
Abstract
Background
Rice (Oryza sativa) is the main food for half of the world’s population, and is considered the model for molecular biology studies of monocotyledon species. Although the rice genome was completely sequenced about 15 years ago, the function of most rice genes is still unknown.
Results
In this study, we developed a vector toolkit that contains 42 vectors for transient expression studies in rice protoplasts and stable expression analysis in transgenic rice. These vectors have been successfully used to study protein subcellular localization, protein-protein interaction, gene overexpression, and the CRISPR/Cas9-mediated gene editing. A novel feature of these vectors is that they contain a universal multiple cloning site, which enables more than 99% of the rice coding sequences to be conveniently transferred between vectors.
Conclusions
The versatile vectors represent a highly efficient and high-throughput toolkit for functional analysis of rice genes.
Similar content being viewed by others


Background
Rice (Oryza sativa) is one of the most important food crops, as well as a model plant for studies of agronomic traits. The RICE2020 project was proposed by rice researchers in 2008 to characterize the functions of all rice genes (~ 32,000) by the year 2020 (Zhang et al. 2008). In the past 10 years, significant progress has been made with the availability of several gene-indexed rice mutant resources (Jung et al. 2008; Krishnan et al. 2009; Wang et al. 2013b), the development of large-scale omics profiles (Han et al. 2017; Lakshmanan et al. 2016; Wang et al. 2015), as well as advances in forward and reverse genetic technologies (Jiang et al. 2013; Schneeberger and Weigel 2011). To date, however, the functions of only a small proportion of rice genes have been reported. Elucidating the biological functions of the remaining genes is a daunting task for the rice research community (Li et al. 2018).
Vectors for gene ectopic expression, silencing, knock-out, promoter activity assay, protein subcellular localization, protein-protein interaction, and enzyme activity analysis are essential tools for gene characterization (Prelich 2012). The classic binary pCAMBIA vectors have been widely used for rice transformation in the past 20 years (Komori et al. 2007). Many specialized vectors for particular purposes have been derived from the pCAMBIA vectors, including the protein subcellular localization vector pGDG/pGDR (Goodin et al. 2002), the transposon Ac-Ds tagging vector pSQ5 (Qu et al. 2008), and the gene editing vectors such as the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system (Jiang et al. 2013; Xing et al. 2014; Zhou et al. 2014). Other vectors, such as pGreenII cloning vectors and pSAT vectors, have also been used for localization and for transient and stable expression of genes in plants (Hellens et al. 2005; Tzfira et al. 2005). In addition to the traditional restriction enzyme cloning strategy, the T-A cloning system was developed for gene cloning when appropriate restriction sites are lacking in the vector (Chen et al. 2009; Wang et al. 2013a). The T-A cloning system, however, is inconvenient for shuttling DNA fragments from one vector to another. At the same time, the cloning efficiency of the T-A cloning system is greatly reduced when the insert size is increased (Wang et al. 2013a). The Gateway system has also been widely applied in plants for over 15 years (Curtis and Grossniklaus 2003; Earley et al. 2006; Nakagawa et al. 2007). For example, the widely used rice RNA interference (RNAi) vector pANDA is based on the Gateway LR recombination (Miki and Shimamoto 2004; Wang et al. 2016). However, Gateway cloning requires an entry vector that matches with a full set of destination vectors, and the clonase used for the recombination reaction is expensive.
Because of the limitations of the TA and Gateway cloning systems, most laboratories still use classic restriction enzyme cloning. Unfortunately, multiple cloning sites (MCS) in the vectors that are suitable for the cloning of most rice genes are unavailable. To facilitate the transfer of rice fragments between vectors, we analyzed the restriction enzyme recognition sites in the rice coding sequences (CDSs) based on the MSU RGAP database Version7 (http://rice.plantbiology.msu.edu/), and we then synthesized a powerful universal MCS suitable for cloning of more than 99% of rice CDSs. At the same time, we optimized the individual components to generate small vectors with high efficiency. A total of 42 vectors were constructed for different applications, including gene transient expression, localization, Co-IP (co-immunoprecipitation), BiFC (bimolecular fluorescence complementation); as well as rice transgenic stable expression for gene editing, tissue or cell localization detection, promoter activity and gene complementation. We confirmed the utilities of these vectors with several assays in rice protoplasts, and stably transformed lines. Because the new vector toolkit is highly efficient, cost-effective, and easy to use, it will accelerate functional studies of genes in rice and other monocots.
Results
Construction of Vectors for Gene Functional Analysis
To construct an efficient vector system (Fig. 1a), we analyzed the efficiency of different expression elements in rice. Compared to the widely used cauliflower mosaic virus 35S promoter in dicots, the maize (Zea mays) ubiquitin-1 (Ubi) promoter is more efficient in monocots (Chen et al. 2009). However, the Ubi promoter contains many frequently used restriction sites, such as BglII, EcoRI, SalI, XbaI, and XhoI (Additional file 1: Figure S1). We therefore systematically analyzed the restriction map of rice CDSs and selected seven abundant restriction sites that are not present in the Ubi promoter; at least two of the sites are absent in 99.55% of the rice CDSs (Table 1). As a consequence, these restriction sites can be used to clone 99.55% of the rice genes into these vectors using the classic double digestion-ligation strategy. We then synthesized these seven restriction sites in the order of BamHI-SmaI-SacI-KpnI-HindIII-SpeI-NotI to form a universal MCS (Fig. 1b). To reduce the vector size, we used PCR to amplify the precise sequence of each element, and we then assembled the high copy number pUC replicon, kanamycin resistance gene, Ubi promoter, TagRFP, MCS, and Nos terminator (NosT). This generated the first vector, named pRTVnRFP, which is an abbreviation of the plasmid for rice transient overexpression with the N-terminal TagRFP tag. This vector contains only 4837 bp, which is suitable for gene expression and subcellular localization analysis in rice protoplasts (Fig. 1d).

