
Abstract
Infection and inflammation account for approximately 25% of cancer-causing factors. Inflammation-related cancers are characterized by mutagenic DNA lesions, such as 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG) and 8-nitroguanine. Our previous studies demonstrated the formation of 8-oxodG and 8-nitroguanine in the tissues of cancer and precancerous lesions due to infection (e.g., Opisthorchis viverrini-related cholangiocarcinoma, Schistosoma haematobium-associated bladder cancer, Helicobacter pylori-infected gastric cancer, human papillomavirus-related cervical cancer, Epstein-Barr virus-infected nasopharyngeal carcinoma) and pro-inflammatory factors (e.g., asbestos, nanomaterials, and inflammatory diseases such as Barrett’s esophagus and oral leukoplakia). Interestingly, several of our studies suggested that inflammation-associated DNA damage in cancer stem-like cells leads to cancer development with aggressive clinical features. Reactive oxygen/nitrogen species from inflammation damage not only DNA but also other biomacromolecules, such as proteins and lipids, resulting in their dysfunction. We identified oxidatively damaged proteins in cancer tissues by 2D Oxyblot followed by MALDI-TOF/TOF. As an example, oxidatively damaged transferrin released iron ion, which may mediate Fenton reactions and generate additional reactive oxygen species. Dysfunction of anti-oxidative proteins due to this damage might increase oxidative stress. Such damage in biomacromolecules may form a vicious cycle of oxidative stress, leading to cancer development. Epigenetic alterations such as DNA methylation and microRNA dysregulation play vital roles in carcinogenesis, especially in inflammation-related cancers. We examined epigenetic alterations, DNA methylation and microRNA dysregulation, in Epstein-Barr virus-related nasopharyngeal carcinoma in the endemic area of Southern China and found several differentially methylated tumor suppressor gene candidates by using a next-generation sequencer. Among these candidates, we revealed higher methylation rates of RAS-like estrogen-regulated growth inhibitor (RERG) in biopsy specimens of nasopharyngeal carcinoma more conveniently by using restriction enzyme-based real-time PCR. This result may help to improve cancer screening strategies. We profiled microRNAs of nasopharyngeal carcinoma tissues using microarrays. Quantitative RT-PCR analysis confirmed the concordant downregulation of miR-497 in cancer tissues and plasma, suggesting that plasma miR-497 could be used as a diagnostic biomarker for nasopharyngeal carcinoma. Chronic inflammation promotes genetic and epigenetic aberrations, with various pathogeneses. These changes may be useful biomarkers in liquid biopsy for early detection and prevention of cancer.
Similar content being viewed by others


Background
It is estimated that infectious diseases and chronic inflammation account for approximately 25% of cancer-causing factors [1]. Under chronic inflammation, reactive oxygen/nitrogen species (ROS/RNS) are produced from not only inflammatory cells but also epithelial cells [2]. ROS/RNS cause DNA damage in organs under inflammation, leading to cancer development. We demonstrated the importance of mutagenic DNA lesions, especially 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG, also known as 8-hydroxy dG; 8-OHdG) and 8-nitroguanine, as a common cancer-causing molecular mechanism [3].
The Khon Kaen region, in the northeastern part of Thailand, is a local endemic area for liver fluke (Opisthorchis viverrini; OV) infection and its related cholangiocarcinoma. OV infection is considered “carcinogenic to humans” (group 1) by the International Agency for Research on Cancer (IARC). In a collaboration with Khon Kaen University, Thailand, we firstly reported 8-nitroguanine and 8-oxodG formation in the livers of hamsters treated orally with the liver fluke in an animal model for OV-related cholangiocarcinoma [4]. In our previous studies based on DNA lesions, we investigated the molecular mechanisms of cancers due to infection (e.g., OV-related cholangiocarcinoma [2, 4,5,6], Schistosoma haematobium-associated bladder cancer [7, 8], Helicobacter pylori-infected gastric cancer [9], human papillomavirus-related cervical cancer [10], Epstein-Barr (EB) virus-infected nasopharyngeal carcinoma [11]), and pro-inflammatory factors (e.g., asbestos [12, 13], nanomaterials [14,15,16,17], and inflammatory diseases such as Barrett’s esophagus [18, 19], and oral leukoplakia [20]). In addition, inflammation plays a role in the epigenetic alterations in cancer. In this paper, we review the roles of RNS-/ROS-mediated DNA damage and epigenetic changes in inflammation-related carcinogenesis.
Oxidative and nitrative damage of biomacromolecules in inflammation-related carcinogenesis
DNA damage
In our early studies on inflammation-related carcinogenesis, we detected the formation of 8-nitroguanine and 8-oxodG in inflammatory and cancer tissues by immunohistochemistry (IHC) and an electrochemical detector coupled to HPLC (HPLC-ECD). HPLC-ECD analyses also demonstrated that urinary 8-oxodG levels were significantly higher in OV-related cholangiocarcinoma patients than in OV-infested patients and healthy subjects and higher in OV-infested patients than in healthy subjects [21]. The 8-oxodG levels of OV-infested patients significantly decreased 2 months after anti-parasite therapy and were comparable with the levels in healthy subjects 1 year after treatment. Therefore, urinary 8-oxodG can be used as a convenient biomarker of inflammation-related cancer.
