
Abstract
The next-generation sequencing (NGS) technology has revolutionized our previous understanding of the plant genomes, relying on its innate advantages, such as high throughput and deep sequencing depth. In addition to the protein-coding gene loci, massive transcription signals have been detected within intergenic or intragenic regions. Most of these signals belong to non-coding ones, considering their weak protein-coding potential. Generally, these transcripts could be divided into long non-coding RNAs and small non-coding RNAs (sRNAs) based on their sequence length. In addition to the well-known microRNAs (miRNAs), many plant endogenous sRNAs were collectively referred to as small interfering RNAs. However, an increasing number of unclassified sRNA species are being discovered by NGS. The high heterogeneity of these novel sRNAs greatly hampered the mechanistic studies, especially on the clear description of their biogenesis and action pathways. Fortunately, public databases, bioinformatics softwares and NGS datasets are increasingly available for plant sRNA research. Here, by summarizing these valuable resources, we proposed a general workflow to decipher the RDR (RNA-dependent RNA polymerase)- and DCL (Dicer-like)-dependent biogenesis pathways, and the Argonaute-mediated action modes (such as target cleavages and chromatin modifications) for specific sRNA species in plants. Taken together, we hope that by summarizing a list of the public resources, this work will facilitate the plant biologists to perform classification and functional characterization of the interesting sRNA species.
Similar content being viewed by others


Review
Introduction
Ten years after the accomplishment of the first plant genome project (Mozo et al., 1999), the advent of the next-generation sequencing (NGS) technology has uncovered an unprecedentedly intricate scene of genome-wide transcription in plants (Varshney et al., 2009; Kelly and Leitch, 2011; Jain, 2012). In addition to the already annotated protein-coding genes, the fact is emerging that millions of the non-coding RNAs (ncRNAs) are transcribed from the intergenic or the intragenic regions (Jiang, 2015; Wendel et al., 2016). These non-coding transcripts could be roughly classified into the long non-coding RNAs (lncRNAs; > 200 nt) (Chekanova, 2015) and the small non-coding RNAs (sRNAs; < 200 nt) (Chen, 2009). Owing to the relatively short read length of NGS, the sRNAs were easier to be cloned at the beginning of the plant ncRNA research. Expectedly, the explosive sRNA world immediately became a hot research topic for the plant biologists. Notably, some of these small transcription “noises”, which were once regarded as the degraded remnants, have been demonstrated to be generated through specific pathways and play essential roles in plant development (Chen, 2009).
One of the well studied sRNA species is microRNA (miRNA) (Jones-Rhoades et al., 2006; Voinnet, 2009). In plants, the transcription of most miRNA genes is driven by RNA polymerase II (Pol II), resulting in the production of the 5′ capped and 3′ poly(A) (polyadenylation)-tailed transcripts called primary microRNAs (pri-miRNAs). Relying on the highly complementary base pairing, stable hairpin-like structures could form within the specific regions of the pri-miRNAs. These local hairpin structures are the featured substrates of Dicer-like 1 (DCL1). Followed by the DCL1-mediated two-step cropping in the nucleus, the pri-miRNAs are sequentially processed into the secondary precursors named as precursor microRNAs (pre-miRNAs), and then into the miRNA/miRNA* duplexes. After exporting to the cytoplasm, the mature miRNAs are selectively loaded into specific Argonaute (AGO)-centered protein complexes. In most cases, the miRNAs will be recruited by AGO1, although some exceptional cases have been reported for “miR172—AGO10” and “miR165/166—AGO10” in Arabidopsis (Arabidopsis thaliana), “miR168—AGO18” in rice (Oryza sativa), and “miR390—AGO7” in both plants (Fang and Qi, 2016). The AGO complex is guided by the recruited miRNA to bind onto a specific target transcript containing a region highly complementary to the miRNA. There are two major action modes of miRNA-guided gene silencing in plants. One is target cleavage which is considered as the most common mode (Voinnet, 2009), and the other is translational repression which has been observed in several studies (Chen, 2004; Gandikota et al., 2007; Li et al., 2013). Another class of plant sRNAs is collectively referred to as the small interfering RNAs (siRNAs), which could be further classified into heterochromatic small interfering RNAs (hc-siRNAs), trans-acting small interfering RNAs (ta-siRNAs), natural antisense transcript-derived small interfering RNAs (nat-siRNAs), and phased small interfering RNAs (phasiRNAs). Specifically, hc-siRNAs are encoded within the heterochromatic loci transcribed by RNA Pol IV. The single-stranded Pol IV transcripts are converted to double-stranded precursors through the RDR2 (RNA-dependent RNA polymerase 2)-dependent pathway. Then, the precursors are processed by DCL3 for the production of the 24-nt hc-siRNAs. The hc-siRNAs are incorporated into AGO4 to perform site-specific chromatin modifications (Xie et al., 2004; Qi et al., 2005; Henderson et al., 2006). In Arabidopsis, there are four TAS gene loci encoding ta-siRNAs. MiRNA-mediated