
Abstract
Background
Selective breeding for genetic improvement is expected to leave distinctive selection signatures within genomes. The identification of selection signatures can help to elucidate the mechanisms of selection and accelerate genetic improvement. Fighting chickens have undergone extensive artificial selection, resulting in modifications to their morphology, physiology and behavior compared to wild species. Comparing the genomes of fighting chickens and wild species offers a unique opportunity for identifying signatures of artificial selection.
Results
We identified selection signals in 100-kb windows sliding in 10-kb steps by using two approaches: the pooled heterozygosity \(({\text{H}}_{\text{p}} )\) and the fixation index \((F_{\text{ST}} )\) between Xishuangbanna fighting chicken (YNLC) and Red Jungle Fowl. A total of 413 candidate genes were found to be putatively under selection in YNLC. These genes were related to traits such as growth, disease resistance, aggressive behavior and energy metabolism, as well as the morphogenesis and homeostasis of many tissues and organs.
Conclusions
This study reveals mechanisms and targets of artificial selection, which will contribute to improve our knowledge about the evolution of fighting chickens and facilitate future quantitative trait loci mapping.
Similar content being viewed by others

Background
Domesticated chickens have long been bred for entertainment and consumption [1]. Fighting chickens are a group of ancient breeds that have been bred for the purpose of cock fighting and have played an important role in the development of human culture [2]. The Xishuangbanna fighting chicken (YNLC) is a typical fighting chicken breed that has been subjected to strong artificial selection, which has led to remarkable phenotypic characteristics in morphology, physiology, and behavior. The YNLC represents an excellent model that can provide new insights into the influence of artificial selection on genetic variation and how this shapes phenotypic diversity.
Selection leads to specific changes in the patterns of variation among selected loci and in neutral loci linked to them. These genomic footprints of selection are known as selection signatures and can be used to identify loci that have been subjected to selection [3]. Various statistical approaches have been proposed for the detection of selection signatures [4–7]. The pooled heterozygosity \(( {\text{H}}_{\text{p}} )\) statistic is a variability estimator based on allele counts across sliding windows of adjacent loci and can be used to identify regions that deviate from the norm [8]. The fixation index \((F_{\text{ST}} )\) can be used to quantify the degree of genetic differentiation among populations based on differences in allele frequencies [9]. Both \({\text{H}}_{\text{p}}\) and \(F_{\text{ST}}\) statistics are useful for the detection of selection signatures [10]. In this study, we used two outlier approaches (\({\text{H}}_{\text{p}}\) and \(F_{\text{ST}}\)) to detect signatures of selection in YNLC and provide insights into the mechanisms of evolution of this specific breed.
Methods
Re-sequencing of chicken samples, mapping and SNP calling
We downloaded the genomic data for eight YNLC and six wild Red Jungle Fowl (RJF) from the EMBL-EBI database (see Additional file 1: Table S1). Details about the sequenced samples and method of sequencing are in [11, 12]. As mentioned in these two papers, individual DNA libraries with an insert size of 500 bp were constructed and sequenced by using the Illumina HiSeq 2000 platform. The samples were sequenced at a genome coverage of 11.1X to 36.6X (see Additional file 1: Table S1) which is appropriate for analysis of selective sweeps [13, 14]. All reads were preprocessed for quality control and filtered using our in-house script in Perl. Before aligning reads onto the reference genome, we performed the following quality checks [15]:
-
1.
If there were more than 10 % unidentified nucleotides (N) or 10-nucleotide adaptors (<10 % mismatch) in either of the paired reads, the reads were removed.
-
2.
If there were more than 50 % low-quality bases (Q ≤ 5) in either of the paired reads, the reads were removed.
-
3.
Duplicated reads were also removed, only paired-end reads were kept for subsequent analyses.
