
Abstract
Background
Three-line Oryza sativa (ssp. japonica) hybrids have been developed mainly using Chinsurah Boro II (BT)-type cytoplasmic male sterility (CMS). The Rf1 gene restores the fertility of BT-type CMS lines, and is the only fertility restorer gene (Rf) that has been used to produce three-line japonica hybrids. Using more Rf genes to breed BT-type restorer lines may broaden the genetic diversity of the restorer lines, and represents a viable approach to improve the heterosis level of BT-type japonica hybrids.
Results
We identified two major Rf genes from ‘93-11’ that are involved in restoring the fertility of BT-type CMS plants. These genes were identified from resequenced chromosome segment substitution lines derived from a cross between the japonica variety ‘Nipponbare’ and the indica variety ‘93-11’. Molecular mapping results revealed that these genes were Rf5 and Rf6, which are the Rf genes that restore fertility to Honglian-type CMS lines. The BT-type F1 hybrids with either Rf5 or Rf6 exhibited normal seed setting rates, but F1 plants carrying Rf6 showed more stable seed setting rates than those of plants carrying Rf5 under heat-stress conditions. Furthermore, the seed setting rates of F1 hybrids carrying both Rf5 and Rf6 were more stable than that of F1 plants carrying only one Rf gene.
Conclusion
Rf6 is an important genetic resource for the breeding of BT-type japonica restorer lines. Our findings may be useful for breeders interested in developing BT-type japonica hybrids.
Similar content being viewed by others

Background
Cytoplasmic male sterility (CMS) is caused by chimeric open reading frames in the mitochondrial genome, and is common in higher plants. It is a maternally inherited trait that results in an inability to produce functional pollen. Male sterility can be restored by the fertility restorer gene (Rf) in the nuclear genome (Hanson and Bentolila 2004). The CMS/Rf system has been widely used for hybrid seed production, and has helped to clarify the interactions between mitochondrial and nuclear genomes in plants (Chase 2007; Havey 2004).
The grain yields from three-line hybrid rice developed using the CMS/Rf system are 15–30% higher than those of inbred rice varieties (Fujimura et al. 1996; Yuan 1994). A CMS line, maintainer line, and a restorer line that carries the Rf gene are combined to develop three-line hybrids. For three-line hybrid rice, wild abortive (WA) and Honglian (HL) are the representative CMS types used for commercial indica hybrid seed production, and Chinsurah Boro II (BT) is the representative CMS type used for generating japonica hybrids (Chen and Liu 2014; Huang et al. 2014; Li et al. 2007; Yuan 1994). The BT-type cytoplasm has been identified from Oryza sativa ssp. indica Chinsurah Boro II and the BT-type CMS/Rf system is gametophytic. The BT-type CMS is caused by orf79 in mitochondria (Wang et al. 2006), and the recovery of pollen fertility is regulated by Rf1, a fertility restorer gene on chromosome 10 (Shinjyo 1975; Wang et al. 2006). The Rf1 gene has been cloned, and two alleles, Rf1a and Rf1b, have been identified at the Rf1 locus (Kazama and Toriyama 2003; Akagi et al. 2004; Komori et al. 2004; Wang et al. 2006). Rf1a and Rf1b both encode pentatricopeptide-repeat containing proteins. RF1A can promote endonucleolytic cleavage of the atp6–orf79 mRNA, whereas RF1B promotes degradation of atp6–orf79 mRNA (Wang et al. 2006). Although progress has been made toward characterizing the mechanisms underlying the development of BT-type CMS and the restoration of fertility by Rf genes, only Rf1 has been used to breed BT-type restorers. This has resulted in limited genetic diversity among BT-type restorers, and this is a major obstacle for the further development of BT-type japonica hybrids (Chen and Liu 2014). Thus, three-line japonica hybrids are not extensively cultivated in China (Deng et al. 2006). Also, F1 hybrids developed using the gametophytic CMS/Rf systems with multiple restorer gene loci can produce more than 50% normal pollen grains and exhibit good seed setting rates (Huang et al. 2012; Komori and Imaseki 2005), which may be useful for breeding BT-type japonica hybrids.