Schematic diagrams of the vectors. a Type and naming system of the 42 vectors. b The sequence and restriction enzymes in the multiple cloning site (MCS). c-e Vectors for rice transient overexpression. c pRTVs vectors with or without multiple N-/C- terminal HA tags or cMyc tags. d pRTVs vectors with N-/C- terminal fusion EGFP, mVenus, or TagRFP for subcellular localization in protoplasts. e Vectors for BiFC assays. mVN(1–155) and mVC(156–238) are the N and C terminal of mVenus, respectively. f-j Binary vectors for Agrobacterium-mediated transformation experiments. f and g, Vectors without the Ubi promoter for rice stable expression with hygromycin or G418 selection marker, respectively. h and i, Vectors for rice stable overexpression with hygromycin or G418 selection marker, respectively. j CRISPR/Cas9 vectors with hygromycin or G418 selection marker. hptII, the hygromycin selection marker gene; nptII, the G418 or kanamycin selection marker gene; LB, T-DNA left border; RB, T-DNA right border; NLS, nuclear localization signal; intron, second intron (IV2) of the potato gene ST-LS1
We subsequently constructed a series of transient expression vectors, including the overexpression vectors with or without the N-/C-terminal epitope tag HA or cMyc (Fig. 1c), the fluorescent fusion expression vectors with the EGFP, mVenus or TagRFP tag (Fig. 1d), as well as the BiFC vectors with the mVenus (mVN(1–155) and mVC(156–238)) halves (Fig. 1e). At the same time, we generated two vectors without the Ubi promoter for regulable expression (e.g., for application in promoter activity analysis and native expression and inducible expression assays) with or without the C-terminal mVenus (Additional file 1: Table S1).
Next, we constructed a set of binary transformation vectors using the transient expression vectors as backbones. The following were added to the transient expression vectors to make 21 transformation vectors: T-DNA border LB and RB, selection marker gene HptII (for hygromycin) or NptII (for G418/kanamycin) under the control of the 35S promoter, and the sequence ori-REP-STA from pVS1 for plasmid stabilization and replication in A. tumefaciens. Among the 21 transformation vectors, eight (four pRHEs and four pRGEs) did not contain the Ubi promoter and were suitable for gene regulable expression (Fig. 1f, g), and 13 vectors (8 pRHVs and 5 pRGVs) for gene overexpression (Fig. 1h, i, Additional file 1: Table S1). These vectors were classified and named according to their selection markers (with H standing for hygromycin and G standing for G418/kanamycin) and promoters (with E standing for regulable expression without the Ubi promoter, and V standing for overexpression with the Ubi promoter). Two CRISPR vectors, pRHCas9 and pRGCas9, were constructed for Cas9-mediated rice genome editing (Fig. 1j). The genome editing tool suites contain two additional compatible entry vectors, pEntry A and pEntry B, for packing the sgRNA cassette (Fig. 1j). These binary vectors can be used for almost all rice transgenic experiments.
Applications for Protein Localization in Rice Protoplasts
To investigate the localization or co-localization of proteins in rice protoplasts, we constructed three kinds of N-/C-terminal fluorescent protein tags driven by the Ubi promoter, and we generated six overexpression vectors (pRTVnGFP, pRTVcGFP, pRTVnYFP, pRTVcYFP, pRTVnRFP, and pRTVcRFP) (Fig. 1d), as well as one vector (pRTEcYFP) that did not have the Ubi promoter but did have one of the brightest fluorescent tags, mVenus (Shaner et al. 2007) (Additional file 1: Table S1).
To confirm the utility of these vectors in protein subcellular localization, we tested pRTVnGFP, pRTVnYFP, and pRTVnRFP with the representative rice proteins OsRac1, WRKY45, and SPIN6 in localization assays. OsRac1, a member of Rac/Rop small GTPases and a key member of the rice defensome, was previously shown to localize in the plasma membrane (Akamatsu et al. 2013; Ono et al. 2001). The results showed that strong yellow fluorescence signals occurred on the protoplast membrane when transfected in rice protoplasts with pRTVnYFP-OsRac1 (Fig. 2a, middle column). Similarly, rice protoplasts transfected with pRTVnRFP-WRKY45, which contained the transcriptional factor WRKY45, a positive regulator of rice defense signaling (Shimono et al. 2007), showed specific red fluorescence signals in the nuclear region (Fig. 2a, right column). In contrast, the empty vector pRTVnGFP produced whole-cell localized GFP signals (Fig. 2a, left column).