Various death signals including ROS and DNA damage induce apoptosis via the mitochondrial pathway. Melatonin is a candidate chemopreventive agent for inhibiting cholangiocarcinoma development. The mechanisms of anti-cancer activity by melatonin remain unclear. We examined the induction of apoptosis in human cholangiocarcinoma cell lines treated with melatonin [22]. Flowcytometric analyses revealed that melatonin increased intracellular ROS levels and induced apoptosis. Immunocytochemistry showed melatonin-induced mitochondrial DNA damage by double staining of 8-oxodG and mitoTracker (a mitochondria-specific fluorescent staining dye). Cancer cells are in a state of high oxidative stress due to their elevated metabolism, and the additional ROS from melatonin may break the balance of anti- and pro-oxidants and damage mitochondrial DNA, leading to apoptosis. The findings offer useful information for chemoprevention and therapeutic strategies.
Chronic inflammation causes various types of damage to nucleic acids, proteins, and lipids via ROS/RNS generation, resulting in tissue damage. The tissue injury may activate progenitor/stem cells for tissue regeneration. Stem cells are damaged by ROS/RNS from inflammation, and the resulted mutations can accumulate, which could generate cancer stem cells [3]. In OV-related cholangiocarcinoma patients, positive staining of stem cell markers, CD133 and/or Oct3/4, showed a significantly higher amount of 8-oxodG than in negative-stained cases, and these positive cases had poor prognoses. Inflammation-associated DNA damage in cancer stem-like cells may thus lead to cholangiocarcinoma development with aggressive clinical features [23].
We established a hydrogen peroxide-resistant cholangiocyte cell line, as a novel model of oxidative stress-related cholangiocarcinoma genesis [24]. Parental immortalized cholangiocyte cells (MMNK1) were treated with a relatively low concentration of hydrogen peroxide (25 μM H2O2) every day for more than 2 months, to be H2O2-resistant cells (ox-MMNK1-L, IC50 for H2O2 350 μM; parental MMNK1 cells, IC50 for H2O2 75 μM). The resistant cells grew more rapidly and had higher expressions of anti-oxidant enzymes such as catalase than the original parental cells. Recently, we found downregulation of early B cell factor 1 (EBF1), a tumor suppressor gene, in cholangiocarcinoma-cultured cells and ox-MMNK1-L cells compared to normal cholangiocyte cells (MMNK1) [25]. Interestingly, EBF1 knockdown in MMNK1 cells upregulated CD133 and Oct3/4 expression with a higher ability of cell migration. EBF1 downregulation was also found in OV-related cholangiocarcinoma tissues. Cholangiocarcinoma patients with low EBF1 expression and high formation of 8-oxodG had poor survival. Thus, prolonged oxidative stress due to chronic inflammation may suppress EBF1 function, leading to cholangiocarcinoma genesis via stem cell DNA damage.
Epstein-Barr (EB) virus infection is assessed as group 1 (carcinogenic to humans) by IARC. Nasopharyngeal carcinoma is a human epithelial tumor with a high prevalence in Southeast Asia and Southern China and has a strong association with EB virus infection [11, 26]. We examined stemness markers (CD44v6, ALDH1A1, CD24) in human nasopharyngeal carcinoma tissues [27]. IHC staining of CD44v6 and ALDH1A1 was observed in cancer tissues, but that of CD24 was not. Formation of the inflammation-specific DNA lesion marker 8-nitroguanine was detected in CD44v6- or ALDH1A1-positive cancer cells. Flow cytometric analysis confirmed both CD44v6- and ALDH1A1-positive cells in a small population of a human nasopharyngeal carcinoma cell line. These findings suggest the importance of 8-nitroguanine formation in nasopharyngeal cancer stem cells. In stromal areas of nasopharyngeal carcinoma tissues, α-smooth muscle actin (α-SMA)-positive cells and CD133-positive cells were detected by IHC, indicating the involvement of cancer-associated fibroblasts and hematopoietic stem/progenitor cells in neoangiogenesis [28].
Protein damage (carbonylation)
Inflammation-derived ROS/RNS damage not only DNA but also proteins. Carbonylation is an irreversible and irreparable protein modification induced by oxidative stress. Carbonylated proteins can be detected by treatment with 2,4-dinitrophenylhydrazine (DNPH), leading to a 2,4-dinitrophenyl (DNP) hydrazone adduct [29], which is analyzed with a specific antibody for DNP hydrazine by immunoblot in 2D gel electrophoresis (2D Oxyblot) [30, 31]. We identified highly carbonylated proteins in OV-related cholangiocarcinoma tissues compared to non-cancerous tissues by 2D Oxyblot, followed by MALDI-TOF/TOF. We confirmed the highly carbonylated proteins including transferrin by using immune-precipitation and Western blotting. Interestingly, iron ion was detected in cancer tissues coincident with oxidatively damaged transferrin by Prussian blue staining and IHC. Iron release from damaged transferrin may mediate Fenton reactions and generate ROS, which may contribute to all steps of carcinogenesis [32].