Several genetic loci related to the restoration of fertility in different types of CMS lines have been mapped in rice. Rf3 and Rf4 are two major fertility restorer genes for WA-type CMS, and have been mapped on chromosomes 1 and 10, respectively (Ahmadikhah and Karlov 2006; Tang et al. 2014; Zhang et al. 1997). Rf5 and Rf6, which have been cloned and mapped on chromosomes 10 and 8, respectively, are the two major fertility restorer genes for HL-type CMS (Hu et al. 2012; Huang et al. 2000, 2012, 2015). Rf17 (chromosome 4) and Rf2 (chromosome 2) are the fertility restorer genes for Chinese wild rice-type CMS (Fujii and Toriyama 2009) and Lead rice-type CMS (Itabashi et al. 2011), respectively. Identifying new Rf genes and/or using previously reported Rf genes (except Rf1) to breed BT-type japonica fertility restorers may accelerate the development and application of BT-type japonica hybrids. This potentially represents an effective way to increase rice yields in China.
In this study, we used 57 chromosome segment substitution lines (CSSLs) derived from a cross between the japonica variety ‘Nipponbare’ (NIP; recipient parent) and the indica restorer ‘93-11’ (donor parent) as males to detect Rf genes for BT-type CMS. To identify possible Rf genes for BT-type CMS in ‘93-11’, a population of 127 CSSLs from this cross were reconstructed and resequenced. We then mapped the detected Rf genes and evaluated their ability to restore fertility to BT-type japonica CMS lines. Our results will be useful for breeding BT-type japonica restorer lines and for the development of three-line japonica hybrids.
Results
Construction of CSSLs and whole-genome resequencing
Between 2004 and 2010, a set of CSSLs was developed via crossing and backcrossing with the aid of 140 molecular markers, and the genotypes of 56 CSSLs (L1–L56) were determined using a high-throughput resequencing strategy. Based on the resequencing data, 152 substituted segments derived from ‘93-11’ were identified. These segments covered about 89.27% of the rice genome (Zhang et al. 2011). We observed that most lines contained five or more introgressed chromosome segments with many gaps. To obtain additional lines with a single segment and a high coverage rate, we reconstructed the CSSLs from the previously generated lines using backcrosses and marker-assisted selection (MAS) with 357 polymorphic molecular markers distributed relatively evenly across the 12 rice chromosomes. We ultimately obtained 351 CSSLs, and 127 lines were selected for resequencing. According to the resequencing results, the 127 CSSLs harbored 362 substituted segments derived from line ‘93-11’. The segments were 0.02–24.60 Mb long (average length: 5.39 Mb). For each CSSL, the number of substituted chromosomal segments varied from one to seven. Ninety-seven CSSLs contained two or fewer introgressed chromosomal segments, with 53 lines carrying only one segment. The total length of these substituted segments was 2023.8 Mb, which was 5.08 times the length of the rice genome. Furthermore, the CSSLs covered 94.93% of the ‘93-11’ genome (Table 1).
Two major genes restore fertility of BT-type CMS lines
In the summer of 2011, the seed setting rate of 52 testcross F1 lines was analyzed. Six F1 hybrids derived from crosses between NIPA, a BT-type japonica CMS line with the nuclear background of NIP and L1, L18–20, L30, and L47 exhibited bagged and natural seed setting rates of 9.16–63.46% and 45.74–92.67%, respectively (Table 2). The other F1 hybrids showed bagged seed setting rates of zero and extremely low natural seed setting rates (i.e., < 5%). Therefore, we considered that these lines were all sterile and the natural seed setting rates of these lines were caused by outcrossing.
To detect quantitative trait loci (QTLs) associated with the fertility restoration of NIPA, we constructed a bin map based on 56 introgression lines, two parents, and the high-throughput resequencing data. A total of 366 bins were generated, with sizes of 0.01–11.92 Mb. The QTL analysis was conducted using the SARS program, and three QTLs for natural spikelet fertility were detected (Table 3). Among these QTLs, qSF8-1 was located in bin 222 on chromosome 8, qSF10-1 was located in bin 314 on chromosome 10, and qSF12-1 was located in bin 349 on chromosome 12. Bin 222 was present in L1, L18, L19, and L20, while L30 and L47 harbored bin 314, and L47 harbored bin 349 (Table 3). The QTL mapping results revealed that bins 349 and 314 were present in L47. The chromosome interval related to bin 314 covered the Rf1 locus (Akagi et al. 1996; Komori et al. 2004; Wang et al. 2006). Thus, we considered the possibility that qSF12-1 does not exist.