Applications for protein localization in rice protoplasts. a Subcellular localization assay. Plasmids pRTVnGFP, pRTVnYFP-OsRac1, and pRTVnRFP-WRKY45 were used for subcellular localization assays. Bar = 20 μm. b Co-localization assay. Plasmids pRTVnGFP-OsRac1, pRTVnRFP-SPIN6, and the corresponding empty vectors were used for co-localization assays. Bar = 20 μm
These vectors were also used for the co-localization assay of SPIN6 and OsRac1. SPIN6 is a Rho GTPase-activating protein (RhoGAP) involved in inactivating OsRac1 and suppressing rice innate immunity (Liu et al. 2015). In the co-localization assay, we co-transfected rice protoplasts with plasmids pRTVnRFP-SPIN6 and pRTVnGFP-OsRac1. Compared with the EGFP and TagRFP controls whose signals were distributed throughout the cell, the red fluorescence signals of TagRFP-SPIN6 were mainly detected in the cytoplasm and plasma membrane, while the green fluorescence signals of EGFP-OsRac1 were detected in the plasma membrane, the signals of the two proteins merged and formed a yellow signal on the membrane (Fig. 2b). These experiments demonstrated that our vectors are suitable for protein localization and co-localization assays in rice protoplasts.
Applications in Protein Transient Expression and Protein-Protein Interaction Assays
Analysis of protein expression and protein-protein interaction is essential for understanding the molecular and biochemical functions of candidate genes (Hakes et al. 2008). Co-IP and BiFC assays are widely used for analysis of protein-protein interactions in vivo (Berggard et al. 2007; Liu et al. 2017; Wang et al. 2016). In this study, we constructed four vectors with multiple HA or cMyc epitope tags at their N-/C-terminals under the control of the Ubi promoter (pRTVnHA, pRTVnMyc, pRTVcHA, and pRTVcMyc) to facilitate immunoblotting, affinity purification, and Co-IP assay (Fig. 1c). To confirm the efficacy of our vectors for assessing protein expression and protein-protein interactions, we first carried out a Co-IP assay in rice protoplasts using these vectors with SPIN6 and OsRac1. The DNA fragments of the two genes were cloned into the pRTVnHA and pRTVnMyc vectors, respectively. We co-transfected rice protoplasts with the two plasmids and isolated total protein for western blot analysis. We detected the ~ 130-kDa HA-SPIN6 and ~ 40-kDa cMyc-OsRac1 proteins, as well as the control cMyc-cLUC (The C terminal 417–568 of luciferase) in the input (Fig. 3a). Moreover, the Co-IP result showed SPIN6 specifically binds to OsRac1 but not to cLUC (Fig. 3a), confirming the in vivo interaction between SPIN6 and OsRac1 as previously reported (Liu et al. 2015).

Applications in protein transient expression and protein-protein interactions. a Application for protein transient expression and Co-IP assay. Plasmids pRTVnHA-SPIN6, pRTVnMyc-OsRac1, and control pRTVnMyc-cLUC were used to express HA- or cMyc-tagged protein. cLUC(417–568), C terminal of luciferase. b Application for BiFC assay. BiFC plasmids pRTVnVN-SPIN6, pRTVnVC-OsRac1, and control pRTVcVC-LUC were used for the interaction assay between SPIN6 and OsRac1. LUC, luciferase. Bar = 20 μm
For BiFC assays, the two mutation sites V150A and I152L in the N-terminal of mVenus (mVN(1–155)) were introduced into our BiFC system to increase the signal-to-noise ratio (Kodama and Hu 2010; Nakagawa et al. 2011). Since the split-gene fragments in both plasmids are required to be expressed in the same cell in BiFC assay, we added the marker genes ECFP and mCherry into the VN (pRTVnVN and pRTVcVN) and VC (pRTVnVC and pRTVcVC) vectors, respectively, so that plasmid transfection and gene expression efficiency in the transfected cells can be assessed. We also added a fusion tag HA or cMyc to facilitate protein immunoblotting detection in the BiFC assay (Fig. 1e). We used these vectors to analyze the SPIN6 and OsRac1 interaction. SPIN6 and OsRac1 DNA fragments were fused with the split mVenus N-terminal and C-terminal in the vectors of pRTVnVN and pRTVnVC, respectively. After co-transfection, we detected clear yellow fluorescence signals on the plasma membrane of the transfected protoplasts, indicating the formation of reconstituted mVenus through the SPIN6-OsRac1 interaction (Fig. 3b). The ECFP and mCherry fluorescence in the control, in contrast, indicated successful transfection and expression of the two plasmids but did not indicate reconstitution of mVenus, which demonstrated the specific interaction between SPIN6 and OsRac1 (Fig. 3b). Together, these results suggested that our vectors are adequate for protein expression and protein-protein interaction assays in rice protoplasts.
Applications for Generating Stable Transgenic Rice
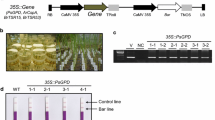
We generated a series of vectors for stable expression in rice with hygromycin or G418/kanamycin selection markers. Epitope tags HA and cMyc, as well as fluorescent protein tags EGFP, TagRFP, and mVneus can be used for protein detection in transgenic plants. To check whether these vectors work well in rice, we selected the empty vector pRHVnGFP for Agrobacterium-mediated rice transformation. We obtained more than 20 transgenic plants in the Nipponbare (NPB) background. Confocal microscopy revealed strong, specific, green fluorescence signals in the epidermis, stomata guard cells, and mesophyll cells of the hygromycin-positive transgenic line #1 (Fig. 4a). We then confirmed the EGFP protein accumulation by western blot. We checked three independent hygromycin-positive lines (#1, #3, and #4). A strong, specific GFP band was detected in the three lines by western blot analysis compared to the wild type, NPB (Fig. 4b). The results indicated that these binary vectors worked properly in rice, and that they can be used in rice transgenic experiments, including gene overexpression assays, tissue or cell localization assays, promoter activity assays, and complementation assays.