High-quality paired-end reads were mapped to the chicken reference genome sequence (ftp://ensembl.org/pub/release-67/fasta/gallus_gallus/dna/) using the BWA software [16] and the command ‘mem -t 4 -k 32 -M’. Duplicated reads were removed using the picard package [16]. After alignment, we performed single nucleotide polymorphism (SNP) calling on a population scale for the two groups (YNLC and RJF) using SAMtools [17]. The ‘mpileup’ command was used to identify SNPs with the parameters ‘-m 2 -F 0.002 -d 1000’. Putative functional effects of SNPs were annotated using the ANNOVAR package [18]. To exclude SNP calling errors caused by incorrect mapping, only high-quality SNPs (root-mean-square mapping quality ≥20, coverage depth ≥4 and ≤1000, distance between adjacent SNPs ≥5 bp and rate of missing data within each group <50 %) were retained for subsequent analyses.
Analysis of selection signatures
We used allele frequencies at variable sites to identify signatures of selection in 100-kb windows with a step size of 10 kb by using two approaches. For each window, we calculated \({\text{H}}_{\text{p}}\) and \(F_{\text{ST}} .\) At each detected SNP position, we counted the number of reads corresponding to the most and least frequently observed allele (nMAJ and nMIN, respectively) for each breed pool (i.e. all eight samples for YNLC and all six samples for RJF were combined, respectively). For each window, we calculated \({\text{H}}_{\text{p}}\) as follows [6]:
Subsequently, individual \({\text{H}}_{\text{p}}\) values were \({\text{Z}}\)-transformed as follows:
\(F_{\text{ST}}\) was calculated from the allele frequencies (not the allele counts) using the standard equation according to the principles of population genetics [19]:
where \({\text{P}}_{\text{i}} \_{\text{within}} = \left( {{\text{P}}_{\text{i}} \_{\text{population}}1 + {\text{P}}_{\text{i}} \_{\text{population}}2} \right)/2,\) and \({\text{P}}_{\text{i}} = 1 - {\text{fA}}^{2} - {\text{fT}}^{2} - {\text{fC}}^{2} - {\text{fG}}^{2}\) with \({\text{fN}}\) being the frequency of nucleotide \({\text{N }}\left( {{\text{i}}.{\text{e}}. {\text{A}}, {\text{T}}, {\text{C or G}}} \right),\,{\text{P}}_{\text{i}} \_{\text{total}}\) is the total \({\text{P}}_{\text{i}}\) for which allele frequencies in both populations are averages and \({\text{P}}_{\text{i}}\) is calculated as above.
The \(F_{\text{ST}}\) values were Z-transformed as follows:
where \({\upmu }F_{\text{ST}}\) is the mean \(F_{\text{ST}} ,\) and \(\sigma F_{\text{ST}}\) is the standard deviation of \(F_{\text{ST}}\) [20]. \({\text{H}}_{\text{p}}\) and \(F_{\text{ST}}\) were calculated by using our in-house script in Perl. The major challenge of such analyses is to exclude signals caused by demographic events and population structure. It is difficult to assign strict thresholds to distinguish selection and drift. We surveyed published literature and used an empirical procedure according to previous studies [21, 22]. Putatively selected regions were located in fully overlapping windows with an extremely low \({\text{ZH}}_{\text{p}}\) value (top 5 % level) and extremely high \({\text{Z}}F_{\text{ST}}\) values (top 5 % level).
Functional enrichment analysis
The genes putatively under selection were submitted to g:profiler (http://biit.cs.ut.ee/gprofiler/. Version: r1488_e83_eg30.) for enrichment analysis of the Gene Ontology (GO) and KEGG pathways. All chicken genes that are annotated in Ensembl were used as the background set. Benjamini–Hochberg FDR (false discovery rate) was used for correcting the P values. Only terms with a P value <0.05 were considered as significant and listed.
Results
Detection of SNPs
A total of 16.40 × 106 SNPs were identified from the genomes of 14 individuals, i.e. eight YNLC (13.15 × 106 SNPs) and six RJF individuals (13.87 × 106 SNPs) (see Additional file 2: Table S2). Most SNPs identified for the YNLC individuals were located in intergenic and intron regions (57.16 and 38.77 %, respectively); 1.36 % of these 13.15 × 106 SNPs were predicted to be within protein-coding regions and 0.40 % as amino acid altering mutations (non-synonymous and stop gain/loss); 1.30 % of the 13.15 × 106 SNPs were within 1-kb regions upstream or downstream of the transcription start or end sites, and thus may have a possible role in transcriptional regulation, with 510 SNPs of these residing within splice sites (Table 1; see Additional file 2: Table S2).