Because of the gametophytic manner in which Rf genes restore fertility in the BT-type CMS/Rf system, plants with the rfrf genotype were not present in the F2 populations. Therefore, most plants likely carried homozygous ‘93-11’ or heterozygous genotypes at the linkage marker loci if there was only one Rf gene that restored the fertility of BT-type CMS. Plants carrying the homozygous NIPA genotype were considered to be recombinants. In the spring of 2012, six F2 populations derived from the fertile F1 hybrids were planted, with each population consisting of 100 individuals. We developed five simple sequence repeat markers on chromosome 8 (1–688,039 bp) and two insertion/deletion markers on chromosome 10 (16,949,055–18,486,880 bp). The markers were used to detect polymorphisms between NIP and ‘93-11’. RM1019 and RM407 on chromosome 8 and STS10-16 and STS10-27 on chromosome 10 were determined to be polymorphic. RM407 was subsequently used to detect the genotypes of plants in the F2 populations derived from the following crosses: NIPA/L1, NIPA/L18, NIPA/L19, and NIPA/L20. All 400 analyzed plants carried homozygous ‘93-11’ or heterozygous genotypes. STS10-16 and STS10-27 were used to detect the genotypes of plants in the F2 populations derived from the NIPA/L30 and NIPA/L47 crosses, respectively. Almost all of the 200 examined plants exhibited homozygous ‘93-11’ or heterozygous genotypes. The exception was one plant that carried a homozygous NIPA genotype at STS10-27. These results indicated that all of L1, L18–20, L30, and L47 carried only one Rf gene, and that qSF8-1 and qSF10-1 were two major QTLs conferring the ability to restore fertility to BT-type CMS lines.
In the summer of 2014, natural spikelet fertility was assessed in 123 F1 lines derived from the cross between NIPA and the reconstructed CSSLs. Among the F1 lines, the populations generated from crosses between NIPA and four CSSLs (i.e., N91, N93, N116, and N119) exhibited natural seed setting rates of 90.95–94.51%, and the other lines were sterile. According to the high-throughput resequencing data, N91 and N93 carried a substituted segment covering qSF8-1, whereas N116 and N119 carried a substituted segment spanning qSF10-1. These results were confirmed by molecular detection with RM407, STS10-16, and STS10-27. Based on these results, we concluded that there were two non-allelic nuclear genes in ‘93-11’ that restored the fertility of BT-type CMS japonica lines.
Molecular mapping of qSF10-1 and qSF8-1
qSF10-1 is located in a region that includes the Rf1 locus, which contains Rf1a, or is tightly linked with Rf1a and Rf1b on chromosome 10 in BT-type japonica restorers (Akagi et al. 1996; Komori et al. 2004; Wang et al. 2006). Rf5, which is a dominant Rf gene associated with HL-type CMS in indica rice, is the same gene as Rf1a (Hu et al. 2012). To determine whether qSF10-1 was Rf1a (Rf5), we sequenced the Rf1a allele in ‘93-11’, L37, and L47. Comparison of the nucleotide sequences of Rf1a and Rf5 with those previously cloned from different restorers revealed that the sequence of the Rf1a allele from ‘93-11’is the same as the sequences of the corresponding genes in IR8 (Akagi et al. 2004) and Miyang 23 (Hu et al. 2012). This result confirmed that qSF10-1 is Rf1a (Rf5).
To clarify the precise genomic position of qSF8-1, we developed 51 markers in the region containing RM407, and 14 polymorphic markers were obtained for mapping (Additional file 1: Table S1). In the summer of 2012, two markers (i.e., RM1019 and RM22271) were used to screen recombinant individuals from more than 4000 plants in the F2 and F3 populations. We identified 23 and 17 recombinants using RM1019 and RM22271, respectively. Another four markers (i.e., STS8-4, STS8-23, RM407, and STS8-32) were used to genotype the 40 recombinants. Among these recombined plants, two were detected by RM407 and one was detected by STS8-32. Thus, based on the reference sequence of the NIP genome, qSF8-1 was localized to an approximately 35.5-kb region between the markers RM407 and STS8-32 on the short arm of chromosome 8, within which an open reading frame (ORF) (Os08g0110200) encoding PPR-containing protein might be the candidate gene for qSF8-1. We sequenced this ORF from 93-11 to L18, respectively. The nucleotide sequences were identical between 93-11 and L18, and 93-11 contains 16 nucleotide substitutions and one insertions of 2 bp at position +2315 compared with that of NIP. During our mapping work, Huang et al. (2012) reported the mapping results of the Rf6 gene, which is also from ‘93-11’, and restores the fertility of HL-type indica CMS lines. Our mapping study essentially produced the same results. Furthermore, the cloning of Rf6 indicated that Rf6 is the ORF of Os08g0110200 and is also capable of restoring the fertility of BT-type CMS plants (Huang et al. 2015). Therefore, we concluded that qSF8-1 is Rf6.