cleavages (miR173 for TAS1 and TAS2, miR390 for TAS3, and miR828 for TAS4) of the primary TAS transcripts are the prerequisite for initiating ta-siRNA production. Through the RDR6-dependent pathway, the cleaved TAS transcripts are converted to double-stranded precursors which will be subject to ta-siRNA processing by DCL4. Finally, most of the ta-siRNAs are loaded into AGO1 silencing complexes to guide target cleavages (Peragine et al., 2004; Vazquez et al., 2004; Allen et al., 2005; Gasciolli et al., 2005; Xie et al., 2005; Yoshikawa et al., 2005; Rajagopalan et al., 2006). For the nat-siRNAs, genome-wide studies in both Arabidopsis and rice showed that they were originated from the overlapping regions of the natural antisense transcript (NAT) pairs through the DCL1-dependent pathway or through the Pol IV-, RDR2-, and DCL3-dependent pathway (Zhang et al., 2012). Moreover, in Borsani et al.’s study (2005), 21- and 24-nt nat-siRNAs were demonstrated to be produced from a cis-NAT pair through the RDR6- and DCL1/2-dependent pathway (Borsani et al., 2005). A major class of phasiRNAs was identified in the reproductive tissues of Gramineae species, such as rice (Johnson et al., 2009) and maize (Zea mays) (Zhai et al., 2015). Notably, in rice, the processing of 21-nt phasiRNAs was highly dependent on DCL4, while the processing of 24-nt ones required the activity of DCL3b (Song et al., 2012). Komiya and his colleagues (2014) reported that a portion of the DCL4-dependent, 21-nt phasiRNAs preferentially associated with MEL1 (the ortholog of Arabidopsis AGO5), and these 5′ C-started phasiRNAs originated from hundreds of lincRNA (long intergenic non-coding RNA) loci. In addition to the above mentioned sRNAs, some non-canonical sRNA species have been discovered, such as the AGO4-associated long siRNAs of 25-nt in length (Zilberman et al., 2003), the DCL3-dependent, AGO4-associated, 24-nt miRNAs called long miRNAs (Wu et al., 2010), the Pol IV- and DCL2/3/4-dependent, AGO2-associated double-strand break-induced sRNAs (Wei et al., 2012), the DCL-independent, AGO4-associated, 20- to 60-nt siRNAs (Ye et al., 2016), and the intron-derived, DCL2/3/4-dependent siRNAs (Chen et al., 2011). For a clear summarization, Fig. 1a provides a brief framework of the biogenesis and action pathways of the plant sRNAs. However, all of the recent discoveries just witnessed the emergence of the unexpectedly huge and complicated RNA world. It is still far from thorough understanding of the biogenesis and action pathways of the enormous sRNA population.

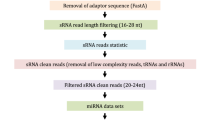
A general workflow for public bioinformatics resource-based investigation of small RNA (sRNA) biogenesis and action pathways in plants. a Presents a brief framework of the biogenesis and action pathways of plant sRNAs. AGO: Argonaute; Pol: RNA polymerase; RDR: RNA-dependent RNA polymerase; DCL: Dicer-like; sRNA: small RNA. b The analysis is divided into five sections according to the step-by-step instructions in the main text, including “genomic features”, “transcription”, “precursors and processing”, “sRNA action modes” and “functional studies”
Fortunately, the valuable public resources have become increasingly available for the mechanistic studies on the plant sRNAs. Here, by taking the two model plants Arabidopsis and rice as an example, we provided a list of the currently available resources to the plant biologists, including the public databases, the bioinformatics softwares and the NGS datasets. Notably, most of the bioinformatics softwares listed here are online tools with user-friendly interface. By proposing a workflow for analyzing the biogenesis and action pathways of the plant sRNAs, we made a clear description for the specific applications of different sequencing datasets and bioinformatics toolkits at each analytical step. Finally, we anticipate that this workflow along with the list could advance the efficiency of data analysis and interpretation, thus facilitating the experimental design for the functional studies on the plant sRNAs. Below, we will introduce the public resources step by step according to the workflow shown in Fig. 1b.
Genomic features and transcription
By using the BLAST tool provided by the plant genomic databases, such as TAIR (the Arabidopsis information resource) (Huala et al., 2001) for Arabidopsis and RGAP (rice genome annotation project) (Kawahara et al., 2013) or RAP-DB (the rice annotation project database) (Ohyanagi et al., 2006) for rice (Table 1), the genomic positions of the sRNA-coding loci could be obtained, facilitating the researchers to tell whether the sRNA loci are intergenic or intragenic. For mapping huge sRNA sequencing (sRNA-seq) datasets onto a plant genome, Bowtie should be selected as one of the powerful tools (Langmead et al., 2009). miRBase (the microRNA database) (Griffiths-Jones et al., 2006) and PLncDB (plant long non-coding RNA database) (Jin et al., 2013) are useful to check whether the sRNA is originated from a miRNA precursor or a lncRNA. Besides, ShortStack should be a useful tool to analyze the sRNA-seq data based on the available reference genomes (Axtell, 2013) (Table 2). It can output reports showing sRNA size distributions, repetitiveness, hairpin-association and phasing. One of its shortage is the requirement of bioinformatics experts for local installation and running.