Genome-wide selective sweep signals
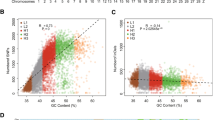
To detect selection signatures, we searched the genome of the YNLC chicken for regions with reduced \({\text{H}}_{\text{p}}\) and increased genetic distances to the RJF genome \((F_{\text{ST}} ).\) Putatively selected genes were located by extracting windows that simultaneously presented extremely low \({\text{ZH}}_{\text{p}}\) (top 5 % level, \({\text{ZH}}_{\text{p}} = - 1.75\)) and extremely high \({\text{Z}}F_{\text{ST}}\) (top 5 % level, \({\text{Z}}F_{\text{ST}} = 1.82\)). A total of 413 candidate genes (Fig. 1 , Additional file 3: Table S3, Additional file 4: Figure S1) were identified in the YNLC genome, which should harbor genes that underwent selection for fighting aptitude. We compared our results with those previously reported by Rubin et al. [6] and detected 91 overlapping genes between the two studies (see Additional file 3: Table S3). We searched for significantly overrepresented GO terms and KEGG pathways among the candidate genes that are specific to YNLC. The most enriched clusters were related to immunity, disease resistance, organ development, response to stimulus, and metabolic processes (see Additional file 5: Table S4, Additional file 6: Table S5).

Distribution of \({\text{ZH}}_{\text{p}}\) and \({\text{Z}}F_{\text{ST}}\) calculated for 100-kb windows sliding in 10-kb steps. Blue points identify Xishuangbanna game chicken (YNLC) genomic regions with both an extremely low \({\text{ZH}}_{\text{p}}\) value (top 5 % level) and an extremely high \({\text{Z}}F_{\text{ST}}\) value (top 5 % level)
Discussion
Four hundred and thirteen genes were discovered in our study, of which 91 overlapped with those reported in [6] and among these, we identified several notable domestication-related genes i.e.: IGF1 (insulin-like growth factor 1), which encodes a peptide that has a similar molecular structure to that of insulin and is a candidate gene for avian growth [6]; BCO2 (β-carotene oxygenase 2), which is associated with yellow skin in domestic chickens [23]; and NELL1 (NEL-like 1) which is assumed to be related to skeletal integrity in chickens [24]. Positive selection on these genes in the YNLC was expected since domestic chickens collectively share morphology and physiology shifts that accompanied domestication [25].
Many genes that were putatively under selection and identified in our study were not reported by Rubin et al. [6]. Functional enrichment analysis of genes that are specific to the YNLC breed revealed that many candidate genes are related to immunity and disease resistance (see Additional file 5: Table S4), which may reflect artificial selection for individuals with improved innate immunity and disease resistance.
Among the identified candidate genes, quite a few are involved in organ development (see Additional file 5: Table S4), e.g. CBFB (core-binding factor subunit beta) and GRHL3 (grainyhead-like 3), which are critical for growth and development of the craniofacial skeleton [26, 27]. These genes may explain why fighting chickens have a wider mandibular joint and frontal bone as compared to other breeds [28]. Many of the genes identified are related to limb development, i.e. Gli3 (transcriptional activator Gli3) and PTCH1 (patched 1), which are involved in the hedgehog (Hh) signal transduction pathway that controls the patterning, growth, morphogenesis and homeostasis of many tissues [29], such as digit patterning [30] and limb development [31]; EFNA5 (ephrin-A5), a GPI-anchored ephrin-Aligand that binds to the Eph receptors, is pivotal in cell migration in the avian forelimb [32]. Compared with other breeds, fighting chickens exhibit larger hindlimb and forelimb muscles, especially for triceps surae and biceps brachii [33], which may reflect adaptation to running and jumping that are essential traits in this breed. The triceps surae muscles assist in extending the foot joints, while the large triceps surae muscles allow fighting chickens to have a high level of jumping performance. The biceps brachii muscles facilitate strong flapping of the wings and act as powerful flexors of the elbow joint to support both jumping and hitting actions. In addition, fighting chickens have long legs, an extended hip joint, and a curved knee joint [33, 34], which indicate that they have adapted to running and upright posture.