Ability of Rf6 to restore fertility is stable and two non-allelic restorer genes enhance heat stress tolerance in F1 hybrids
Among the samples sown in the field on May 10 (S1) in Yangzhou, most of the testcross F1 lines from the CSSLs headed on August 6–9, similar to the heading dates of NIP plants. In 2011, of the six fertile testcross F1 lines, the NIPA/L30 F1 plants headed on August 24. The daily maximum temperature during the flowering period was 24.0–28.0 °C (Additional file 2: Figure S1a), which was lower than the temperature during the flowering periods for the five other testcross F1 lines. The NIPA/L30 F1 plants exhibited a low seed setting rate (Table 2). These results indicated that exposure to low temperatures may influence the seed setting rates of F1 plants. Considering the temperature fluctuations during the flowering period of the testcross F1 lines, two sowing dates were used in 2012 [May 10 (S1) and May 20 (S2)], and 2013 [May 10 (S1) and June 5 (S3)]. In 2012, the testcross F1 plants and the F2 plants with different genotypes headed on August 6 and August 9, respectively. They also exhibited normal spikelet fertility (i.e., > 80%), indicating that Rf5 and Rf6 were able to restore fertility to BT-type CMS lines (Table 4). In 2013, the F1 plants and F3 plants carrying different genotypes headed on August 6 and August 20, respectively. The highest temperature (i.e., 37.0 °C) was first recorded on August 6, and high daytime temperatures continued for more than 1 week (Additional file 2: Figure S1b). Among the plants that headed on August 6, the seed setting rates of plants carrying heterozygous genotypes were relatively low, while the plants carrying homozygous genotypes exhibited normal seed setting rates (Table 4). In contrast, the seed setting rate was normal for all plants that headed on August 20. These results implied that heat stress affects the ability of Rf5 and Rf6 to restore fertility to BT-type CMS plants.
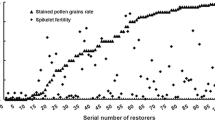
To further evaluate the ability of F1 hybrids with one or two Rf genes to adapt to heat stress, we developed the following three F1 populations: NIPA/L18 (Rf6rf6), NIPA/L47 (Rf5rf5), and (NIPA/L18) F2//L47 (Rf5rf5Rf6rf6). We also selected the plants harboring homozygous genotypes in the NIPA/L18 and NIPA/L47 F4 populations, which were exposed to high temperatures (25 °C nights, and maximum daytime temperatures of 37 °C) in 2014. These plants headed on August 6 when grown in the field, and on August 1 when grown in the greenhouse. Compared with the untreated plants, the F1 hybrids with only one Rf gene exhibited poorer seed setting rates, and the seed setting rate of plants with Rf6 was higher than that of plants with Rf5 (Fig. 1). In contrast, F1 plants carrying two restorer genes and plants with homozygous genotypes at Rf loci exhibited normal seed setting rates (Fig. 1). These results revealed that the presence of two non-allelic restorer genes in F1 hybrids increased the stability of their seed setting rate during exposure to environmental stresses such as high temperature.

Effects of high temperatureson seed setting rates of plants harboring different genotypes. Error bars of seed setting rate is the mean value ± SD (n = 10). The heat stress is treated with the maximum temperatures of 37 °C
Discussion
Three-line japonica hybrids developed mainly with BT-type CMS lines have contributed greatly to rice production in China (Huang et al. 2014; Li et al. 2007). The BT-type CMS lines can be developed by nuclear substitution using japonica varieties from Japan and China as the recurrent paternal parent in backcrosses. Most BT-type japonica restorers contain only the Rf1 locus (Akagi et al. 1996; Chen and Liu 2014; Huang et al. 2014; Komori et al. 2004; Wang et al. 2006), which was first transferred from indica varieties cultivated in Southeast Asia (Li et al. 2007). The F1 hybrids derived from BT-type japonica restorers exhibit a normal seed setting rate, with 50% of the produced pollen grains being fertile. Because BT-type japonica restorers contain only Rf1, the genetic diversity of restorers is relatively low. Additionally, the systematic use of heterosis for breeding BT-type japonica hybrid rice has been limited. Therefore, exploiting other Rf genes may be an effective way to increase the genetic diversity of BT-type restorers, which will likely enhance the development of japonica hybrids in China.