Summary of biomacromolecule damage by inflammation
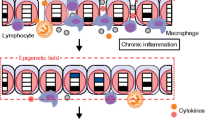
In the inflammatory microenvironment, ROS/RNS damage biomacromolecules, including DNA, proteins, and lipids, as shown in Fig. 1. Inflammation factors, such as infectious organisms and physicochemical and endogenous factors, recruit inflammatory cells to induce respiratory bursts and inflammatory cytokines. NADH oxidase and inducible nitric oxide synthase (iNOS) in inflammatory cells and epithelial cells generate superoxide (O2•−) and NO. NO reacts with O2•− to form highly reactive peroxynitrite (ONOO−), causing 8-oxodG and 8-nitroguanine. H2O2 generated from dismutation of O2•− reacts with Fe(II) to produce hydroxyl radical (•OH) in the Fenton reaction. Highly reactive •OH can attack DNA, proteins, and lipids. In addition to the above mentioned DNA and protein oxidation, inflammation also contributes to lipid peroxidation, for example, higher plasma levels of isoprostanes and malonaldehyde in cholangiocarcinoma patients [33]. Lipid peroxidation is a complex reaction; free radical attacks on polyunsaturated fatty acids yield unstable intermediates (L• and LOO•), resulting in a chain reaction that leads to a loss of membrane properties. Relatively, reactive LO• can further interact with other molecules causing protein and DNA damage. Similar to the case of iron ion from damaged transferrin, as mentioned above, oxidative stress can cause dysfunction of various molecules, forming a vicious cycle. These inflammation-related damages may play critical roles not only in cancer but also in other non-communicable diseases including cardiovascular and neurodegenerative diseases [34].

Biomacromolecule damage by inflammation
Epigenetic alterations in inflammation-related carcinogenesis
Epigenetics is the phenomenon of heritable changes in gene expression that occur without a change in DNA sequence [35], through histone modification, non-coding RNA including microRNA, and DNA methylation. Under the normal microenvironment (Fig. 2, left), the anti-oxidant system works to balance pro-oxidants and anti-oxidants. Promoter cytosine-phosphate-guanine (CpG) sites of tumor suppressor genes and tumor suppressor microRNA-targeting oncogenes are hypomethylated and normally expressed. The epigenome is influenced by environmental factors such as inflammation and aging [36]. Accumulating evidence makes it increasingly clear that epigenetic silencing plays an important role in carcinogenesis via the downregulation of tumor suppressor genes and microRNAs. Under an inflammatory microenvironment (Fig. 2, right), exposure to ROS/RNS or pro-inflammatory cytokines such as interleukin 6 (IL-6) transcriptionally affects the DNA methyltransferase 1 (DNMT1) protein, resulting in enhanced DNA methylation of tumor suppressor genes and microRNAs [37]. ROS/RNS also induce global DNA hypomethylation, resulting in genomic instability. Multistage carcinogenesis consists of three steps: tumor initiation, promotion, and progression. Similar to loss-of-function by mutation, downregulation of a tumor suppressor gene by promoter DNA methylation can be the first step of carcinogenesis, “initiation.” We previously reported on ROS/RNS and IL-6 in EB virus-related human nasopharyngeal carcinoma [11]. To gain further insight regarding epigenetic changes in inflammation-related carcinogenesis, we explored the epigenetic alterations in nasopharyngeal carcinoma.

Epigenetic alterations under an inflammatory microenvironment. White circles indicate unmethylated CpG sites, and black circles show methylated CpG sites in the promoter region
Aberrant DNA methylation
Human malignant tumors are characterized by changes in the patterns of DNA methylation, which include a globally hypomethylated tumor cell genome and the focal hypermethylation of numerous CpG islands; many of them are associated with gene promoters [38]. Promoter hypermethylation events can lead to silencing of genes functioning in tumor-relevant pathways. Together with our collaborators at Guangxi Medical University, we examined aberrant DNA methylation of nasopharyngeal carcinoma tissues of patients in the endemic area of Southern China. We used mRNA microarrays to analyze gene expression changes in human nasopharyngeal carcinoma cell lines treated with a demethylation reagent, 5-aza-2′-deoxycytidine. Among the upregulated candidate genes, we confirmed promoter DNA hypermethylation of RRAD (RAS associated with diabetes) in biopsy specimens and cancer cell lines. Transfection of RRAD in cancer cells suppressed cell proliferation, colony formation, and migration, suggesting that RRAD has tumor-suppressive functions in nasopharyngeal carcinoma [39]. We also detected downregulation of RERG (RAS-like estrogen-regulated growth inhibitor) by DNA methylation in the cancer tissues. RERG-overexpressing cells showed significantly slower growth and less angiogenesis in tumor xenografts in nude mice [40]. DNA methylation occurs in Helicobacter pylori infection-related gastric cancer [41, 42] and ulcerative colitis-associated colorectal cancer [43], indicating that DNA methylation is a key event of inflammation-related carcinogenesis.