Fighting chickens are bred specifically for cockfighting and fighting cocks possess congenital aggression towards all males of the same species. Several of the identified genes are related to aggressive behavior (Table 2). For example, the brain-derived neurotrophic factor (BDNF) gene (Fig. 2), a member of the nerve growth factor gene family, plays a major role in neuronal growth, proliferation, differentiation and neuronal survival [35]. A mutation in the human BDNF gene has been reported to be correlated with aggressive behavior in humans [36]. Furthermore, BDNF loss-of-function mice have been used as a model to study animal aggression [37]. Another gene neurotensin/neuromedin N precursor (NTS) encodes a common precursor for neurotensin (NT) and neuromedin N (NN). NT is involved in interactions with dopamine [38] and corticotropin-releasing factor (CRF) signaling [39], two neurotransmitter systems known to modulate aggressive behavior [40, 41]. Furthermore, NT mRNA levels were shown to be significantly reduced in high maternal aggression mice [42].

Example of the BDNF gene (arrow) with selection signals in Xishuangbanna game chicken (YNLC). \({\text{Z}}F_{\text{ST}}\) (blue) and \({\text{ZH}}_{\text{p}}\) (red)
Cockfighting is a very toilsome and furious form of exercise. The ability to sustain and effectively allocate fuel substrates for oxidative metabolism is critical for cockfighting. Energy metabolism-related genes were found to be under selection in YNLC (see Additional file 5: Table S4). The RICTOR (RPTOR independent companion of MTOR complex 2) gene encodes an essential subunit of the target of the rapamycin (mTOR) complex (mTORC) 2. In fat cells, RICTOR/mTORC2 plays an important role in whole-body energy homeostasis [44]. The SDHB (succinate dehydrogenase (SDH) subunit B) gene encodes a crucial metabolic enzyme that is involved in the respiratory chain and Krebs cycle [45]. Positive selection of these genes may represent adaptation of the energy metabolism in fighting chickens.
Conclusions
In this work, we used two distinct methods to detect selection signatures across the genomes of YNLC and RJF chicken. Our analyses identified genes under positive selection in YNLC, which included genes related to aggressive behavior, immunity, energy metabolism and tissue and organ development. Our data will help improve our understanding of the mechanisms and identify the targets of artificial selection in fighting chickens and facilitate future quantitative trait loci (QTL) mapping.
References
Komiyama T, Iwama H, Osada N, Nakamura Y, Kobayashi H, Tateno Y, et al. Dopamine receptor genes and evolutionary differentiation in the domestication of fighting cocks and long-crowing chickens. PLoS One. 2014;9:e101778.
Zhu W, Li H, Song W, Xu W, Shu J, Han W, et al. The genetic diversity and origin of Chinese game chicken. Avian Biol Res. 2009;2:197–202.
Kreitman M. Methods to detect selection in populations with applications to the human. Annu Rev Genom Hum Genet. 2000;1:539–59.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72.
Rubin CJ, Zody MC, Eriksson J, Meadows JRS, Sherwood E, Webster MT, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–91.
Wang GD, Fan RX, Zhai W, Liu F, Wang L, Zhong L, et al. Genetic convergence in the adaptation of dogs and humans to the high-altitude environment of the Tibetan plateau. Genome Biol Evol. 2014;6:2122–8.
Qanbari S, Strom TM, Haberer G, Weigend S, Gheyas AA, Turner F, et al. A high resolution genome-wide scan for significant selective sweeps: an application to pooled sequence data in laying chickens. PLoS One. 2012;7:e49525.
Lewontin RC, Krakauer J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973;74:175–95.
Axelsson E, Ratnakumar A, Arendt ML, Maqbool K, Webster MT, Perloski M, et al. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495:360–4.
Yan YY, Yi GQ, Sun CQ, Qu LJ, Yang N. Genome-wide characterization of insertion and deletion variation in chicken using next generation sequencing. PLoS One. 2014;9:e104652.