In this study, two Rf genes (i.e., qSF8-1 and qSF10-1) involved in restoring the fertility of NIPA plants were identified from ‘93-11’, which is an elite indica fertility restorer for HL-type CMS. Rf5 and Rf6, the two non-allelic fertility restorer genes in ‘93-11’ associated with HL-type CMS, have been mapped and cloned (Huang et al. 2012, 2015). Rf5 and Rf1a are the same gene, and Rf6 is also capable of restoring the fertility of BT-type CMS plants (Huang et al. 2015). We mapped qSF10-1 to a region on chromosome 10 containing Rf5 (Rf1a) and mapped qSF8-1 to a region on chromosome 8 containing Rf6. The ‘93-11’ Rf5 and Rf6 allele were sequenced. The sequencing results confirmed that qSF10-1 is Rf5 (Rf1a) and qSF8-1 is Rf6. Therefore, the two ‘93-11’ Rf genes associated with BT-type CMS are Rf5 and Rf6. Indeed, rice breeding experiments have revealed similarities in the restoration and maintenance relationship between BT-type CMS and HL-type CMS (Li et al. 2007; Zhang et al. 2016), consistent with the mapping results of this study.
The Rf5 and Rf6 genes exhibit similar abilities to restore the fertility of HL-type indica CMS lines (Huang et al. 2012). We observed that only a major gene (i.e., Rf5 or Rf6) can restore normal fertility to BT-type CMS lines. However, this study revealed for the first time that the ability to restore fertility to BT-type japonica CMS lines is more stable with Rf6 than with Rf5 under heat-stress conditions. In the HL-type CMS/Rf system, Rf6 and Rf5 function via distinct mechanisms to rescue the sterility of HL-type CMS (Huang et al. 2012, 2015). Therefore, we hypothesize that the molecular mechanism underlying the fertility restoration of BT-type CMS plants differs between Rf5 and Rf6, leading to the observed differences in the restorer activities of Rf5 and Rf6. Additional studies are required to test this hypothesis.
In this study, F2 plants harboring Rf5Rf5 or Rf6Rf6 and plants harboring Rf5rf5Rf6rf6 in the F1 population of a three-way cross exhibited the most stable fertility levels. These observations imply that breeding BT-type japonica restorers with multiple dominant Rf genes may increase the stability of seed setting in BT-type hybrid japonica rice. Therefore, using Rf6 to restore fertility to BT-type CMS plants may facilitate the exploitation of heterosis in japonica breeding.
Conclusion
We determined that Rf5 and Rf6 are the major Rf genes associated with BT-type CMS, and that the restorer activity of Rf6 is more stable than that of Rf5 under heat-stress conditions. The Rf6 gene in ‘93-11’ may be an important resource for breeding BT-type japonica restorers to broaden the genetic diversity of BT-type rice. Also, combining Rf6 with Rf5 (Rf1a) in most pre-existing BT-type restorers may be an effective way to breed new BT-type restorers that exhibit stable seed setting abilities.
Methods
Plant materials
The donor parent used in this study was ‘93-11’, which is a typical indica cultivar and fertility restorer for BT-type and HL-type CMS. The recipient parent was NIP, which is a japonica cultivar and maintainer for BT-type CMS. The genomes of both parents have been sequenced. Between 2004 and 2010, a set of CSSLs derived from the cross between NIP and ‘93-11’ was developed by crossing and backcrossing with MAS, of which 56 lines were genotyped in 2010 using a resequencing method (Zhang et al. 2011). Based on the resequencing results, another 127 CSSLs from the same cross were further developed by backcrossing with MAS and then genotyped by resequencing (Additional file 3: Figure S2). In the spring of 2011 and 2014, these CSSLs were used as males for crosses with ‘NipponbareA’ (NIPA), which has the BT-type sterile cytoplasm with the nuclear background of NIP. We generated 52,123 F1 hybrids.
In 2011 and 2012, several F2 and F3 populations were generated from plants harboring the heterozygous genotype for Rf genes in the NIPA/CSSL testcross population. The corresponding progenies were used for the fine mapping of genes. To evaluate the ability of identified Rf genes to restore the fertility of BT-type CMS lines, we crossed NIPA with the CSSLs carrying various Rf genes. Plants harboring the homozygous or heterozygous genotypes for Rf genes in the F2–F4 populations were identified by MAS between 2012 and 2014. Plants harboring the homozygous genotype for Rf genes were crossed with each other in 2013 and 2014.
Field experiment
All plant materials were sown on three dates [i.e., May 10 (S1), May 20 (S2), and June 5 (S3)] between 2011 and 2014 at the experimental field of Yangzhou University in Yangzhou, Jiangsu, China. Additionally, in the summer of 2014, plants carrying different genotypes for Rf genes were exposed to heat stress (25 °C nights, and maximum daytime temperatures of 37 °C) in a greenhouse for 10 days during the flowering period. Each field plot consisted of four rows separated by 25 cm, with each row consisting of five plants separated by 20 cm. Plants were grown using normal rice cultivation practices.