Profiling of DNA methylation across the genome is extremely important to understanding the influence of epigenetics. There has been a revolution in DNA methylation analysis technology over the past decade, and analyses that previously were restricted to specific loci can now be performed on a genome scale [44]. The main principles of DNA methylation analysis are restriction enzyme-based, affinity-based, and bisulfite deamination-based pretreatments, followed by the analytical methods such as endpoint PCR, real-time PCR, microarray, and next-generation sequencing [44]. In our early studies, we used bisulfite deamination-based methods, such as methylation-specific PCR (MSP) and bisulfite genomic sequencing (BGS) [39, 45]. Genomic DNA was treated by sodium bisulfite to deaminate unmethylated cytosine to uracil (finally detected as thymine), but methylated cytosine (5-methylcytosine) was not affected (detected as cytosine). Based on this sequence change, DNA methylation was analyzed by endpoint PCR (MSP) or Sanger sequencing of amplified bisulfited DNA (BGS). Nowadays, whole-genome bisulfite sequencing can bioinformatically lift out regions of interest, and sequencing costs are reduced. However, it remains expensive to achieve sufficient sequencing depth for high accuracy, and it is difficult to apply this technique to small samples. We recently explored methylated tumor suppressor genes using affinity-based enrichment with a methyl-CpG-binding domain protein, followed by a next-generation sequencer (methyl-capture sequencing) [46]. This method can be applied to relatively small samples at less cost. Combined with the gene expression microarray, we identified candidate genes that are silenced by promoter DNA methylation in cancer tissues. We quantified the methylation rates of the candidate genes by bisulfite amplicon sequencing (BAS, also known as amplicon bisulfite sequencing: AmpliconBS). BAS involves targeted sequencing of PCR amplicons generated from bisulfite-deaminated DNA. It is a flexible, cost-effective way to study methylation of a sample at single CpG resolution and to perform subsequent multi-target, multi-sample comparisons [47]. The BLUEPRINT consortium evaluated BAS as one of the best all-round methods for use in DNA methylation assays in large-scale validation studies, biomarker development, and clinical diagnostics [48]. We found several differentially methylated candidate genes in nasopharyngeal carcinoma tissues compared to normal nasopharynx tissues that may be useful biomarkers for cancer screening [46]. Among these candidates, we measured the methylation rates of RERG in nasopharynx biopsy specimens by using restriction enzyme-based real-time PCR, which is more convenient than BAS. The methylation rate of RERG in cancer tissues was significantly higher than that in normal tissues, with 78% sensitivity and 100% specificity to screen nasopharyngeal carcinoma [40]. It is advantageous to apply this biomarker for less-invasive specimens.
Dysregulation of microRNA
MicroRNAs are a broad class of non-coding RNAs, 18–25 nucleotides in length in mature form, which control gene expression post-transcriptionally through binding to the 3′-untranslated region (3′-UTR) of mRNA transcripts, stimulating translational suppression or breakage of the target gene. MicroRNAs are involved in a wide range of biological processes including cell proliferation, differentiation, and apoptosis. Various microRNAs are frequently dysregulated in human cancers. We profiled microRNAs of nasopharyngeal carcinomas using microarrays and confirmed the results by quantitative RT-PCR. Among the dysregulated microRNAs in the tissues, plasma miR-497 was lower in cancer patients relative to non-cancerous control patients. Functional analyses revealed that miR-497 mimic-transfected cancer cells suppressed cell growth and migration and induced apoptosis, and showed slower tumor growth in subcutaneous xenografts. These results indicate that miR-497 has a potent tumor suppressor function and may be used as a diagnostic biomarker for nasopharyngeal carcinoma [49].
Head and neck squamous cell carcinoma (HNSCC) is a prevalent malignancy worldwide, and its risk factors such as smoking, alcohol consumption, and HPV infection, are related to inflammation. In a collaborative study with the Department of Otolaryngology and Head and Neck Surgery, Mie University, we compared microRNA expression levels between HNSCC tissues and the adjacent non-cancerous tissues by microarray. Let-7c was one of the downregulated microRNAs in HNSCC tissues. To elucidate the molecular mechanisms underlying the progression of HNSCC, we investigated the function of let-7c as a tumor suppressor. In vitro and in vivo studies revealed that let-7c negatively regulated cancer cell proliferation, migration, and epithelial-mesenchymal transition (EMT) via dysregulation of its direct target genes, insulin-like growth factor 1 receptor (IGF1R), and the high mobility group AT-hook 2 (HMGA2) [50]. Furthermore, we examined the circulating microRNA in plasma before and 6 months after treatment, revealing that plasma miR-21, miR-223, and miR-99a may serve as biomarkers to evaluate the efficacy of therapy and the prognosis of HNSCC [51]. We are currently attempting to measure circulating microRNAs at many time points after treatment to detect cancer recurrence earlier.
