-breed populations of cattle and sheep. BMC Genet. 2014;15:34.
Bomba L, Nicolazzi EL, Milanesi M, Negrini R, Mancini G, Biscarini F, et al. Relative extended haplotype homozygosity signals across breeds reveal dairy and beef specific signatures of selection. Genet Sel Evol. 2015;47:25.
Bosma M, Sparks LM, Hooiveld GJ, Jorgensen JA, Houten SM, Schrauwen P, et al. Overexpression of PLIN5 in skeletal muscle promotes oxidative gene expression and intramyocellular lipid content without compromising insulin sensitivity. Biochim Biophys Acta. 2013;1831:844–52.
Puig-Oliveras A, Ramayo-Caldas Y, Corominas J, Estellé J, Pérez-Montarelo D, Hudson NJ, et al. Differences in muscle composition transcriptome among pigs phenotypically extreme for fatty acid composition. PLoS One. 2014;9:e99720.
Tsang WY, Wang L, Chen Z, Sanchez I, Dynlacht BD. SCAPER, a novel cyclin A-interacting protein that regulates cell cycle progression. J Cell Biol. 2007;178:621–33.
Mashima R, Okuyama T. The role of lipoxygenasas in pathophysiology; new insights and future perspectives. Redox Biol. 2015;6:297–310.
Randhawa IAS, Khatkar MS, Thomson PC, Raadsma HW. Composite selection signals for complex traits exemplified through bovine stature using multibreed cohorts of European and African Bos taurus. G3 (Bethesda). 2015;5:1391–401.
Noyes H, Brass A, Obara I, Anderson S, Archibald AL, Bradley DG, et al. Genetic and expression analysis of cattle identifies candidate genes in pathways responding to Trypanosoma congolense infection. Proc Natl Acad Sci USA. 2011;108:9304–9.
Pareek CS, Michno J, Smoczynski R, Tyburski J, Golebiewski M, Piechocki K, et al. Identification of predicted genes expressed differentially in pituitary gland tissue of young growing bulls revealed by the cDNA-AFLP technique. Czech J Anim Sci. 2013;58:147–58.
Yao C, Spurlock DM, Armentano LE, Page CD Jr, VandeHaar MJ, Bickhart DM, et al. Random Forests approach for identifying additive and epistatic single nucleotide polymorphisms associated with residual feed intake in dairy cattle. J Dairy Sci. 2013;96:6716–29.
Flori L, Fritz S, Jaffrézic F, Boussaha M, Gut I, Heath S, et al. The genome response to artificial selection: a case study in dairy cattle. PLoS One. 2009;4:e6595.
Fellius M, Beerling ML, Buchanan DS, Theunissen B, Kollmees PA, Lenstra JA. On the history of cattle genetic resources. Diversity. 2014;6:705–50.
Authors’ contributions
LV conceived the study; CD, JP and JA contributed to the study design; AG, SM, EFM, JJC and LV developed and applied the methods; AG, CD, JP and LV drafted the manuscript; CD, JP, JA, AM and JAB contributed with the generation of experimental data. All authors read and approved the final manuscript.
Acknowledgements
The research leading to these results received funding from a Ministerio de Ciencia e Innovación AGL 2010-15903 Grant from the Spanish Government, from the European Union’s Seventh Framework Programme for research, technological development and demonstration under Grant Agreement No. 289592—Gene2Farm, from FEDER funds and from the regional government of Aragon (DGA) through its research group A51. The collaboration of Breed Societies in collecting samples and the support of FEAGAS are also acknowledged. The authors wish to thank JJ Arranz (Universidad de Leon) for facilitating the sampling of the Morucha population. J. J. Cañas-Álvarez acknowledges the financial support provided by COLCIENCIAS through the Francisco José de Caldas fellowship 497/2009 and A González-Rodríguez acknowledges the financial support from the Spanish Government BES-2011-045434.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
12711_2016_258_MOESM1_ESM.pdf
Additional file 1: Figure S1. Manhattan plots of the results along the autosomal genome obtained with the Tajima procedure.
12711_2016_258_MOESM2_ESM.pdf
Additional file 2: Figure S2. Manhattan plots of the results along the autosomal genome obtained with the Fu and Li procedure.
12711_2016_258_MOESM3_ESM.pdf
Additional file 3: Figure S3. Manhattan plots of the results along the autosomal genome obtained with the Fay and Wu procedure.
12711_2016_258_MOESM4_ESM.pdf
Additional file 4: Figure S4. Manhattan plots of the results along the autosomal genome obtained with the SelEstim procedure.
12711_2016_258_MOESM5_ESM.pdf
Additional file 5: Figure S5. Manhattan plots of the results along the autosomal genome obtained with the XP-CLR procedure.
12711_2016_258_MOESM7_ESM.pdf
Additional file 7: Figure S7. Manhattan plots of the results along the autosomal genome obtained with the iHS procedure.
12711_2016_258_MOESM8_ESM.pdf
Additional file 8: Figure S8. Manhattan plots of the results along the autosomal genome obtained with the nSL procedure.
12711_2016_258_MOESM9_ESM.pdf
Additional file 9: Figure S9. Manhattan plots of the results along the autosomal genome obtained with the F ST procedure.
12711_2016_258_MOESM10_ESM.pdf
Additional file 10: Figure S10. Manhattan plots of the results along the autosomal genome obtained with the XP-EHH procedure.
12711_2016_258_MOESM11_ESM.pdf
Additional file 11: Figure S11. Manhattan plots of the results along the autosomal genome obtained with the VarLD procedure.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
González-Rodríguez, A., Munilla, S., Mouresan, E.F. et al. On the performance of tests for the detection of signatures of selection: a case study with the Spanish autochthonous beef cattle populations. Genet Sel Evol 48, 81 (2016). https://doi.org/10.1186/s12711-016-0258-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12711-016-0258-1