
Abstract
Background
Autoantibodies are a hallmark of autoimmune diseases. Autoantibody screening by indirect immunofluorescence staining of HEp-2 cells with patient sera is a current standard in clinical practice. Differential diagnosis of autoimmune disorders is based on commonly recognizable nuclear and cytoplasmic staining patterns. In this study, we attempted to identify as many autoantigens as possible from HEp-2 cells using a unique proteomic DS-affinity enrichment strategy.
Methods
HEp-2 cells were cultured and lysed. Total proteins were extracted from cell lysate and fractionated with DS-Sepharose resins. Proteins were eluted with salt gradients, and fractions with low to high affinity were collected and sequenced by mass spectrometry. Literature text mining was conducted to verify the autoantigenicity of each protein. Protein interaction network and pathway analyses were performed on all identified proteins.
Results
This study identified 107 proteins from fractions with low to high DS-affinity. Of these, 78 are verified autoantigens with previous reports as targets of autoantibodies, whereas 29 might be potential autoantigens yet to be verified. Among the 107 proteins, 82 can be located to nucleus and 15 to the mitotic cell cycle, which may correspond to the dominance of nuclear and mitotic staining patterns in HEp-2 test. There are 55 vesicle-associated proteins and 12 ribonucleoprotein granule proteins, which may contribute to the diverse speckled patterns in HEp-2 stains. There are also 32 proteins related to the cytoskeleton. Protein network analysis indicates that these proteins have significantly more interactions among themselves than would be expected of a random set, with the top 3 networks being mRNA metabolic process regulation, apoptosis, and DNA conformation change.
Conclusions
This study provides a proteomic repertoire of confirmed and potential autoantigens for future studies, and the findings are consistent with a mechanism for autoantigenicity: how self-molecules may form molecular complexes with DS to elicit autoimmunity. Our data contribute to the molecular etiology of autoimmunity and may deepen our understanding of autoimmune diseases.
Similar content being viewed by others

Background
Autoimmune diseases occur when an immune system starts to attack its own body. Any tissue may become a target of autoimmune attacks, which is why autoimmune diseases constitute a wide spectrum of symptoms, with more than 100 autoimmune diseases having been classified thus far. The underlying mechanism of autoimmunity has been rather intriguing. Normally, the immune system reserves immune responses to attack invading microorganisms and protect the body. Due to unclear circumstances, however, the immune response can sometimes go astray and mistakenly attack its own tissue. From a molecular point of view, certain self-molecules are targeted, as if they are non-self, by autoreactive cells, autoantibodies (autoAbs), or other factors, which subsequently leads to damage of the tissue where the autoantigens (autoAgs) reside.
Among the hundreds of thousands of human molecules, only a small portion have been reported to be autoAgs. Moreover, the autoAgs appear to be a random collection of molecules that are expressed in different parts of human body and exhibit various biological functions. Thus, it is mysterious how these different molecules can all trigger a similar cascade of autoimmune responses. A key, missing mechanism is how non-antigenic self-molecules become autoantigenic non-self. Theoretically, a molecule may change itself in many ways by alternating its chemical composition or biochemical properties. A protein may change by mutation, glycosylation, phosphorylation, methylation, citrullination, or fusion with another protein. While simple compositional or structural changes can certainly generate a huge random mix of altered molecules, it cannot explain how they may induce autoimmune responses, let alone similar ones.
Based on our studies, we have proposed a uniform autoantigenicity mechanism by which autoAgs share a common biochemical property in their affinity to dermatan sulfate (DS), and by which autoAgs forming a molecular complex with DS to induce autoreactive B cell responses [1,2,3,4,5]. DS, a glycosaminoglycan polysaccharide, is expressed most abundantly in the skin and other connective tissues [6], and its expression has been reported to increase during high cellular turnaround, such as wound healing [7,8,9]. It is possible that DS is upregulated to facilitate dead cell clearance and new cell growth for tissue regeneration. We found that, when cells are dying or under stress, they express certain self-molecules to which DS has peculiar affinity [1]. By forming complexes with DS, these self-molecules are transformed from non-antigenic singular molecules to antigenic complexes. Furthermore, these DS-autoAg complexes are capable of stimulating autoreactive CD5 B cell proliferation and differentiation [1], likely via PKC- and PI3K-dependent signaling pathways [10, 11].
To relate our findings to clinical utilities, this study examined proteins from HEp-2 cells, the classic substrate in the routine clinical tests for antinuclear autoantibodies (ANAs) [12]. HEp-2 cells were chosen for clinical tests because of their human origin, high mitotic activity, and ability to induce expression of clinically important autoantigens such as mitotic nuclear autoantigens. AutoAbs in patient sera are typically detected by indirect fluorescence staining of HEp-2 cells, and clinical diagnoses are based on about 30 commonly recognizable staining patterns, e.g., nuclear, cytoplasmic, homogenous, or speckled [12]. Despite its clinical popularity, this test lacks molecular details of the autoAg targets, as it is not known exactly which autoAgs are recognized by the autoAbs or whether a single or several autoAgs are recognized. Further tests of specific autoAgs by molecular assays such as ELISA are often performed using proteins extracted from HEp-2 cells or other means. This study was thus pursued to identify the repertoire of autoAgs produced by HEp-2 cells for development of future molecular clinical tests.