Here, the workflow for analyzing the sRNA biogenesis and action pathways is proposed based on the scenario that the sRNAs are processed from their precursors transcribed from specific genomic loci (Fig. 1). If the sRNA precursor was experimentally cloned by using fine-scale methods such as RACE (rapid amplification of cDNA ends), the transcription boundary of the precursor-coding locus could be defined. In this case, the upstream region of user-defined length could be retrieved from the above mentioned genomic database, and be treated as the promoter region of this gene locus for cis-element analysis by using PlantCARE (a plant cis-acting regulatory element database) (Rombauts et al., 1999), PLACE (Plant cis-acting regulatory DNA elements database) (Higo et al., 1998), or the newly updated tools JASPAR (Mathelier et al., 2016) and the MEME suite (Bailey et al., 2015) (Table 2). The prediction results from these online tools might provide some valuable hints to infer the basic transcriptional features of this gene locus. For example, the coexistence of CAAT-box and TATA-box within the upstream region near to the transcription start site indicates the Pol II-drived transcription of the host gene (Lewin, 1990). Of course, we should acknowledge that the fine-scale cloning of the sRNA precursors is time consuming and laborious. The high-throughput solution is by analyzing the publicly available RNA sequencing (RNA-seq) data. Notably, distinct types of RNA-seq libraries were prepared with different purposes. For example, in a recent study, the poly(A)-tailed RNA-seq libraries were constructed for the detection of Pol II-dependent transcripts, while the rRNA-depleted total RNA-seq libraries were prepared for the identification of Pol IV-dependent transcripts (Li et al., 2015). After mapping such kind of RNA-seq data (Table 3) onto the plant genome by using a high-throughput alignment tool, Bowtie 2 (Langmead and Salzberg, 2012) for example, the transcription boundaries of the sRNA precursor-coding loci could be delineated. Also based on the mapping result, the RNA polymerase dependence could be partially determined for the loci. Moreover, some of the sRNA-seq datasets, such as those originated from the nrpd1 mutant (a Pol IV mutant) (Table 3), could also be used to investigate the polymerase dependence of the sRNA-coding loci. Notably, compared to Bowtie, Bowtie 2 is particularly efficient for long read (up to hundreds of nucleotides in length) mapping. Thus, Bowtie 2 is recommended to be employed for RNA-seq data analysis, while Bowtie is more suitable for sRNA-seq read mapping as mentioned above.
Precursor formation and processing
As introduced above, there are two major forms of sRNA precursors that could be processed by DCLs, i.e. the long double-stranded RNA (dsRNA) precursors and the single-stranded RNAs with short internal double-stranded regions (Fig. 1). The former ones could be synthesized either through the RDR-dependent (such as the precursors of the hc-siRNAs or the ta-siRNAs) pathway or through the RDR-independent (such as the NATs) pathway. However, the later ones are unexceptionally generated through the RDR-independent pathways (such as the pri-miRNAs and the sirtrons). Thus, distinct bioinformatics toolkits should be selected to identify the sRNA precursors belonging to the two different types, respectively.
PlantNATsDB (plant natural antisense transcripts database) (Chen et al., 2012) provides the users with the genome-wide prediction results of both cis- and trans-NAT pairs of 70 plant species. Gene locus ID could be used as a query to see the possibility of this gene to form NAT pairs with other genes. Optionally, “batch download” could be selected to obtain the complete list of the predicted NAT pairs of a plant species. In the other way, the researchers could perform large-scale NAT prediction by using the program NATpipe (Yu et al., 2016a). The genome-independent feature of this software allows users to carry out NAT prediction solely based on the RNA-seq data. If the genomic information is available, then the predicted NATs could be classified into cis and trans ones. Additionally, if the sRNA-seq data is available, phase-distributed nat-siRNAs could be identified from the predicted NATs by using NATpipe.
RNAfold (Hofacker, 2003) and RNAshapes (Steffen et al., 2006) are both easy-to-use tools for local RNA secondary structure predictions. RNAfold is a web server allowing a query sequence of up to 10,000 nt in length, but its graphic outputs are difficult to be modified according to the users’ requirements. RNAshapes is a locally installed program with a strict length limitation (up to ~400 nt based our experience) of an input sequence. However, the outputs of RNAshapes could be graphically edited.
Recently, NGS-based, transcriptome-wide strategies have been developed to probe the RNA secondary structures, such as dsRNA sequencing (dsRNA-seq) (Kwok et al., 2015). The dsRNA-seq library is prepared by treating the total RNAs with the ribonuclease specific for the single-stranded RNAs, thus enabling researchers to detect the annealed region within an RNA sequence, or between two transcripts. Currently, the plant dsRNA-seq data is only available for Arabidopsis. These dsRNA-seq datasets were prepared not only from the wild type (WT) plants, but also from the nrpd1 and rdr6 mutants (Table 3). Thus, the Pol IV and RDR6 dependence of the dsRNA precursors could be interrogated by using these datasets. In addition to the dsRNA-seq data, sRNA-seq data of nrpd1, rdr2 and rdr6 should also be valuable to investigate the biogenesis pathways of the sRNA precursors (Table 3).
DCLs have been demonstrated to be widely implicated in the processing of diverse sRNA precursors in plants (Chen, 2009). Thus, by comparing to the sequencing data of WT, the public sRNA-seq data of the dcl mutants could be used to investigate the specific DCL-mediated sRNA processing pathways.
sRNA action modes and network construction
According to the current understanding, target cleavages and chromatin modifications are the two major action modes of the plant sRNAs. And, these two distinct regulatory pathways are largely predetermined by the association of the sRNAs with specific AGO complexes (Fang and Qi, 2016). Thus, AGO enrichment analysis is necessary for functional studies on the plant sRNAs. To date, sequencing data of the AGO-associated sRNA populations has been reported by several research groups (Table 3). In Arabidopsis, AGO1-, AGO2-, AGO4-, AGO5-, AGO7-, AGO9- and AGO10-associated sRNA sequencing datasets are available (Mi et al., 2008; Montgomery et al., 2008; Havecker et al., 2010; Wang et al., 2011; Zhu et al., 2011). And in rice, AGO1-, AGO4-, MEL1-, AGO16- and AGO18-associated sRNA sequencing datasets have been published (Wu et al., 2009; Wu et al., 2010; Komiya et al., 2014). By comparing the level of a sRNA in a specific AGO complex to that in the total RNA extract, whether this sRNA is enriched in the AGO complex could be determined. The result could facilitate the researchers to deduce the action mode of this sRNA.