Fertility scoring
In 2011, we analyzed the spikelet fertility in natural and bagged panicles of five plants from the CMS lines and each line in the testcross population. Between 2012 and 2014, only the spikelet fertility in natural panicles of five plants was assessed in the testcross F1 population, F2–F4 populations, and the three-way-cross F1 population. We counted the filled and unfilled grains of two panicles from one plant harvested at 20 days after flowering, and the spikelet fertility of one plant was measured as the average seed setting rate.
Genotyping CSSLs by whole-genome resequencing
We used the high-throughput genotyping method developed by Huang et al. (2009). The CSSLs were genotyped based on single nucleotide polymorphisms (SNPs) generated from whole-genome resequencing as previously described (Xu et al. 2010). Briefly, at least 5 μg genomic DNA from each sample was randomly fragmented by sonication and electrophoretically size-fractionated. The DNA fragments approximately 500 bp long were then purified. Adapters were ligated to the purified fragments, which were then clustered and sequenced using the Illumina HiSeq 2000 system according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). According to the published NIP genome sequence (http://rgp.dna.affrc.go.jp/IRGSP/Build4/build4.html), the detected SNPs were arranged along the chromosomes according to their physical locations. The genotypes of these CSSLs were evaluated by a group of consecutive SNPs using a sliding window approach, and different window sizes were used according to the SNP density of the CSSLs. The physical length of the substituted segments in each CSSL was estimated based on the resequencing results for each CSSL. The overlapping donor segments between or among different lines were used to divide the fragments into smaller segments (i.e., bins).
Quantitative trait locus mapping and data analysis
Statistical analyses of the fertility of the plant materials and populations were conducted using the analysis of variance package in MATLAB (version 7.0). Commonly used bin mapping schemes were constructed and the contributions of the target bins to phenotypic variability were determined as described by Xu et al. (2010). The nomenclature of QTLs for natural spikelet fertility was according to the standard procedure described elsewhere (McCouch et al. 2002).
DNA extraction, polymerase chain reaction, and sequencing
Genomic DNA was isolated from fresh leaves using cetyltrimethylammonium bromide (Rogers and Bendich 1985). Simple sequence repeat markers were identified using the Gramene database (http://www.gramene.org/). New insertion/deletion markers for the NIP (japonica) and ‘93-11’ (indica) genome sequences (http://www.ncbi.nlm.nih.gov/) were identified using the BLAST online tool. Primers were synthesized by Shanghai Sangon Inc. (Shanghai, China). Molecular marker analyses were conducted in 1× reaction buffer containing 0.1 mM dNTP, 1.0 U Taq polymerase, 0.2 μM primer, and 20 ng template DNA. The final volume was adjusted to 20 μL with ultra-pure water. The polymerase chain reaction (PCR) program was as follows: 94 °C for 4 min; 30 cycles of 94 °C for 45 s, 55 °C for 45 s, and 72 °C for 50 s; with final extension at 72 °C for 5 min. The amplification products were separated by 3% (w/v) agarose gel electrophoresis, detected with ethidium bromide, and visualized with a Gel Doc 1000 system (Bio-Rad Laboratories, Hercules, CA, USA).
According to the mapping results, the Rf1a allele and Rf6 allele in ‘93-11’ were sequenced. The gene fragments were amplified by PCR using PrimeSTAR® GXL DNA polymerase (Takara, Dalian, China). The PCR products were purified using a TIANGEN PCR purification kit, and then ligated into the pEASY-Blunt cloning vector. The plasmid DNA was sequenced by GENEWIZ (Suzhou, China), and the correct DNA sequence was identified by comparing five plasmids. Sequence alignment was conducted with BLAST tools provided by the National Center for Biotechnology Information. The primers used to sequence the Rf1a and Rf6 allele are listed in Additional file 1: Table S1.
Accession number
Sequence data generated in this study have been deposited in the GenBank/EMBL database (accession numbers [KX517796, KY387609]).