Liquid biopsy
Liquid biopsy is an approach to determine the genomic profile of patients with cancer for monitoring treatment responses and to assess the emergence of therapy resistance [52]. Body fluids such as blood, urine, and cerebrospinal fluid have been shown to contain tumor-derived genetic materials and invasive tumor cells. Blood samples, as shown in Fig. 3, contain materials including circulating cell-free DNA/RNA (ccf DNA/RNA), vesicles (such as exosomes), proteins, and cells that can originate from different tissues, including cancers. The rapid turnover of cancer cells is postulated to result in the constant release of tumor-derived DNA/RNA and vesicles into the circulation, and viable tumor cells can also enter the bloodstream (circulating tumor cells, CTC). Liquid biopsy allows for repeated minimally invasive monitoring of tumor clonal evolution (mutation, epigenome alteration) during multistep carcinogenesis. In the future, liquid biopsy may be applied for early detection of cancer, if possible, at the tumor initiation step.

Liquid biopsy and multistage carcinogenesis
Conclusion
We elucidated the molecular mechanisms of inflammation-related carcinogenesis from the aspects of DNA damage and epigenetic alteration and found several candidate biomarkers. Inflammation plays pivotal roles not only in cancer but also in neurodegenerative diseases [53], cardiovascular diseases [54], diabetes [55], and other conditions. Our previous studies indicated that oxidative stress due to inflammation causes oxidative damage to biomacromolecules and induces apoptosis in neural cells, leading to neurodegenerative diseases [31, 34, 56]. Chronic inflammation promotes genetic and epigenetic aberrations in various pathogeneses. ccf DNA/RNA, including DNA methylation and microRNA, can be biomarkers of lifestyle diseases. In the future, liquid biopsy might be a useful tool to improve preventive strategies.
Abbreviations
- 3′-UTR:
-
3′-Untranslated region
- 8-OxodG:
-
8-Oxo-7,8-dihydro-2′-deoxyguanosine
- BAS:
-
Bisulfite amplicon sequencing
- BGS:
-
Bisulfite genomic sequencing
- ccf DNA/RNA:
-
Circulating cell-free DNA/RNA
- CpG:
-
5′-Cytosine-phosphate-guanine-3′
- CTC:
-
Circulating tumor cells
- DNMT1:
-
DNA methyltransferase 1
- DNP:
-
2,4-Dinitrophenyl
- DNPH:
-
2,4-Dinitrophenylhydrazine
- EB:
-
Epstein-Barr
- EBF1:
-
Early B cell factor 1
- EMT:
-
Epithelial-mesenchymal transition
- HMGA2:
-
High-mobility group AT-hook 2
- HNSCC:
-
Head and neck squamous cell carcinoma
- HPLC-ECD:
-
An electrochemical detector coupled to HPLC
- IARC:
-
International Agency for Research on Cancer
- IGF1R:
-
Insulin-like growth factor 1 receptor
- IHC:
-
Immunohistochemistry
- IL-6:
-
Interleukin 6
- iNOS:
-
Inducible nitric oxide synthase
- MSP:
-
Methylation-specific PCR
- O2 •− :
-
Superoxide
- OH:
-
Hydroxyl radical
- ONOO− :
-
Peroxynitrite
- OV:
-
Opisthorchis viverrini
- RERG:
-
RAS-like estrogen-regulated growth inhibitor
- ROS/RNS:
-
Reactive oxygen/nitrogen species
- RRAD:
-
RAS associated with diabetes
- α-SMA:
-
α-Smooth muscle actin
References
Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121:2373–80. https://doi.org/10.1002/ijc.23173.
Pinlaor S, Ma N, Hiraku Y, Yongvanit P, Semba R, Oikawa S, et al. Repeated infection with Opisthorchis viverrini induces accumulation of 8-nitroguanine and 8-oxo-7,8-dihydro-2′-deoxyguanine in the bile duct of hamsters via inducible nitric oxide synthase. Carcinogenesis. 2004;25:1535–42. https://doi.org/10.1093/carcin/bgh157.
Ohnishi S, Ma N, Thanan R, Pinlaor S, Hammam O, Murata M, et al. DNA damage in inflammation-related carcinogenesis and cancer stem cells. Oxidative Med Cell Longev. 2013;387014:2013. https://doi.org/10.1155/2013/387014.
Pinlaor S, Yongvanit P, Hiraku Y, Ma N, Semba R, Oikawa S, et al. 8-Nitroguanine formation in the liver of hamsters infected with Opisthorchis viverrini. Biochem Biophys Res Commun. 2003;309:567–71.
Pinlaor S, Hiraku Y, Ma N, Yongvanit P, Semba R, Oikawa S, et al. Mechanism of NO-mediated oxidative and nitrative DNA damage in hamsters infected with Opisthorchis viverrini: a model of inflammation-mediated carcinogenesis. Nitric Oxide. 2004;11:175–83. https://doi.org/10.1016/j.niox.2004.08.004.
Pinlaor S, Sripa B, Ma N, Hiraku Y, Yongvanit P, Wongkham S, et al. Nitrative and oxidative DNA damage in intrahepatic cholangiocarcinoma patients in relation to tumor invasion. World J Gastroenterol. 2005;11:4644–9.