A large portion of the miRNAs and some of the siRNAs such as the ta-siRNAs are incorporated into AGO1. These AGO1-associated sRNAs can recognize the highly complementary regions on the target transcripts, and inhibit gene expression through target cleavages. The high complementarity between the sRNA and its target forms an essential basis for the development of the target prediction tools. For plants, there are several user-friendly online tools for target prediction (Table 2), such as psRNATarget (a plant small RNA target analysis server) (Dai and Zhao, 2011), Small RNA Target Prediction (Jones-Rhoades and Bartel, 2004), and TAPIR (target prediction for plant microRNAs) (Bonnet et al., 2010). Compared to TAPIR, the former two tools are more flexible for different users’ purposes. By using psRNATarget or Small RNA Target Prediction, the users can select one of the cDNA libraries provided by the tools, or can upload their own cDNA sequences for target prediction. However, TAPIR does not provide the pre-stored cDNA libraries for the users. Additionally, much more parameters are adjustable before performing analysis by using the former two tools. Thus, psRNATarget and Small RNA Target Prediction should be the efficient and easy-to-use tools for plant sRNA target prediction.
The 3′ cleavage remnants from the target transcripts are relatively stable in vivo, and could be detected by sequencing. This kind of high-throughput sequencing technology was called GMUCT (global mapping of uncapped and cleaved transcripts) (Willmann et al., 2014) or PARE (parallel analysis of RNA ends) (German et al., 2008; German et al., 2009), which is collectively referred to as degradome sequencing (degradome-seq) here. As listed in Table 3, there are many degradome-seq datasets available to perform large-scale validation of the predicted sRNA targets. For analyzing the degradome-seq data, comPARE (PARE validated miRNA targets) and sPARTA-Web (small RNA-PARE target analyzer) (Kakrana et al., 2014) might be the easy-to-use online tools for the wet-lab researchers (Table 2). The difference between comPARE and sPARTA-Web is that the former was designed specifically for the miRNA target validation whereas the latter was developed for all of the sRNA target candidates. CleaveLand4 (Addo-Quaye et al., 2009) is also suitable for degradome-seq data-based validation of the sRNA targets. However, it is a Perl program, which requires extensive support from bioinformatics experts for local installation and running. Besides, our previously proposed workflow could also be referenced for degradome-seq data-based sRNA target validation (Meng et al., 2011b).
The AGO4-associated sRNAs, such as the hc-siRNAs, repress gene expression through chromatin modifications (Chen, 2009; Fang and Qi, 2016). By using BLAST or Bowtie, the genomic regions highly complementary to the AGO4-associated sRNAs could be identified with a genome-wide scale. Then, it will be interesting to investigate the chromatin status surrounding the complementary sites. Several epigenome databases are available for Arabidopsis, such as Arabidopsis epigenome maps (Lister et al., 2008), the SIGnAL Arabidopsis Methylome Mapping Tool (Zhang et al., 2006), and the Arabidopsis epigenome data displayed in the UCSC Genome Browser (Zhang et al., 2006; Zhang et al., 2007; Stroud et al., 2013). In some databases, in addition to the WT data, the epigenomes of diverse mutants are also available, which might be valuable to inspect the sRNA-guided chromatin modification pathways in detail. Although the rice epigenome data was reported nearly ten years ago (Li et al., 2008), and the database was established at that time, the web link is no long active. Fortunately, Plant Methylome DB provides researchers with the WT epigenomes of 40 species including Arabidopsis and rice.
“Target mimicry” was reported as a novel pathway for the regulation of the miRNA activities by the target mimics (Franco-Zorrilla et al., 2007). Although the online server TAPIR is not superior in sRNA target prediction, it provides a unique functional module for target mimic prediction (Bonnet et al., 2010). Besides, the recently established PceRBase (plant ceRNA database) stores the lists of the competing endogenous RNAs (similar to the target mimics) of 26 plant species for the users (Yuan et al., 2017).
Finally, researchers could construct a sRNA-centered regulatory network involving sRNA targets and target mimics by using Cytoscape (Shannon et al., 2003) or Gephi (Bastian et al., 2009). The expression levels of the sRNA precursors, the sRNA target genes and the target mimics could be partially uncovered by visiting PmiRKB (plant microRNA knowledge base) (Meng et al., 2011a), mirEX (Arabidopsis pri-miRNA expression atlas) (Bielewicz et al., 2012), AVT (AtGenExpress visualization tool) (Kilian et al., 2007; Goda et al., 2008), and Arabidopsis eFP Browser (Winter et al., 2007). The biological functions of the sRNA target genes could be analyzed by using agriGO (Du et al., 2010).
Conclusions
In the present work, we proposed a general workflow for deciphering the biogenesis and action pathways of the plant sRNAs by using a series of publicly available resources. Most of the recently reported sRNA-seq and dsRNA-seq datasets of Arabidopsis and rice were summarized in Table 3, emphasizing their importance for elucidating the RDR- and DCL-dependent biogenesis pathways of the plant endogenous sRNAs. However, we should acknowledged that several useful toolkits have not been included in the list of softwares for plant small RNA research. For example, the UEA sRNA workbench, that is downloadable for local installation, provides an user-friendly platform for sRNA-seq data processing (Stocks et al., 2012). It contains several useful tools, such as “adaptor remover” and “Filter” for sRNA-seq data pre-treatment, “miRCat” and “hairpin annotation” for miRNA prediction, and the ta-siRNA prediction tool. Besides, as noticed in Tables 1 and 2, several valuable databases and bioinformatics tools, including Rice epigenome maps and PNRD, are currently terminated for unknown reason. We hope that these useful resources could be activated again for the plant biologists. Summarily, more research efforts, from both the bioinformaticians and the experimental practitioners, are anticipated to devote to the plant sRNA research.