Abbreviations
- BT:
-
Chinsurah Boro II
- CMS:
-
Cytoplasmic male sterility
- CSSL:
-
Chromosome segment substitution lines
- HL:
-
Honglian
- Nipponbare:
-
NIP
- NipponbareA:
-
NIPA
- PCR:
-
Polymerase chain reaction
- QTLs:
-
Quantitative trait loci
- Rf :
-
Fertility restorer gene
References
Ahmadikhah A, Karlov GI (2006) Molecular mapping of the fertility-restoration gene Rf4 for WA-cytoplasmic male sterility in rice. Plant Breed 125:363–367
Akagi H, Yokozeki Y, Inagaki A, Nakamura A, Fujimura T (1996) A codominant DNA marker closely linked to the rice nuclear restorer gene, Rf1, identified with inter-SSR fingerprinting. Genome 39:1205–1209
Akagi H, Nakamura A, Yokozeki-Misono Y, Inagaki A, Takahashi H, Mori K, Fujimura T (2004) Positional cloning of the rice Rf-1 gene, a restorer of BT-type cytoplasmic male sterility that encodes a mitochondria-targeting PPR protein. Theor Appl Genet 108:1449–1457
Chase CD (2007) Cytoplasmic male sterility: a window to the world of plant mitochondrial–nuclear interactions. Trends Genet 23:81–90
Chen LT, Liu YG (2014) Male sterility and fertility restoration in crops. Annu Rev Plant Biol 65:579–606
Deng HF, He Q, Shu F, Zhang WH, Yang F, Jing YH, Dong L, Xie H (2006) Status and technical strategy on development of japonica hybrid rice in china. Hybrid Rice 21:1–6
Fujii S, Toriyama K (2009) Suppressed expression of retrograde-regulated male sterility restores pollen fertility in cytoplasmic male sterile rice plants. Proc Natl Acad Sci U S A 106:9513–9518
Fujimura T, Akagi H, Oka M, Nakamura A, Sawada R (1996) Establishment of a rice protoplast culture and application of an asymmetric protoplast fusion technique to hybrid rice breeding. Plant Tissue Cult Lett 13:243–247
Hanson MR, Bentolila S (2004) Interactions of mitochondrial and nuclear genes that affect male gametophytic development. Plant Cell 16:S154–S169
Havey M (2004) The use of cytoplasmic male sterility for hybrid seed production. In: Daniell H, Chase C (eds) Molecular biology and biotechnology of plant organelles. Springer Publishers, Berlin, pp 617–628
Hu J, Wang K, Huang WC, Liu G, Gao Y, Wang JM, Huang Q, Ji YX, Qin XJ, Wan L, Zhu RS, Li SQ, Yang DC, Zhu YG (2012) The rice pentatricopeptide repeat protein RF5 restores fertility in Hong-Lian cytoplasmic male-sterile lines via a complex with the glycine-rich protein GRP162. Plant Cell 24:109–122
Huang QY, He YQ, Jing RC, Zhu RS, Zhu YG (2000) Mapping of the nuclear fertility restorer gene for HL cytoplasmic male sterility in rice using microsatellite markers. Chin Sci Bull 45:430–432
Huang XF, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, Guan JP, Fan DL, Weng QJ, Huang T, Dong GJ, Sang T, Han B (2009) High-throughput genotyping by whole-genome resequencing. Genome Res 19:1068–1076
Huang WC, Hu J, Yu CC, Huang Q, Wan L, Wang LL, Qin XJ, Ji YX, Zhu RS, Li SQ, Zhu YG (2012) Two non-allelic nuclear genes restore fertility in a gametophytic pattern and enhance abiotic stress tolerance in the hybrid rice plant. Theor Appl Genet 124:799–807
Huang JZ, E ZG, Zhang HL, Shu QY (2014) Workable male sterility systems for hybrid rice: Genetics, biochemistry, molecular biology, and utilization. Rice 7:1–14
Huang WC, Yu CC, Hu J, Wang LL, Dan ZW, Zhou W, He CL, Zeng YF, Yao GX, Qi JZ, Zhang ZH, Zhu RS, Chen XF, Zhu YG (2015) Pentatricopeptide-repeat family protein RF6 functions with hexokinase 6 to rescue rice cytoplasmic male sterility. Proc Natl Acad Sci U S A 112:14984–14989
Itabashi E, Iwata N, Fujii S, Kazama T, Toriyama K (2011) The fertility restorer gene, Rf2, for Lead Rice-type cytoplasmic male sterility of rice encodes a mitochondrial glycine-rich protein. Plant J 65:359–367
Kazama T, Toriyama K (2003) A pentatricopeptide repeat-containing gene that promotes the processing of aberrant atp6 RNA of cytoplasmic male-sterile rice. FEBS Lett 544:99–102
Komori T, Imaseki H (2005) Transgenic rice hybrids that carry the Rf-1 gene at multiple loci show improved fertility at low temperature. Plant Cell Environ 28:425–431