Application of overexpression vectors in generating GFP transgenic rice. a GFP signals in transgenic line #1 tissues. The arrow indicates the stoma. Vector pRHVnGFP was used for generating transgenic rice plants by Agrobacterium-mediated transformation. Bar = 10 μm. b Western blot detection of the GFP protein in transgenic lines. Three independent transgenic lines and the wild type were used for western blot detection. The HSP (heat shock protein) indicates the loading amount of each sample
Applications of CRISPR/Cas9 Vectors for Generating Rice Mutants
CRISPR/Cas9-mediated gene editing has been wildly used for functional studies of rice genes (Belhaj et al. 2015; Bi and Yang 2017; Ma et al. 2016). Most of the CRISPR/Cas9 vectors developed for rice are based on the pCAMBIA1300 vector (Ma et al. 2015; Piatek et al. 2015; Zhou et al. 2014). It is difficult to construct target sites in these CRISPR/Cas9 vectors through the Gateway cloning or the complicated Golden Gate Assembly strategy (Ma et al. 2015; Xie et al. 2015). In this study, we built a concise and accessible CRISPR/Cas9 system based on the pRHV and pRGV vectors. The two core parts, rice codon-optimized SpCas9 and sgRNA cassette, were cloned from a highly efficient genome editing system in a previous report (Zhou et al. 2014). To eliminate the toxicity of Cas9 in Agrobacteria, the second intron (IV2) of the ST-LS1 gene (Thole et al. 2007) from potato was amplified and inserted into the Cas9 HNH nuclease domain. We confirmed the expression of the Cas9-intron in rice protoplasts by western blot analysis, and we detected the same size protein band with Cas9, indicating that the IV2 intron was correctly spliced in rice cells (Fig. 5a). The construction of a new CRISPR/Cas9 vector is simple because only a BsaI digestion is needed to insert the 20 bp gene-specific target fragments (target sequences should followed by a PAM sequence NGG, Fig. 5b) to pEntry A or pEntry B (Fig. 5c, d). Four pairs of isocaudamers were used for assembling the U6P-sgRNA cassette in the pEntry vectors (Additional file 1: Figure S2, Additional file 1: Figure S3). The two entry vectors should be used in turns when targeting multiple sites. Two strategies, one-step ligation and step-by-step ligation, could be used to assemble single or multiple targets (Fig. 5e, f). By using an one-step ligation strategy, we easily ligated three targets for SPIN6 into pRHCas9.

Diagram of how CRISPR/Cas9 vectors were constructed and used to edit the rice IPA1 gene. a Western blot analysis of the Cas9-intron expression in rice protoplasts. HSP indicates the loading amount of each sample. b Target site selection for candidate genes in the rice genome. A 20-bp specific sequence followed by the PAM “NGG” structure is required. c and d Target cloning to the entry vectors. Synthesis of the primer pairs of the 20-bp specific target with the 4-bp adapters, and ligation with the BsaI linearized pEntry A or pEntry B vector. e One-step ligation and f step-by-step ligation of multiple targets to pRHCas9/pRGCas9. Four pairs of isocaudamers, PstI(E1)-NsiI(E1’), XbaI(E2)-SpeI(E2’), BamHI(E3)-BglI(E3’), and SalI(E4)-XholI(E4’) are marked. The sgRNA cassettes with U6P1 and U6P2 in pEntry A and B should be used in turn. g Representative sequencing chromatogram of the CRISPR-IPA1 transgenic lines. Line #7, wild-type genotype; line #5, mutant genotype. h Representative gene editing results in the CRISPR-IPA1 transgenic lines
To test our CRISPR/Cas9 vectors for generating rice mutants, we tried to edit the rice IPA1 (Ideal Plant Architecture 1) gene, which is involved in the regulation of plant architecture and grain yield (Jiao et al. 2010). A previously reported CRIPSPR/Cas9 target sequence with complementary NGG PAM structure in IPA1 gene (Li et al. 2016) was cloned into the pEntry A vector and then sub-cloned into the binary vector pRHCas9. Over 30 independent transgenic plants were generated through Agrobacterium-mediated rice transformation. We then randomly selected 20 plants for genotyping by PCR and sequencing. Among them, 16 (80%) showed gene editing at the target site of the IPA1 gene (Fig. 5g), which was much higher than the editing rate (27.5%) of IPA1 in the previous report (Li et al. 2016). Additionally, most of the edits appeared in a bi-allelic manner, and the most abundant editing pattern was one base pair insertion at 3 bp downstream of the complementary NGG PAM site (Fig. 5h). This result demonstrated that our CRISPR/Cas9 system has a high gene editing efficiency and is a useful tool for generating rice mutants.
Discussion and Conclusions
To establish a highly efficient rice cell and plant experimental platform, we constructed a series of 41 vectors in this study. We added different modules from other systems, deleted most redundant sequences, and mutated the NotI recognition site between the connection region of pVS1 REP and STA from pCAMBIA1300. Furthermore, we tested these vectors with several genes in rice protoplasts and transgenic plants and demonstrated the usefulness of the system for the functional analysis of rice genes.