Abbreviations
- AGO:
-
Argonaute
- AVT:
-
AtGenExpress visualization tool
- DCL:
-
Dicer-like
- degradome-seq:
-
degradome sequencing
- dsRNA:
-
double-stranded RNA
- dsRNA-seq:
-
double-stranded RNA sequencing
- GMUCT:
-
Global mapping of uncapped and cleaved transcripts
- hc-siRNA:
-
heterochromatic small interfering RNA
- lincRNA:
-
long intergenic non-coding RNA
- lncRNA:
-
long non-coding RNA
- miRNA:
-
microRNA
- NAT:
-
Natural antisense transcript
- nat-siRNA:
-
natural antisense transcript-derived small interfering RNA
- ncRNA:
-
non-coding RNA
- NGS:
-
Next-generation sequencing
- PARE:
-
Parallel analysis of RNA ends
- PceRBase:
-
Plant ceRNA database
- phasiRNA:
-
phased small interfering RNA
- PlantCARE:
-
a plant cis-acting regulatory element database
- PlantNATsDB:
-
Plant natural antisense transcripts database
- PLncDB:
-
Plant long non-coding RNA database
- PmiRKB:
-
Plant microRNA knowledge base
- Pol II:
-
RNA polymerase II
- poly(A):
-
polyadenylation
- pri-miRNA:
-
primary transcript of microRNA
- psRNATarget:
-
a plant small RNA target analysis server
- RACE:
-
rapid amplification of cDNA ends
- RAP-DB:
-
the rice annotation project database
- RDR:
-
RNA-dependent RNA polymerase
- RGAP:
-
Rice genome annotation project
- RNA-seq:
-
RNA sequencing
- rRNA:
-
ribosomal RNA
- siRNA:
-
Small interfering RNA
- sRNA:
-
Small RNA
- sRNA-seq:
-
sRNA sequencing
- TAIR:
-
the Arabidopsis information resource
- TAPIR:
-
Target prediction for plant microRNAs
- ta-siRNA:
-
trans-acting small interfering RNA
- WT:
-
Wild type
References
Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ (2008) Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol 18:758–762
Addo-Quaye C, Miller W, Axtell MJ (2009) CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 25:130–131
Allen E, Xie Z, Gustafson AM, Carrington JC (2005) microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121:207–221
Axtell MJ (2013) ShortStack: comprehensive annotation and quantification of small RNA genes. RNA 19:740–751
Bailey TL, Johnson J, Grant CE, Noble WS (2015) The MEME suite. Nucleic Acids Res 43:W39–W49
Baldrich P, Campo S, Wu MT, Liu TT, Hsing YI, San Segundo B (2015) MicroRNA-mediated regulation of gene expression in the response of rice plants to fungal elicitors. RNA Biol 12:847–863
Bastian M., Heymann S., Jacomy M (2009) Gephi: an open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media
Bielewicz D, Dolata J, Zielezinski A, Alaba S, Szarzynska B, Szczesniak MW, Jarmolowski A, Szweykowska-Kulinska Z, Karlowski WM (2012) mirEX: a platform for comparative exploration of plant pri-miRNA expression data. Nucleic Acids Res 40:D191–D197
Bonnet E, He Y, Billiau K, Van de Peer Y (2010) TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 26:1566–1568
Borsani O, Zhu J, Verslues PE, Sunkar R, Zhu JK (2005) Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell 123:1279–1291
Chekanova JA (2015) Long non-coding RNAs and their functions in plants. Curr Opin Plant Biol 27:207–216
Chen D, Meng Y, Yuan C, Bai L, Huang D, Lv S, Wu P, Chen LL, Chen M (2011) Plant siRNAs from introns mediate DNA methylation of host genes. RNA 17:1012–1024
Chen D, Yuan C, Zhang J, Zhang Z, Bai L, Meng Y, Chen LL, Chen M (2012) PlantNATsDB: a comprehensive database of plant natural antisense transcripts. Nucleic Acids Res 40:D1187–D1193
Chen X (2004) A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303:2022–2025
Chen X (2009) Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol 25:21–44
Creasey KM, Zhai J, Borges F, Van Ex F, Regulski M, Meyers BC, Martienssen RA (2014) miRNAs trigger widespread epigenetically activated siRNAs from transposons in Arabidopsis. Nature 508:411–415
Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14:1188–1190
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39:W155–W159
Du Z, Zhou X, Ling Y, Zhang Z, Su Z (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38:W64–W70
Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210
Fahlgren N, Sullivan CM, Kasschau KD, Chapman EJ, Cumbie JS, Montgomery TA, Gilbert SD, Dasenko M, Backman TW, Givan SA et al (2009) Computational and analytical framework for small RNA profiling by high-throughput sequencing. RNA 15:992–1002
Fang X, Qi Y (2016) RNAi in plants: an Argonaute-centered view. Plant Cell 28:272–285
Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, Leyva A, Weigel D, Garcia JA, Paz-Ares J (2007) Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet 39:1033–1037
Gandikota M, Birkenbihl RP, Hohmann S, Cardon GH, Saedler H, Huijser P (2007) The miRNA156/157 recognition element in the 3′ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J 49:683–693
Gasciolli V, Mallory AC, Bartel DP, Vaucheret H (2005) Partially redundant functions of Arabidopsis DICER-like enzymes and a role for DCL4 in producing trans-acting siRNAs. Curr Biol 15:1494–1500
German MA, Luo S, Schroth G, Meyers BC, Green PJ (2009) Construction of parallel analysis of RNA ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat Protoc 4:356–362
German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R et al (2008) Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol 26:941–946