Wang MS, Li Y, Peng MS, Zhong L, Wang ZJ, Li QY, et al. Genomic analyses reveal potential independent adaptation to high altitude in Tibetan chickens. Mol Biol Evol. 2015;32:1880–9.
Qiu Q, Wang LZ, Wang K, Yang YZ, Ma T, Wang ZF, et al. Yak whole-genome resequencing reveals domestication signatures and prehistoric population expansions. Nat Commun. 2015;6:10283.
Chen C, Liu ZG, Pan Q, Chen X, Wang HH, Guo HK, et al. Genomic analyses reveal demographic history and temperate adaptation of the newly discovered honey bee subspecies Apis mellifera sinisxinyuan n. ssp. Mol Biol Evol. 2016;33:1337–48.
Li MZ, Tian SL, Yeung CKL, Meng XH, Tang QZ, Niu LL, et al. Whole-genome sequencing of Berkshire (European native pig) provides insights into its origin and domestication. Sci Rep. 2014;4:4678.
Li H, Durbin R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Wang K, Li MY, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Hartl DL, Clark AG. Principles of population genetics. 4th ed. Sunderland: Sinauer Associates; 2007.
Karlsson EK, Baranowska I, Wade CM, Salmon Hillbertz NHC, Zody MC, Anderson N, et al. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat Genet. 2007;39:1321–8.
Li M, Tian S, Jin L, Zhou G, Li Y, Zhang Y, et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat Genet. 2013;45:1431–8.
Qu Y, Tian SL, Han NJ, Zhao HW, Gao B, Fu J, et al. Genetic responses to seasonal variation in altitudinal stress: whole-genome resequencing of great tit in eastern Himalayas. Sci Rep. 2015;5:14256.
Eriksson J, Larson G, Gunnarsson U, Bed’hom B, Tixier-Boichard M, Stromstedt L, et al. Identification of the yellow skin gene reveals a hybrid origin of the domestic chicken. PLoS Genet. 2008;4:e1000010.
Elferink MG, Megens HJ, Vereijken A, Hu X, Crooijmans RPMA, Groenen MAM. Signatures of selection in the genomes of commercial and non-commercial chicken breeds. PLoS One. 2012;7:e32720.
Wang MS, Zhang RW, Su LY, Li Y, Peng MS, Liu HQ, et al. Positive selection rather than relaxation of functional constraint drives the evolution of vision during chicken domestication. Cell Res. 2016;26:556–73.
Kundu M, Javed A, Jeon JP, Horner A, Shum L, Eckhaus M, et al. Cbf beta interacts with Runx2 and has a critical role in bone development. Nat Genet. 2002;32:639–44.
Dworkin S, Simkin J, Darido C, Partridge DD, Georgy SR, Caddy J, et al. Grainyhead-like 3 regulation of endothelin-1 in the pharyngeal endoderm is critical for growth and development of the craniofacial skeleton. Mech Dev. 2014;133:77–90.
Kudo K, Tsunekawa N, Ogawa H, Endo H. Comparative functional morphology of skulls among Japanese breeds of domestic fowls. J Poult Sci. 2016;53:43–50.
Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–87.
Litingtung Y, Dahn RD, Li YN, Fallon JF, Chiang C. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature. 2002;418:979–83.
Büscher D, Bosse B, Heymer J, Rüther U. Evidence for genetic control of Sonic hedgehog by Gli3 in mouse limb development. Mech Dev. 1997;62:175–82.
Swartz ME, Eberhart J, Pasquale EB, Krull CE. EphA4/ephrin-A5 interactions in muscle precursor cell migration in the avian forelimb. Development. 2001;128:4669–80.
Endo H, Mori K, Hosojima M, Mekwichai W, Ogawa H, Tsunekawa N, et al. Functional-morphological characteristics in the musculoskeletal system of standing-type cocks Including some game breeds. Jpn J Zoo Wildl Med. 2012;17:131–8.
Vasinopas L, Meckvichai W. The difference of morphological characteristics between fighting and meat type Thai native chickens. Khon Kaen Agr J. 2015;43:70–3.