Ma N, Thanan R, Kobayashi H, Hammam O, Wishahi M, El Leithy T, et al. Nitrative DNA damage and Oct3/4 expression in urinary bladder cancer with Schistosoma haematobium infection. Biochem Biophys Res Commun. 2011;414:344–9. https://doi.org/10.1016/j.bbrc.2011.09.073.
Thanan R, Murata M, Ma N, Hammam O, Wishahi M, El Leithy T, et al. Nuclear localization of COX-2 in relation to the expression of stemness markers in urinary bladder cancer. Mediat Inflamm. 2012;165879:2012. https://doi.org/10.1155/2012/165879.
Ma N, Adachi Y, Hiraku Y, Horiki N, Horiike S, Imoto I, et al. Accumulation of 8-nitroguanine in human gastric epithelium induced by Helicobacter pylori infection. Biochem Biophys Res Commun. 2004;319:506–10. https://doi.org/10.1016/j.bbrc.2004.04.193.
Hiraku Y, Tabata T, Ma N, Murata M, Ding X, Kawanishi S. Nitrative and oxidative DNA damage in cervical intraepithelial neoplasia associated with human papilloma virus infection. Cancer Sci. 2007;98:964–72. https://doi.org/10.1111/j.1349-7006.2007.00497.x.
Ma N, Kawanishi M, Hiraku Y, Murata M, Huang GW, Huang Y, et al. Reactive nitrogen species-dependent DNA damage in EBV-associated nasopharyngeal carcinoma: the relation to STAT3 activation and EGFR expression. Int J Cancer. 2008;122:2517–25. https://doi.org/10.1002/ijc.23415.
Hiraku Y, Kawanishi S, Ichinose T, Murata M. The role of iNOS-mediated DNA damage in infection- and asbestos-induced carcinogenesis. Ann N Y Acad Sci. 2010;1203:15–22. https://doi.org/10.1111/j.1749-6632.2010.05602.x.
Hiraku Y, Sakai K, Shibata E, Kamijima M, Hisanaga N, Ma N, et al. Formation of the nitrative DNA lesion 8-nitroguanine is associated with asbestos contents in human lung tissues: a pilot study. J Occup Health. 2014;56:186–96.
Afroz T, Hiraku Y, Ma N, Ahmed S, Oikawa S, Kawanishi S, et al. Nitrative DNA damage in cultured macrophages exposed to indium oxide. J Occup Health. 2018;60:148–55. https://doi.org/10.1539/joh.17-0146-OA.
Guo F, Ma N, Horibe Y, Kawanishi S, Murata M, Hiraku Y. Nitrative DNA damage induced by multi-walled carbon nanotube via endocytosis in human lung epithelial cells. Toxicol Appl Pharmacol. 2012;260:183–92. https://doi.org/10.1016/j.taap.2012.02.010.
Hiraku Y, Guo F, Ma N, Yamada T, Wang S, Kawanishi S, et al. Multi-walled carbon nanotube induces nitrative DNA damage in human lung epithelial cells via HMGB1-RAGE interaction and Toll-like receptor 9 activation. Part Fibre Toxicol. 2016;13:16. https://doi.org/10.1186/s12989-016-0127-7.
Hiraku Y, Nishikawa Y, Ma N, Afroz T, Mizobuchi K, Ishiyama R, et al. Nitrative DNA damage induced by carbon-black nanoparticles in macrophages and lung epithelial cells. Mutat Res. 2017;818:7–16. https://doi.org/10.1016/j.mrgentox.2017.04.002.
Thanan R, Ma N, Hiraku Y, Iijima K, Koike T, Shimosegawa T, et al. DNA damage in CD133-positive cells in Barrett’s esophagus and esophageal adenocarcinoma. Mediat Inflamm. 2016;2016:7937814. https://doi.org/10.1155/2016/7937814.
Thanan R, Ma N, Iijima K, Abe Y, Koike T, Shimosegawa T, et al. Proton pump inhibitors suppress iNOS-dependent DNA damage in Barrett’s esophagus by increasing Mn-SOD expression. Biochem Biophys Res Commun. 2012;421:280–5. https://doi.org/10.1016/j.bbrc.2012.03.152.
Ma N, Tagawa T, Hiraku Y, Murata M, Ding X, Kawanishi S. 8-Nitroguanine formation in oral leukoplakia, a premalignant lesion. Nitric Oxide. 2006;14:137–43. https://doi.org/10.1016/j.niox.2005.09.012.
Thanan R, Murata M, Pinlaor S, Sithithaworn P, Khuntikeo N, Tangkanakul W, et al. Urinary 8-oxo-7,8-dihydro-2′-deoxyguanosine in patients with parasite infection and effect of antiparasitic drug in relation to cholangiocarcinogenesis. Cancer Epidemiol Biomark Prev. 2008;17:518–24. https://doi.org/10.1158/1055-9965.EPI-07-2717.