Goda H, Sasaki E, Akiyama K, Maruyama-Nakashita A, Nakabayashi K, Li W, Ogawa M, Yamauchi Y, Preston J, Aoki K et al (2008) The AtGenExpress hormone and chemical treatment data set: experimental design, data evaluation, model data analysis and data access. Plant J 55:526–542
Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N et al (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40:D1178–D1186
Gregory BD, O'Malley RC, Lister R, Urich MA, Tonti-Filippini J, Chen H, Millar AH, Ecker JR (2008) A link between RNA metabolism and silencing affecting Arabidopsis development. Dev Cell 14:854–866
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ (2006) miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34:D140–D144
Gustafson AM, Allen E, Givan S, Smith D, Carrington JC, Kasschau KD (2005) ASRP: the Arabidopsis small RNA project database. Nucleic Acids Res 33:D637–D640
Havecker ER, Wallbridge LM, Hardcastle TJ, Bush MS, Kelly KA, Dunn RM, Schwach F, Doonan JH, Baulcombe DC (2010) The Arabidopsis RNA-directed DNA methylation argonautes functionally diverge based on their expression and interaction with target loci. Plant Cell 22:321–334
Henderson IR, Zhang X, Lu C, Johnson L, Meyers BC, Green PJ, Jacobsen SE (2006) Dissecting Arabidopsis Thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat Genet 38:721–725
Higo K, Ugawa Y, Iwamoto M, Higo H (1998) PLACE: a database of plant cis-acting regulatory DNA elements. Nucleic Acids Res 26(1):358–359
Hofacker IL (2003) Vienna RNA secondary structure server. Nucleic Acids Res 31:3429–3431
Hou CY, Lee WC, Chou HC, Chen AP, Chou SJ, Chen HM (2016) Global analysis of truncated RNA ends reveals new insights into ribosome stalling in plants. Plant Cell 28:2398–2416
Huala E, Dickerman AW, Garcia-Hernandez M, Weems D, Reiser L, LaFond F, Hanley D, Kiphart D, Zhuang M, Huang W et al (2001) The Arabidopsis information resource (TAIR): a comprehensive database and web-based information retrieval, analysis, and visualization system for a model plant. Nucleic Acids Res 29:102–105
Jain M (2012) Next-generation sequencing technologies for gene expression profiling in plants. Brief Funct Genomics 11:63–70
Jeong DH, Park S, Zhai J, Gurazada SG, De Paoli E, Meyers BC, Green PJ (2011) Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 23:4185–4207
Jeong DH, Thatcher SR, Brown RS, Zhai J, Park S, Rymarquis LA, Meyers BC, Green PJ (2013) Comprehensive investigation of microRNAs enhanced by analysis of sequence variants, expression patterns, ARGONAUTE loading, and target cleavage. Plant Physiol 162:1225–1245
Jiang J (2015) The 'dark matter' in the plant genomes: non-coding and unannotated DNA sequences associated with open chromatin. Curr Opin Plant Biol 24:17–23
Jin J, Liu J, Wang H, Wong L, Chua NH (2013) PLncDB: plant long non-coding RNA database. Bioinformatics 29:1068–1071
Johnson C, Bowman L, Adai AT, Vance V, Sundaresan V (2007) CSRDB: a small RNA integrated database and browser resource for cereals. Nucleic Acids Res 35:D829–D833
Johnson C, Kasprzewska A, Tennessen K, Fernandes J, Nan GL, Walbot V, Sundaresan V, Vance V, Bowman LH (2009) Clusters and superclusters of phased small RNAs in the developing inflorescence of rice. Genome Res 19:1429–1440
Jones-Rhoades MW, Bartel DP (2004) Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell 14:787–799
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53
Kakrana A, Hammond R, Patel P, Nakano M, Meyers BC (2014) sPARTA: a parallelized pipeline for integrated analysis of plant miRNA and cleaved mRNA data sets, including new miRNA target-identification software. Nucleic Acids Res 42:e139
Kaminuma E, Mashima J, Kodama Y, Gojobori T, Ogasawara O, Okubo K, Takagi T, Nakamura Y (2010) DDBJ launches a new archive database with analytical tools for next-generation sequence data. Nucleic Acids Res 38:D33–D38
Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S et al (2013) Improvement of the Oryza Sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6:4
Kelly LJ, Leitch IJ (2011) Exploring giant plant genomes with next-generation sequencing technology. Chromosom Res 19:939–953
Kilian J, Whitehead D, Horak J, Wanke D, Weinl S, Batistic O, D'Angelo C, Bornberg-Bauer E, Kudla J, Harter K (2007) The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J 50:347–363
Kodama Y, Shumway M, Leinonen R, International Nucleotide Sequence Database C (2012) The sequence read archive: explosive growth of sequencing data. Nucleic Acids Res 40:D54–D56
Komiya R, Ohyanagi H, Niihama M, Watanabe T, Nakano M, Kurata N, Nonomura K (2014) Rice germline-specific Argonaute MEL1 protein binds to phasiRNAs generated from more than 700 lincRNAs. Plant J 78:385–397
Kwok CK, Tang Y, Assmann SM, Bevilacqua PC (2015) The RNA structurome: transcriptome-wide structure probing with next-generation sequencing. Trends Biochem Sci 40:221–232
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with bowtie 2. Nat Methods 9:357–359
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25
Lewin B (1990) Commitment and activation at pol II promoters: a tail of protein-protein interactions. Cell 61:1161–1164