Komori T, Ohta S, Murai NY, Kuraya Y, Suzuki S, Hiei Y (2004) Map-based cloning of a fertility restorer gene, Rf-1, in rice (Oryza sativa L.). Plant J 37:315–325
Li SQ, Yang DC, Zhu YG (2007) Characterization and use of male sterility in hybrid rice breeding. J Integr Plant Biol 49:791–804
McCouch S, Teytelman L, Xu Y, Lobos K, Clare K, Walton M, Fu B, Maghirang R, Li Z, Xing Y, Zhang Q, Kono I, Yano M, Fjellstrom R, DeClerck G, Schneider D, Cartinhour S, Ware D, Stein L (2002) Development and mapping of 2240 new SSR markers for ricee (Oryza sativa L.). DNA Res 9:199–207
Rogers SO, Bendich AJ (1985) Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant tissues. Plant Mol Biol 5:69–76
Shinjyo C (1975) Genetical studies of cytoplasmic male sterility and fertility restoration in rice (Oryza sativa L). Sci Bull Coll Agric Univ Ryukyus 22:1–57
Tang HW, Luo DP, Zhou DG, Zhang QY, Tian DS, Zheng XM, Chen LT, Liu YG (2014) The rice restorer Rf4 for wild-abortive cytoplasmic male sterility encodes a mitochondrial-localized PPR protein that functions in reduction of WA352 transcripts. Mol Plant 7:1497–1500
Wang ZH, Zou YJ, Li XY, Zhang QY, Chen LT, Wu H, Su DH, Chen YL, Guo JX, Luo D, Long YM, Zhong Y, Liu YG (2006) Cytoplasmic male sterility of rice with boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing. Plant Cell 18:676–687
Xu JJ, Zhao Q, Du PN, Xu CW, Wang BH, Feng Q, Liu QQ, Tang SZ, Gu MH, Han B, Liang GH (2010) Developing high throughput genotyped chromosome segment substitution lines based on population whole-genome re-sequencing in rice (Oryza sativa L.). BMC Genomics 11:656
Yuan LP (1994) Increasing yield potential in rice by exploitation of heterosis. In: Virmanni SS (ed) Hybrid rice technology. New developments and future prospects. IRRI, Manila, pp 1–6
Zhang G, Lu Y, Bharaj TS, Virmani SS, Huang N (1997) Mapping of the Rf-3 nuclear fertility-restoring gene for WA cytoplasmic male sterility in rice using RAPD and RFLP markers. Theor Appl Genet 94:27–33
Zhang H, Zhao Q, Sun ZZ, Zhang CQ, Feng Q, Tang SZ, Liang GH, Gu MH, Han B, Liu QQ (2011) Development and high-throughput genotyping of substitution lines carring the chromosome segments of indica 9311 in the background of japonica Nipponbare. J Genet Genomics 38:603–611
Zhang HG, Zhang LJ, Si H, Ge YS, Liang GH, Gu MH, Tang SZ (2016) Rf5 is able to partially restore fertility to Honglian-type cytoplasmic male sterile japonica rice (Oryza sativa) lines. Mol Breed 36:1–10
Acknowledgments
This study was financially supported by the National Key Research and Development Program (2016YFD0101107), the National Natural Science Foundation (31071384), and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Authors’ contributions
HZ analyzed the data and drafted the manuscript. JC and YG completed the phenotypic evaluations and data analyses. YP and LZ helped construct the populations. QL and MG were involved in designing the study. ST designed the study and revised the manuscript. All authors have read and approved the final manuscript.
Authors’ information
HZ is a lecturer at Yangzhou University (China), JC, YG and LZ are Masters students at Yangzhou University (China), and QL, MG, and ST are professors at Yangzhou University (China).
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Newly developed markers and primers used for gene mapping and sequencing. (DOCX 14 kb)
Additional file 2: Figure S1.
Temperatures during August in 2011 (a) and 2013 (b). (DOCX 132 kb)
Additional file 3: Figure S2.
Breeding strategy to produce chromosome segment substitution lines (CSSLs). Plant genotypes identified by marker-assisted selection (MAS) are indicated. (DOCX 126 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, H., Che, J., Ge, Y. et al. Ability of Rf5 and Rf6 to Restore Fertility of Chinsurah Boro II-type Cytoplasmic Male Sterile Oryza Sativa (ssp. Japonica) Lines. Rice 10, 2 (2017). https://doi.org/10.1186/s12284-017-0142-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-017-0142-9