Except for the CRISPR/Cas9 suite, all of the vectors share the same MCS (Fig. 1b). The synthesized MCS with seven restriction sites is sufficient for cloning 99.55% of the rice genes. In other words, most of the rice genes can be easily cloned into our vectors through a double digestion-ligation strategy. In addition, the universal MCS is compatible with other commercial vectors, including prokaryotic expression with the MBP and GST fusion protein tag vectors pMalc2x and pGEX6p-1, and with the yeast-two-hybrid system pPC86 and pDBLeu vectors. The universal MCS is also helpful for fragment shuttling between vectors if necessary. The small size and high copy number of the vectors are ideal for efficient cloning, transformation, and plasmid preparation. For example, the pRTV vector for transient overexpression is only ~ 4.3 kb (Additional file 1: Figure S2). The plant overexpression vector pRHV is only 8.6 kb (Additional file 1: Figure S2), which is about 30% smaller than the 12-kb T-A cloning-based binary vector pCXUN (Chen et al. 2009). The remaining PstI, EcoRV, NotI, AscI, and PmlI recognition sites between modules may also be used for adding other components to the vectors (Additional file 1: Figure S2).
Functional analysis of candidate genes requires their transient expression in protoplasts and their stable expression in transgenic plants. Our vectors contain several epitope tags or fluorescent tags for protein expression analysis, including HA, cMyc, EGFP, mVenus, and TagRFP (Fig. 1 and Additional file 1: Table S1). BiFC assay is a popular method for determining protein-protein interactions and the interaction location in the cell. Researchers have improved the BiFC signal-to-noise ratio by selecting different fluorescent proteins, by split sites, and by creating point mutations (Kodama and Hu 2012). To increase BiFC reliability in rice protoplasts, we introduced the mutations V150A and I152L in the fluorescent protein Venus (Kodama and Hu 2010; Nakagawa et al. 2011) into the pRTVnVN and pRTVcVN vectors. The independently expressed ECFP and mCherry selection marker genes are used as controls to monitor the efficiency of construct transfection and gene expression in the treated cells in the BiFC system. The results from our subcellular localization, Co-IP, and BiFC assays are consistent with (or even better than) previous results (Akamatsu et al. 2013; Liu et al. 2015; Ono et al. 2001; Shimono et al. 2007) (Figs. 2, 3).
Hygromycin resistance is an effective selection marker in regenerating rice plants. An alternative selection marker such as G418/kanamycin resistance is required, however, when introducing another transgene or the CRISPR/Cas9 cassette into the existing hygromycin-resistant transgenic rice plants (Chakraborty et al. 2016; Park et al. 2016). In the 23 vectors suitable for rice transgenic experiments, 13 contain the HptII gene and 10 contain the NptII gene (Additional file 1: Table S1, Figure S2). To simplify target assembling, we have established the CRISPR/Cas9 system with four pairs of isocaudamers, and we have successfully used them to edit the rice IPA1 gene, with an 80% gene editing efficiency. Therefore, our CRISPR/Cas9 vectors can be used to efficiently generate rice mutants.
Finally, some of the vectors developed in this research have been successfully used in our laboratory for several rice projects in the past few years. For example, we used the pRTVnHA vector to express HA-SPIN6 in protoplasts and to thereby suppress OsRac1 activity (Liu et al. 2015), and we used the pRTVnMyc vector to express cMyc-OsNPR1 in protoplasts and to thereby elucidate the OsCUL3a-OsNPR1 degradation mechanism (Liu et al. 2017). In conclusion, we have established a versatile vector toolkit for high efficiency and throughput gene functional analysis in rice. The vectors will be freely available to the community.
Methods
Plant Materials
Rice (Oryza sativa L. ssp. japonica) cultivar Nipponbare was used for protoplast isolation and transformation experiments. Rice plants were grown at 26–28 °C in greenhouses or growth chambers. For protoplast isolation, rice plants were kept in the dark for about 10 days.
Plasmid Vector Construction
The construction of the vectors was based on PCR amplification, restriction enzyme (NEB and TaKaRa) digestion, and T4 DNA ligase ligation (Thermo Scientific, cat. no. EL0011). T4 PNK (NEB, #M0201 V) and CIP (NEB, #M0290 V) were used to modify the terminals of DNA fragments. The first constructed vector was pRTVnRFP with an N-terminal TagRFP tag. Based on this original vector, a series of transient expression vectors with N-/C-terminal tags of HA, cMyc, EGFP or mVenus or without any tags were generated. Binary vectors for rice stable expression were also generated by adding the T-DNA, selection markers, as well as the replicon and stabilization sequences from Agrobacterium tumefaciens. The detailed scheme of vector construction is shown in Additional file 1: Additional Methods. A final list, maps, and sequence structure around the MCS for all of the vectors are provided in Additional file 1: Table S1, Figure S2, and Figure S3, respectively.