Li S, Liu L, Zhuang X, Yu Y, Liu X, Cui X, Ji L, Pan Z, Cao X, Mo B et al (2013) MicroRNAs inhibit the translation of target mRNAs on the endoplasmic reticulum in Arabidopsis. Cell 153:562–574
Li S, Vandivier LE, Tu B, Gao L, Won SY, Li S, Zheng B, Gregory BD, Chen X (2015) Detection of pol IV/RDR2-dependent transcripts at the genomic scale in Arabidopsis reveals features and regulation of siRNA biogenesis. Genome Res 25:235–245
Li X, Wang X, He K, Ma Y, Su N, He H, Stolc V, Tongprasit W, Jin W, Jiang J et al (2008) High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant Cell 20:259–276
Li YF, Zheng Y, Addo-Quaye C, Zhang L, Saini A, Jagadeeswaran G, Axtell MJ, Zhang W, Sunkar R (2010) Transcriptome-wide identification of microRNA targets in rice. Plant J 62:742–759
Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR (2008) Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133:523–536
Mathelier A, Fornes O, Arenillas DJ, Chen CY, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley-Hunt R et al (2016) JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 44:D110–D115
Meng Y, Gou L, Chen D, Mao C, Jin Y, Wu P, Chen M (2011a) PmiRKB: a plant microRNA knowledge base. Nucleic Acids Res 39:D181–D187
Meng Y, Shao C, Chen M (2011b) Toward microRNA-mediated gene regulatory networks in plants. Brief Bioinform 12:645–659
Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, Wu L, Li S, Zhou H, Long C et al (2008) Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 133:116–127
Montgomery TA, Howell MD, Cuperus JT, Li D, Hansen JE, Alexander AL, Chapman EJ, Fahlgren N, Allen E, Carrington JC (2008) Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell 133:128–141
Mozo T, Dewar K, Dunn P, Ecker JR, Fischer S, Kloska S, Lehrach H, Marra M, Martienssen R, Meier-Ewert S et al (1999) A complete BAC-based physical map of the Arabidopsis Thaliana genome. Nat Genet 22:271–275
Nakano M, Nobuta K, Vemaraju K, Tej SS, Skogen JW, Meyers BC (2006) Plant MPSS databases: signature-based transcriptional resources for analyses of mRNA and small RNA. Nucleic Acids Res 34:D731–D735
Ohyanagi H, Tanaka T, Sakai H, Shigemoto Y, Yamaguchi K, Habara T, Fujii Y, Antonio BA, Nagamura Y, Imanishi T et al (2006) The Rice annotation project database (RAP-DB): hub for Oryza Sativa Ssp. Japonica genome information. Nucleic Acids Res 34:D741–D744
Patel P, Ramachandruni SD, Kakrana A, Nakano M, Meyers BC (2016) miTRATA: a web-based tool for microRNA truncation and tailing analysis. Bioinformatics 32:450–452
Peragine A, Yoshikawa M, Wu G, Albrecht HL, Poethig RS (2004) SGS3 and SGS2/SDE1/RDR6 are required for juvenile development and the production of trans-acting siRNAs in Arabidopsis. Genes Dev 18:2368–2379
Qi Y, Denli AM, Hannon GJ (2005) Biochemical specialization within Arabidopsis RNA silencing pathways. Mol Cell 19:421–428
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP (2006) A diverse and evolutionarily fluid set of microRNAs in Arabidopsis Thaliana. Genes Dev 20:3407–3425
Rombauts S, Dehais P, Van Montagu M, Rouze P (1999) PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res 27:295–296
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Song X, Li P, Zhai J, Zhou M, Ma L, Liu B, Jeong DH, Nakano M, Cao S, Liu C et al (2012) Roles of DCL4 and DCL3b in rice phased small RNA biogenesis. Plant J 69:462–474
Steffen P, Voss B, Rehmsmeier M, Reeder J, Giegerich R (2006) RNAshapes: an integrated RNA analysis package based on abstract shapes. Bioinformatics 22:500–503
Stocks MB, Moxon S, Mapleson D, Woolfenden HC, Mohorianu I, Folkes L, Schwach F, Dalmay T, Moulton V (2012) The UEA sRNA workbench: a suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics 28:2059–2061
Stroud H, Greenberg MV, Feng S, Bernatavichute YV, Jacobsen SE (2013) Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152:352–364
Thatcher SR, Burd S, Wright C, Lers A, Green PJ (2015) Differential expression of miRNAs and their target genes in senescing leaves and siliques: insights from deep sequencing of small RNAs and cleaved target RNAs. Plant Cell Environ 38:188–200
Vandivier LE, Campos R, Kuksa PP, Silverman IM, Wang LS, Gregory BD (2015) Chemical modifications mark alternatively spliced and uncapped messenger RNAs in Arabidopsis. Plant Cell 27:3024–3037
Varshney RK, Nayak SN, May GD, Jackson SA (2009) Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol 27:522–530
Vazquez F, Vaucheret H, Rajagopalan R, Lepers C, Gasciolli V, Mallory AC, Hilbert JL, Bartel DP, Crete P (2004) Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol Cell 16:69–79
Voinnet O (2009) Origin, biogenesis, and activity of plant microRNAs. Cell 136:669–687
Wang H, Zhang X, Liu J, Kiba T, Woo J, Ojo T, Hafner M, Tuschl T, Chua NH, Wang XJ (2011) Deep sequencing of small RNAs specifically associated with Arabidopsis AGO1 and AGO4 uncovers new AGO functions. Plant J 67:292–304
Wei L, Gu L, Song X, Cui X, Lu Z, Zhou M, Wang L, Hu F, Zhai J, Meyers BC et al (2014) Dicer-like 3 produces transposable element-associated 24-nt siRNAs that control agricultural traits in rice. Proc Natl Acad Sci U S A 111:3877–3882
Wei W, Ba Z, Gao M, Wu Y, Ma Y, Amiard S, White CI, Rendtlew Danielsen JM, Yang YG, Qi Y (2012) A role for small RNAs in DNA double-strand break repair. Cell 149:101–112