Laothong U, Hiraku Y, Oikawa S, Intuyod K, Murata M, Pinlaor S. Melatonin induces apoptosis in cholangiocarcinoma cell lines by activating the reactive oxygen species-mediated mitochondrial pathway. Oncol Rep. 2015;33:1443–9. https://doi.org/10.3892/or.2015.3738.
Thanan R, Pairojkul C, Pinlaor S, Khuntikeo N, Wongkham C, Sripa B, et al. Inflammation-related DNA damage and expression of CD133 and Oct3/4 in cholangiocarcinoma patients with poor prognosis. Free Radic Biol Med. 2013;65:1464–72. https://doi.org/10.1016/j.freeradbiomed.2013.07.034.
Thanan R, Techasen A, Hou B, Jamnongkan W, Armartmuntree N, Yongvanit P, et al. Development and characterization of a hydrogen peroxide-resistant cholangiocyte cell line: a novel model of oxidative stress-related cholangiocarcinoma genesis. Biochem Biophys Res Commun. 2015;464:182–8. https://doi.org/10.1016/j.bbrc.2015.06.112.
Armartmuntree N, Murata M, Techasen A, Yongvanit P, Loilome W, Namwat N, et al. Prolonged oxidative stress down-regulates early B cell factor 1 with inhibition of its tumor suppressive function against cholangiocarcinoma genesis. Redox Biol. 2017;14:637–44. https://doi.org/10.1016/j.redox.2017.11.011.
Chua MLK, Wee JTS, Hui EP, Chan ATC. Nasopharyngeal carcinoma. Lancet. 2016;387:1012–24. https://doi.org/10.1016/S0140-6736(15)00055-0.
Wang S, Ma N, Zhao W, Midorikawa K, Kawanishi S, Hiraku Y, et al. Inflammation-related DNA damage and cancer stem cell markers in nasopharyngeal carcinoma. Mediat Inflamm. 2016;2016:9343460. https://doi.org/10.1155/2016/9343460.
Wang S, Ma N, Kawanishi S, Hiraku Y, Oikawa S, Xie Y, et al. Relationships of alpha-SMA-positive fibroblasts and SDF-1-positive tumor cells with neoangiogenesis in nasopharyngeal carcinoma. Biomed Res Int. 2014;2014:507353. https://doi.org/10.1155/2014/507353.
Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, et al. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464–78.
Nakamura A, Goto S. Analysis of protein carbonyls with 2,4-dinitrophenyl hydrazine and its antibodies by immunoblot in two-dimensional gel electrophoresis. J Biochem. 1996;119:768–74.
Oikawa S, Kobayashi H, Kitamura Y, Zhu H, Obata K, Minabe Y, et al. Proteomic analysis of carbonylated proteins in the monkey substantia nigra after ischemia-reperfusion. Free Radic Res. 2014;48:694–705. https://doi.org/10.3109/10715762.2014.901509.
Thanan R, Oikawa S, Yongvanit P, Hiraku Y, Ma N, Pinlaor S, et al. Inflammation-induced protein carbonylation contributes to poor prognosis for cholangiocarcinoma. Free Radic Biol Med. 2012;52:1465–72. https://doi.org/10.1016/j.freeradbiomed.2012.01.018.
Dechakhamphu S, Pinlaor S, Sitthithaworn P, Nair J, Bartsch H, Yongvanit P. Lipid peroxidation and etheno DNA adducts in white blood cells of liver fluke-infected patients: protection by plasma alpha-tocopherol and praziquantel. Cancer Epidemiol Biomark Prev. 2010;19:310–8. https://doi.org/10.1158/1055-9965.EPI-09-0849.
Thanan R, Oikawa S, Hiraku Y, Ohnishi S, Ma N, Pinlaor S, et al. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int J Mol Sci. 2014;16:193–217. https://doi.org/10.3390/ijms16010193.
Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286:481–6.
Wilson AG. Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J Periodontol. 2008;79:1514–9. https://doi.org/10.1902/jop.2008.080172.
Rokavec M, Oner MG, Hermeking H. Inflammation-induced epigenetic switches in cancer. Cell Mol Life Sci. 2016;73:23–39. https://doi.org/10.1007/s00018-015-2045-5.
Pfeifer GP. Defining driver DNA methylation changes in human cancer. Int J Mol Sci. 2018;19. https://doi.org/10.3390/ijms19041166.
Mo Y, Midorikawa K, Zhang Z, Zhou X, Ma N, Huang G, et al. Promoter hypermethylation of Ras-related GTPase gene RRAD inactivates a tumor suppressor function in nasopharyngeal carcinoma. Cancer Lett. 2012;323:147–54. https://doi.org/10.1016/j.canlet.2012.03.042.
Zhao W, Ma N, Wang S, Mo Y, Zhang Z, Huang G, et al. RERG suppresses cell proliferation, migration and angiogenesis through ERK/NF-kappaB signaling pathway in nasopharyngeal carcinoma. J Exp Clin Cancer Res. 2017;36:88. https://doi.org/10.1186/s13046-017-0554-9.