Zhou J, Bradford HF, Stern GM. The stimulatory effect of brain-derived neurotrophic factor on dopaminergic phenotype expression of embryonic rat cortical neurons in vitro. Brain Res Dev Brain Res. 1994;81:318–24.
Spalletta G, Morris DW, Angelucci F, Rubino IA, Spoletini I, Bria P, et al. BDNF Val66Met polymorphism is associated with aggressive behavior in schizophrenia. Eur Psychiatry. 2010;25:311–3.
Ito W, Chehab M, Thakur S, Li J, Morozov A. BDNF-restricted knockout mice as an animal model for aggression. Genes Brain Behav. 2011;10:365–74.
Cáceda R, Kinkead B, Nemeroff CB. Neurotensin: role in psychiatric and neurological diseases. Peptides. 2006;27:2385–404.
Rowe W, Viau V, Meaney MJ, Quirion R. Stimulation of CRH-mediated ACTH secretion by central administration of neurotensin: evidence for the participation of the paraventricular nucleus. J Neuroendocrinol. 1995;7:109–17.
Gammie SC, Negron A, Newman SM, Rhodes JS. Corticotropin-releasing factor inhibits maternal aggression in mice. Behav Neurosci. 2004;118:805–14.
Ricci LA, Schwartzer JJ, Melloni RH Jr. Alterations in the anterior hypothalamic dopamine system in aggressive adolescent AAS-treated hamsters. Horm Behav. 2009;55:348–55.
Gammie SC, Auger AP, Jessen HM, Vanzo RJ, Awad TA, Stevenson SA. Altered gene expression in mice selected for high maternal aggression. Genes Brain Behav. 2007;6:432–43.
Chamero P, Katsoulidou V, Hendrix P, Bufe B, Roberts R, Matsunami H, et al. G protein Gαo is essential for vomeronasal function and aggressive behavior in mice. Proc Natl Acad Sci USA. 2011;108:12898–903.
Kumar A, Lawrence JC, Jung DY, Ko HJ, Keller SR, Kim JK, et al. Fat cell–specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes. 2010;59:1397–406.
Ackrell BAC. Cytopathies involving mitochondrial complex II. Mol Asp Med. 2002;23:369–84.
Authors’ contributions
XG, QF, CM, BZ and YW analysed data and performed bioinformatics. XG and RJ drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by a grant from the National Natural Science Foundation of China (No. 31572395).
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
12711_2016_239_MOESM1_ESM.xls
Additional file 1: Table S1. Summary of the downloaded chicken genome re-sequencing data. Description: The table provides sequencing depth and accession numbers from the downloaded chicken genome.
12711_2016_239_MOESM2_ESM.xls
Additional file 2: Table S2. Summary and annotation of SNPs in Xishuangbanna fighting chicken (YNLC) and Red Jungle Fowl (RJF).
12711_2016_239_MOESM3_ESM.xls
Additional file 3: Table S3. Putatively selected genes (top 5 % level of \({\text{ZH}}_{\text{p}}\) and \({\text{Z}}F_{\text{ST}}\) values) in the Xishuangbanna fighting chicken (YNLC).
12711_2016_239_MOESM4_ESM.docx
Additional file 4: Figure S1. Genome-wide distribution of \({\text{ZH}}_{\text{p}}\) and \({\text{Z}}F_{\text{ST}}\) along chromosomes. This figure presents the putatively selected regions on YNLC chromosomes.
12711_2016_239_MOESM5_ESM.xls
Additional file 5: Table S4. GO analysis for all genes under selection in YNLC. The table provides the significantly overrepresented gene ontology (GO) terms among putatively selected genes in YNLC.
12711_2016_239_MOESM6_ESM.xls
Additional file 6: Table S5. KEGG analysis for all genes under selection in YNLC. The table provides the significantly enriched KEGG pathways among putatively selected genes in YNLC.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Guo, X., Fang, Q., Ma, C. et al. Whole-genome resequencing of Xishuangbanna fighting chicken to identify signatures of selection. Genet Sel Evol 48, 62 (2016). https://doi.org/10.1186/s12711-016-0239-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12711-016-0239-4