Wendel JF, Jackson SA, Meyers BC, Wing RA (2016) Evolution of plant genome architecture. Genome Biol 17:37
Willmann MR, Berkowitz ND, Gregory BD (2014) Improved genome-wide mapping of uncapped and cleaved transcripts in eukaryotes--GMUCT 2.0. Methods 67:64–73
Winter D, Vinegar B, Nahal H, Ammar R, Wilson GV, Provart NJ (2007) An "electronic fluorescent pictograph" browser for exploring and analyzing large-scale biological data sets. PLoS One 2:e718
Wu L, Zhang Q, Zhou H, Ni F, Wu X, Qi Y (2009) Rice MicroRNA effector complexes and targets. Plant Cell 21:3421–3435
Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu C, Qi Y (2010) DNA methylation mediated by a microRNA pathway. Mol Cell 38:465–475
Xie Z, Allen E, Wilken A, Carrington JC (2005) DICER-LIKE 4 functions in trans-acting small interfering RNA biogenesis and vegetative phase change in Arabidopsis Thaliana. Proc Natl Acad Sci U S A 102:12984–12989
Xie Z, Johansen LK, Gustafson AM, Kasschau KD, Lellis AD, Zilberman D, Jacobsen SE, Carrington JC (2004) Genetic and functional diversification of small RNA pathways in plants. PLoS Biol 2:E104
Ye R, Chen Z, Lian B, Rowley MJ, Xia N, Chai J, Li Y, He XJ, Wierzbicki AT, Qi Y (2016) A Dicer-independent route for biogenesis of siRNAs that direct DNA methylation in Arabidopsis. Mol Cell 61:222–235
Yi X, Zhang Z, Ling Y, Xu W, Su Z (2015) PNRD: a plant non-coding RNA database. Nucleic Acids Res 43:D982–D989
Yoshikawa M, Peragine A, Park MY, Poethig RS (2005) A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. Genes Dev 19:2164–2175
Yu D, Meng Y, Zuo Z, Xue J, Wang H (2016b) NATpipe: an integrative pipeline for systematical discovery of natural antisense transcripts (NATs) and phase-distributed nat-siRNAs from de novo assembled transcriptomes. Sci Rep 6:21666
Yu X, Willmann MR, Anderson SJ, Gregory BD (2016a) Genome-wide mapping of uncapped and cleaved transcripts reveals a role for the nuclear mRNA cap-binding complex in Cotranslational RNA decay in Arabidopsis. Plant Cell 28:2385–2397
Yuan C, Meng X, Li X, Illing N, Ingle RA, Wang J, Chen M (2017) PceRBase: a database of plant competing endogenous RNA. Nucleic Acids Res 45:D1009–D1014
Zhai J, Zhang H, Arikit S, Huang K, Nan GL, Walbot V, Meyers BC (2015) Spatiotemporally dynamic, cell-type-dependent premeiotic and meiotic phasiRNAs in maize anthers. Proc Natl Acad Sci U S A 112:3146–3151
Zhai J, Zhao Y, Simon SA, Huang S, Petsch K, Arikit S, Pillay M, Ji L, Xie M, Cao X et al (2013) Plant microRNAs display differential 3′ truncation and tailing modifications that are ARGONAUTE1 dependent and conserved across species. Plant Cell 25:2417–2428
Zhang X, Clarenz O, Cokus S, Bernatavichute YV, Pellegrini M, Goodrich J, Jacobsen SE (2007) Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol 5:e129
Zhang X, Xia J, Lii YE, Barrera-Figueroa BE, Zhou X, Gao S, Lu L, Niu D, Chen Z, Leung C et al (2012) Genome-wide analysis of plant nat-siRNAs reveals insights into their distribution, biogenesis and function. Genome Biol 13:R20
Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW, Chen H, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE et al (2006) Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 126:1189–1201
Zhang X, Zhao H, Gao S, Wang WC, Katiyar-Agarwal S, Huang HD, Raikhel N, Jin H (2011) Arabidopsis Argonaute 2 regulates innate immunity via miRNA393(*)-mediated silencing of a Golgi-localized SNARE gene, MEMB12. Mol Cell 42:356–366
Zheng Q, Ryvkin P, Li F, Dragomir I, Valladares O, Yang J, Cao K, Wang LS, Gregory BD (2010) Genome-wide double-stranded RNA sequencing reveals the functional significance of base-paired RNAs in Arabidopsis. PLoS Genet 6:e1001141
Zhou M, Gu L, Li P, Song X, Wei L, Chen Z, Cao X (2010) Degradome sequencing reveals endogenous small RNA targets in rice (Oryza Sativa L. Ssp. Indica). Front Biol 5:67–90
Zhu H, Hu F, Wang R, Zhou X, Sze SH, Liou LW, Barefoot A, Dickman M, Zhang X (2011) Arabidopsis Argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell 145:242–256
Zilberman D, Cao X, Jacobsen SE (2003) ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 299:716–719
Acknowledgements
The authors would like to apologize to those whose original works could not be cited in this paper. This work was funded by the National Natural Science Foundation of China [31571349] and [31601062], Zhejiang Provincial Natural Science Foundation of China [LQ16C060003] and [LY15C060006], and Hangzhou Scientific and Technological Program [20170432B04].
Author information
Authors and Affiliations
Contributions
YM conceived the idea and prepared the manuscript, DY contributed to drafting and revising the manuscript, XM, ZZ and WS helped in preparing the tables, figures and formatting of the manuscript, HW reviewed and provided comments on the manuscript. All of the authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yu, D., Ma, X., Zuo, Z. et al. Bioinformatics resources for deciphering the biogenesis and action pathways of plant small RNAs. Rice 10, 38 (2017). https://doi.org/10.1186/s12284-017-0177-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-017-0177-y