For testing the feasibility of using those vectors, three representative proteins, OsRac1 (LOC_Os01g12900) (Akamatsu et al. 2013; Ono et al. 2001), WRKY45 (LOC_Os05g25770) (Shimono et al. 2007), and SPIN6 (LOC_Os07g46450) (Liu et al. 2015), as well as the EGFP or TagRFP empty vector and luciferase (LUC) were chosen for fluorescence detection, Co-IP, and BiFC assay in rice protoplasts. The OsRac1 gene was cloned through BamHI and HindIII, generating pRTVnYFP-OsRac1, pRTVnGFP-OsRac1, pRTVnMyc-OsRac1, and pRTVnVC-OsRac1 constructs. The WRKY45 CDS fragment was cloned through BamHI and KpnI, generating the pRTVnRFP-WRKY45 construct. The SPIN6 was cloned through BamHI and NotI, generating the pRTVnRFP-SPIN6, pRTVnHA-SPIN6, and pRTVnVN-SPIN6 constructs. The C terminal of luciferase, cLUC(417–568), was amplified from pCAMBIA1300-cLUC (Chen et al. 2008) and inserted into pRTVnMyc through EcoRV. The LUC fragment was generated by overlap PCR from pCAMBIA1300-nLUC (Chen et al. 2008) and pCAMBIA1300-cLUC, and was ligated through BamHI and HindIII restriction sites, generating the pRTVcVC-LUC construct. The primers used for construction are listed in Additional file 1: Table S2.
Rice Protoplast Isolation and Transfection
Rice protoplast isolation and transfection were performed as described previously (Chen et al. 2006; He et al. 2016). About 3 μg of plasmid DNA was used to transfect ~ 2 × 105 protoplasts by the PEG-mediated method. The fluorescent proteins or epitope-tagged proteins were detected at 14–20 h after transfection.
Fluorescence Detection
Four plasmids, including pRTVnRFP-SPIN6, pRTVnGFP-OsRac1, pRTVnYFP-OsRac1, and pRTVnRFP-WRKY45, as well as the empty vectors pRTVnGFP and pRTVnRFP were used for protein localization and co-localization analysis in rice protoplasts. In the BiFC assay, plasmids pRTVnVN-SPIN6 with pRTVnVC-OsRac1 or pRTVcVC-LUC were co-expressed in rice protoplasts. The fluorescent proteins were observed with a confocal microscope (Carl Zeiss LSM T-PMT 880). The steps of fluorescence observation were executed according to the previous protocol (He et al. 2016). For observation of fluorescence in transgenic plants, the young leaf sheath tissue of pRHVnGFP transgenic T0 plants was used.
Proteins Extraction, Co-IP, and Western Blot Detection
Procedures for protein extraction and Co-IP assay in the rice protoplast system were previously described (He et al. 2016). The plasmid pRTVnHA-SPIN6 was co-transfected with pRTVnMyc-OsRac1 or with the control, pRTVnMyc-cLUC. The anti-HA antibody (Roche, Cat. No. 11867423001) was used to purify the HA-tagged protein in the Co-IP assay. For detection of GFP protein in plants, proteins extracted from young leaves were used western blot detection with the anti-GFP antibody (Roche, Cat. No. 11814460001).
Agrobacterium-Mediated Rice Transformation
For construction of the CRISPR/Cas9 plasmid pRHCas9-IPA1, the specific sequence primer pairs with the complementary PAM site of IPA1 (LOC_Os08g39890) (Jiao et al. 2010) target sequence 5’-ccaCCGACTCGAGCTGTGCTCTC-3′ were synthesized, cloned to pEntry A, and then sub-cloned to pRHCas9. The detailed construction procedure of pRHCas9-IPA1 targeting rice IPA1 is described in Additional file 1: Additional Methods. The pRHVnGFP empty vector and pRHCas9-IPA1 plasmids were introduced into Agrobacterium strain EHA105. Rice calli were induced from the mature seeds of NPB. Transgenic rice plants were generated by the Agrobacterium-mediated method (Qu et al. 2006).
Genomic DNA Extraction for Genotyping
Rice genomic DNA was extracted by the cetyltrimethylammonium bromide (CTAB) method. To detect mutagenesis in the IPA1 gene, target fragments were amplified with specific primer pairs IPA1-CSPID-F/ IPA1-CSPID-R (Additional file 1: Table S2) and were then sequenced.
Change history
07 September 2018
The caption of Fig. 5 and the figure contained an error [1]. The updated figure and caption are published in this correction article.