Maeda M, Moro H, Ushijima T. Mechanisms for the induction of gastric cancer by Helicobacter pylori infection: aberrant DNA methylation pathway. Gastric Cancer. 2017;20:8–15. https://doi.org/10.1007/s10120-016-0650-0.
Matsusaka K, Funata S, Fukayama M, Kaneda A. DNA methylation in gastric cancer, related to Helicobacter pylori and Epstein-Barr virus. World J Gastroenterol. 2014;20:3916–26. https://doi.org/10.3748/wjg.v20.i14.3916.
Emmett RA, Davidson KL, Gould NJ, Arasaradnam RP. DNA methylation patterns in ulcerative colitis-associated cancer: a systematic review. Epigenomics. 2017;9:1029–42. https://doi.org/10.2217/epi-2017-0025.
Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. https://doi.org/10.1038/nrg2732.
Wang S, Xiao X, Zhou X, Huang T, Du C, Yu N, et al. TFPI-2 is a putative tumor suppressor gene frequently inactivated by promoter hypermethylation in nasopharyngeal carcinoma. BMC Cancer. 2010;10:617. https://doi.org/10.1186/1471-2407-10-617.
Zhao W, Mo Y, Wang S, Midorikawa K, Ma N, Hiraku Y, et al. Quantitation of DNA methylation in Epstein-Barr virus-associated nasopharyngeal carcinoma by bisulfite amplicon sequencing. BMC Cancer. 2017;17:489. https://doi.org/10.1186/s12885-017-3482-3.
Pabinger S, Ernst K, Pulverer W, Kallmeyer R, Valdes AM, Metrustry S, et al. Analysis and visualization tool for targeted amplicon bisulfite sequencing on ion torrent sequencers. PLoS One. 2016;11:e0160227. https://doi.org/10.1371/journal.pone.0160227.
The BLUEPRINT consortium. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat Biotechnol. 2016;34:726–37.
Wang S, Mo Y, Midorikawa K, Zhang Z, Huang G, Ma N, et al. The potent tumor suppressor miR-497 inhibits cancer phenotypes in nasopharyngeal carcinoma by targeting ANLN and HSPA4L. Oncotarget. 2015;6:35893–907. https://doi.org/10.18632/oncotarget.5651.
Hou B, Ishinaga H, Midorikawa K, Nakamura S, Hiraku Y, Oikawa S, et al. Let-7c inhibits migration and epithelial-mesenchymal transition in head and neck squamous cell carcinoma by targeting IGF1R and HMGA2. Oncotarget. 2018;9:8927–40. https://doi.org/10.18632/oncotarget.23826.
Hou B, Ishinaga H, Midorikawa K, Shah SA, Nakamura S, Hiraku Y, et al. Circulating microRNAs as novel prognosis biomarkers for head and neck squamous cell carcinoma. Cancer Biol Ther. 2015;16:1042–6. https://doi.org/10.1080/15384047.2015.1045692.
Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol. 2017;14:531–48. https://doi.org/10.1038/nrclinonc.2017.14.
Stephenson J, Nutma E, van der Valk P, Amor S. Inflammation in CNS neurodegenerative diseases. Immunology. 2018;154:204–19. https://doi.org/10.1111/imm.12922.
Becatti M, Mannucci A, Taddei N, Fiorillo C. Oxidative stress and inflammation: new molecular targets for cardiovascular diseases. Intern Emerg Med. 2018. https://doi.org/10.1007/s11739-018-1865-3.
Turkmen K. Inflammation, oxidative stress, apoptosis, and autophagy in diabetes mellitus and diabetic kidney disease: the Four Horsemen of the Apocalypse. Int Urol Nephrol. 2017;49:837–44. https://doi.org/10.1007/s11255-016-1488-4.
Oikawa S, Yamada T, Minohata T, Kobayashi H, Furukawa A, Tada-Oikawa S, et al. Proteomic identification of carbonylated proteins in the monkey hippocampus after ischemia-reperfusion. Free Radic Biol Med. 2009;46:1472–7. https://doi.org/10.1016/j.freeradbiomed.2009.02.029.
Acknowledgements
The author is grateful to Professor Shosuke Kawanishi, Professor Ning Ma, Dr. Shiho Ohnishi (Suzuka University of Medical Science), Dr. Shinji Oikawa, Dr. Yusuke Hiraku, Dr. Kaoru Midorikawa, Ms. Yoshiko Onishi (Mie University Graduate School of Medicine), and all collaborators for their encouragement and help throughout this work.
Funding
This work was supported in part by JSPS KAKENHI (Grant Numbers JP16H05255 and JP16H05829).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Contributions
MM contributed to the writing of the manuscript. The author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The author declares that she has no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
This article is based on research that received the Japanese Society for Hygiene Award and its associated lecture at the 88th annual meeting of the Japanese Society for Hygiene held in Tokyo, Japan, on 22–24 March 2018.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Murata, M. Inflammation and cancer. Environ Health Prev Med 23, 50 (2018). https://doi.org/10.1186/s12199-018-0740-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12199-018-0740-1