References
Akamatsu A, Wong HL, Fujiwara M, Okuda J, Nishide K, Uno K, Imai K, Umemura K, Kawasaki T, Kawano Y, Shimamoto K (2013) An OsCEBiP/OsCERK1-OsRacGEF1-OsRac1 module is an essential early component of chitin-induced rice immunity. Cell Host Microbe 13:465–476
Belhaj K, Chaparro-Garcia A, Kamoun S, Patron NJ, Nekrasov V (2015) Editing plant genomes with CRISPR/Cas9. Curr Opin Biotechnol 32:76–84
Berggard T, Linse S, James P (2007) Methods for the detection and analysis of protein-protein interactions. Proteomics 7:2833–2842
Bi H, Yang B (2017) Gene editing with TALEN and CRISPR/Cas in Rice. Prog Mol Biol Transl Sci 149:81–98
Chakraborty M, Sairam Reddy P, Laxmi Narasu M, Krishna G, Rana D (2016) Agrobacterium-mediated genetic transformation of commercially elite rice restorer line using nptII gene as a plant selection marker. Physiol Mol Biol Plants 22:51–60
Chen H, Zou Y, Shang Y, Lin H, Wang Y, Cai R, Tang X, Zhou JM (2008) Firefly luciferase complementation imaging assay for protein-protein interactions in plants. Plant Physiol 146:368–376
Chen S, Songkumarn P, Liu J, Wang GL (2009) A versatile zero background T-vector system for gene cloning and functional genomics. Plant Physiol 150:1111–1121
Chen S, Tao L, Zeng L, Vega-Sanchez ME, Umemura K, Wang GL (2006) A highly efficient transient protoplast system for analyzing defence gene expression and protein-protein interactions in rice. Mol Plant Pathol 7:417–427
Curtis MD, Grossniklaus U (2003) A gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol 133:462–469
Earley KW, Haag JR, Pontes O, Opper K, Juehne T, Song KM, Pikaard CS (2006) Gateway-compatible vectors for plant functional genomics and proteomics. Plant J 45:616–629
Goodin MM, Dietzgen RG, Schichnes D, Ruzin S, Jackson AO (2002) pGD vectors: versatile tools for the expression of green and red fluorescent protein fusions in agroinfiltrated plant leaves. Plant J 31:375–383
Hakes L, Pinney JW, Robertson DL, Lovell SC (2008) Protein-protein interaction networks and biology--what's the connection? Nat Biotechnol 26:69–72
Han Y, Chu X, Yu H, Ma Y-K, Wang X-J, Qian W, Jiao Y (2017) Single-cell transcriptome analysis reveals widespread monoallelic gene expression in individual rice mesophyll cells. Science Bulletin 62:1304–1314
He F, Chen S, Ning Y, Wang G-L (2016) Rice (Oryza sativa) protoplast isolation and its application for transient expression analysis. Curr Protoc Plant Biol 1:373–383
Hellens RP, Allan AC, Friel EN, Bolitho K, Grafton K, Templeton MD, Karunairetnam S, Gleave AP, Laing WA (2005) Transient expression vectors for functional genomics, quantification of promoter activity and RNA silencing in plants. Plant Methods 1:13
Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP (2013) Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res 41:e188
Jiao Y, Wang Y, Xue D, Wang J, Yan M, Liu G, Dong G, Zeng D, Lu Z, Zhu X, Qian Q, Li J (2010) Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat Genet 42:541–544
Jung KH, An G, Ronald PC (2008) Towards a better bowl of rice: assigning function to tens of thousands of rice genes. Nat Rev Genet 9:91–101
Kodama Y, Hu CD (2012) Bimolecular fluorescence complementation (BiFC): a 5-year update and future perspectives. BioTechniques 53:285–298
Kodama Y, Hu C-D (2010) An improved bimolecular fluorescence complementation assay with a high signal-to-noise ratio. BioTechniques 49:793–805
Komori T, Imayama T, Kato N, Ishida Y, Ueki J, Komari T (2007) Current status of binary vectors and superbinary vectors. Plant Physiol 145:1155–1160
Krishnan A, Guiderdoni E, An G, Hsing YI, Han CD, Lee MC, Yu SM, Upadhyaya N, Ramachandran S, Zhang Q, Sundaresan V, Hirochika H, Leung H, Pereira A (2009) Mutant resources in rice for functional genomics of the grasses. Plant Physiol 149:165–170
Lakshmanan M, Cheung CY, Mohanty B, Lee DY (2016) Modeling rice metabolism: from elucidating environmental effects on cellular phenotype to guiding crop improvement. Front Plant Sci 7:1795
Li M, Li X, Zhou Z, Wu P, Fang M, Pan X, Lin Q, Luo W, Wu G, Li H (2016) Reassessment of the four yield-related genes Gn1a, DEP1, GS3, and IPA1 in rice using a CRISPR/Cas9 system. Front Plant Sci 7:377
Li Y, Xiao J, Chen L, Huang X, Cheng Z, Han B, Zhang Q, Wu C (2018) Rice functional genomics research: past decade and future. Mol Plant 11:359–380
Liu J, Park CH, He F, Nagano M, Wang M, Bellizzi M, Zhang K, Zeng X, Liu W, Ning Y, Kawano Y, Wang GL (2015) The RhoGAP SPIN6 associates with SPL11 and OsRac1 and negatively regulates programmed cell death and innate immunity in rice. PLoS Pathog 11:e1004629
Liu Q, Ning Y, Zhang Y, Yu N, Zhao C, Zhan X, Wu W, Chen D, Wei X, Wang GL, Cheng S, Cao L (2017) OsCUL3a negatively regulates cell death and immunity by degrading OsNPR1 in rice. Plant Cell 29:345–359
Ma X, Zhang Q, Zhu Q, Liu W, Chen Y, Qiu R, Wang B, Yang Z, Li H, Lin Y, Xie Y, Shen R, Chen S, Wang Z, Chen Y, Guo J, Chen L, Zhao X, Dong Z, Liu YG (2015) A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant 8:1274–1284
Ma X, Zhu Q, Chen Y, Liu Y-G (2016) CRISPR/Cas9 platforms for genome editing in plants: developments and applications. Mol Plant 9:961–974
Miki D, Shimamoto K (2004) Simple RNAi vectors for stable and transient suppression of gene function in rice. Plant Cell Physiol 45:490–495
Nakagawa C, Inahata K, Nishimura S, Sugimoto K (2011) Improvement of a Venus-based bimolecular fluorescence complementation assay to visualize bFos-bJun interaction in living cells. Biosci Biotechnol Biochem 75:1399–1401
Nakagawa T, Kurose T, Hino T, Tanaka K, Kawamukai M, Niwa Y, Toyooka K, Matsuoka K, Jinbo T, Kimura T (2007) Development of series of gateway binary vectors, pGWBs, for realizing efficient construction of fusion genes for plant transformation. J Biosci Bioeng 104:34–41
Ono E, Wong HL, Kawasaki T, Hasegawa M, Kodama O, Shimamoto K (2001) Essential role of the small GTPase Rac in disease resistance of rice. Proc Natl Acad Sci U S A 98:759–764
Park CH, Shirsekar G, Bellizzi M, Chen S, Songkumarn P, Xie X, Shi X, Ning Y, Zhou B, Suttiviriya P, Wang M, Umemura K, Wang GL (2016) The E3 ligase APIP10 connects the effector AvrPiz-t to the NLR receptor Piz-t in rice. PLoS Pathog 12:e1005529
Piatek A, Ali Z, Baazim H, Li L, Abulfaraj A, Al-Shareef S, Aouida M, Mahfouz MM (2015) RNA-guided transcriptional regulation in planta via synthetic dCas9-based transcription factors. Plant Biotechnol J 13:578–589
Prelich G (2012) Gene overexpression: uses, mechanisms, and interpretation. Genetics 190:841–854
Qu S, Desai A, Wing R, Sundaresan V (2008) A versatile transposon-based activation tag vector system for functional genomics in cereals and other monocot plants. Plant Physiol 146:189–199
Qu S, Liu G, Zhou B, Bellizzi M, Zeng L, Dai L, Han B, Wang GL (2006) The broad-spectrum blast resistance gene Pi9 encodes a nucleotide-binding site-leucine-rich repeat protein and is a member of a multigene family in rice. Genetics 172:1901–1914
Schneeberger K, Weigel D (2011) Fast-forward genetics enabled by new sequencing technologies. Trends Plant Sci 16:282–288
Shaner NC, Patterson GH, Davidson MW (2007) Advances in fluorescent protein technology. J Cell Sci 120:4247–4260
Shimono M, Sugano S, Nakayama A, Jiang CJ, Ono K, Toki S, Takatsuji H (2007) Rice WRKY45 plays a crucial role in benzothiadiazole-inducible blast resistance. Plant Cell 19:2064–2076
Thole V, Worland B, Snape JW, Vain P (2007) The pCLEAN dual binary vector system for agrobacterium-mediated plant transformation. Plant Physiol 145:1211–1219
Tzfira T, Tian GW, Lacroix B, Vyas S, Li JX, Leitner-Dagan Y, Krichevsky A, Taylor T, Vainstein A, Citovsky V (2005) pSAT vectors: a modular series of plasmids for autofluorescent protein tagging and expression of multiple genes in plants. Plant Mol Biol 57:503–516
Wang C, Yin X, Kong X, Li W, Ma L, Sun X, Guan Y, Todd CD, Yang Y, Hu X (2013a) A series of TA-based and zero-background vectors for plant functional genomics. PLoS One 8:e59576
Wang J, Qi M, Liu J, Zhang Y (2015) CARMO: a comprehensive annotation platform for functional exploration of rice multi-omics data. Plant J 83:359–374
Wang N, Long T, Yao W, Xiong L, Zhang Q, Wu C (2013b) Mutant resources for the functional analysis of the rice genome. Mol Plant 6:596–604
Wang R, Ning Y, Shi X, He F, Zhang C, Fan J, Jiang N, Zhang Y, Zhang T, Hu Y, Bellizzi M, Wang GL (2016) Immunity to rice blast disease by suppression of effector-triggered necrosis. Curr Biol 26:2399–2411
Xie K, Minkenberg B, Yang Y (2015) Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci U S A 112:3570–3575
Xing HL, Dong L, Wang ZP, Zhang HY, Han CY, Liu B, Wang XC, Chen QJ (2014) A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol 14:327
Zhang Q, Li J, Xue Y, Han B, Deng XW (2008) Rice 2020: a call for an international coordinated effort in rice functional genomics. Mol Plant 1:715–719
Zhou H, Liu B, Weeks DP, Spalding MH, Yang B (2014) Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res 42:10903–10914
Acknowledgments
We thank Dr. Lei Yao for providing the fluorescence tags and Dr. Huan-bin Zhou for providing the SpCas9 and sgRNA cassettes.
Funding
This work was supported by the National Key Research and Development Program of China (2016YFD0100600), the National Natural Science Foundation of China (31571944), and the Young Elite Scientist Sponsorship of China Association for Science and Technology (2015QNRC001).
Availability of Data and Materials
The datasets supporting the conclusions of this paper are included within the paper. The vectors described in the paper will be available upon request.
Author information
Authors and Affiliations
Contributions
G-L W, YN and FH designed the work. FH carried out the experiments. FH, FZ, and WS performed the analysis. YN and FH wrote the manuscript. G-L W edited and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Competing Interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Additional Methods. Figure S1. Restriction maps for ubiquitin-1 and 35S promoters. Figure S2. Maps of all the 42 vectors. Figure S3. The sequence structures in the MCS region of the vectors. Table S1. List of the 42 vectors generated in this study. Table S2. List of all primers used in this paper. (PDF 1648 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
He, F., Zhang, F., Sun, W. et al. A Versatile Vector Toolkit for Functional Analysis of Rice Genes. Rice 11, 27 (2018). https://doi.org/10.1186/s12284-018-0220-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-018-0220-7