Methods
HEp-2 cell culture and protein extraction
HEp-2 cell line was obtained from ATCC (Manassas, VA, USA). HEp-2 cells were cultured in Eagle’s Minimum Essential Medium supplemented with 10% fetal bovine serum (ThermoFisher Scientific) and penicillin–streptomycin-glutamine mixture (ThermoFisher Scientific) at 37 °C in 75 cm2 tissue culture flasks. About 100 million cells were harvested and used for protein extraction. Harvested cells were suspended in 10 mL of 50 mM phosphate buffer (pH 7.4) containing the Roche Complete Mini protease inhibitor cocktail (Sigma Aldrich). Cells were homogenized on ice with a microprobe sonicator until the turbid mixture became nearly clear with no visible cells left. The homogenate was centrifuged at 10,000g at 4 °C for 20 min, and the supernatant was collected as the total protein extract. Protein concentrations were measured with the Bio-Rad RC DC protein assay.
DS-Sepharose resin preparation
Dermatan sulfate (DS) (Sigma-Aldrich) were covalently coupled to EAH Sepharose 4B resins (GE Healthcare) as previously described [2,3,4]. Twenty-mL Sepharose 4B resins were washed with distilled water and then with 0.5 M NaCl solution. The resins were mixed with 100 mg of DS that was pre-dissolved in 10 mL of 0.1 M MES buffer (pH 5.0). The mixture was then added 0.58 g of N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (Sigma-Aldrich). The coupling reaction was carried out by end-over-end tube rotation at 25 °C for 24 h. After the first 60 min, the pH of the reaction mixture was adjusted to 5.0 with 0.1 M NaOH. After the coupling, the resins were washed three times, first with a low-pH buffer (0.1 M acetate, 0.5 M NaCl, pH 5.0) and then with a high-pH buffer (0.1 M Tris, 0.5 M NaCl, pH 8.0). The DS-Sepharose resins were packed in 10 mM phosphate buffer (pH 7.4) into a C 16/20 FPLC column (GE Healthcare Life Sciences).
DS-affinity fractionation
The total proteins extracted from HEp-2 cells were fractionated in a DS-Sepharose column with a BioLogic Duo-Flow system (Bio-Rad). About 40 mg of proteins in 40 mL of 10 mM phosphate buffer (pH 7.4; buffer A) were loaded into the DS-Sepharose column at a rate of 1 mL/min. After loading, the column was washed with 60 mL of buffer A to remove excess and non-binding proteins, followed by eluting with 40 mL of 0.2 M NaCl in buffer A to further remove very weakly binding proteins. Proteins with low to high DS-affinity were eluted with sequential 40-mL step gradients of 0.4 M, 0.6 M, and 1.0 M NaCl in buffer A. Fractions were desalted and concentrated to 0.5 mL with 5-kDa cut-off Vivaspin 20 centrifugal filters (Sartorius). The protein concentration of each fraction was measured. Fractionated proteins were further separated by 1-D SDS PAGE using 4–12% NuPAGE Novex Bis–Tris gels with MES running buffer (Invitrogen). Each gel lane was divided into three sections and subjected to protein sequencing.
Mass spectrometry sequencing
Protein sequencing was performed at the Taplin Biological Mass Spectrometry Facility at Harvard Medical School (Boston, MA, USA). Protein gel sections were cut into 1-mm3 pieces, dehydrated with acetonitrile, and dried in a speed-vac. The gel pieces were rehydrated with 50 mM NH4HCO3 containing 12.5 μg/mL modified sequencing-grade trypsin (Promega) at 4 °C for 45 min. Tryptic peptides were separated on a nano-scale C18 HPLC capillary column and analyzed after electrospray ionization in an LTQ linear ion-trap mass spectrometer (Thermo Scientific). Peptide sequences and protein identities were assigned by matching the measured fragmentation patterns with protein or translated nucleotide databases using Sequest software. Peptides were required to be fully tryptic peptides with XCorr values of at least 1.5 for 1+ ion, 1.5 for 2+ ion, or 3.0 for 3+ ion. All data were manually inspected. Only proteins with ≥ 2 peptide matches were considered positively identified.
Literature search for autoantigen confirmation
Extensive literature searches with PubMed were carried out to identify whether proteins identified in this study had been previously reported as autoantibody-targeted autoantigens. Keywords in the searches included the protein name and the MeSH term “autoantibodies.” When a particular protein name did not yield any results, alternative protein names were obtained from the Uniprot database and used as keywords for repeated searches. In the case of no results, gene names and alternative gene names were used as additional keywords in searches. When multiple reports for an autoantigen were found, reports with the most relevance or open access and free text were preferably cited in this paper.
Pathway and process enrichment analysis by Metascape
The collection of DS-affinity enriched proteins identified from this study was analyzed with Metascape, a gene annotation and analysis resource [13]. Pathway and process enrichment analysis was carried out with KEGG Pathway, GO Biological Processes, Reactome Gene Sets, Canonical Pathways, CORUM, TRRUST, DisGeNET, and PaGenBase. All genes in the genome were used as the enrichment background. Terms with a p value < 0.01, a minimum count of 3, and an enrichment factor (ratio between the observed counts and the counts expected by chance) > 1.5 were collected and grouped into clusters based on their membership similarities. p-values were calculated based on the accumulative hypergeometric distribution, and q-values were calculated using the Banjamini-Hochberg procedure to account for multiple testings. Kappa scores were used as the similarity metric when performing hierarchical clustering on the enriched terms, and sub-trees with a similarity of > 0.3 were considered a cluster. The most statistically significant term within a cluster was chosen to represent the cluster. Protein–protein interaction enrichment analysis was carried out with BioGrid, InWeb_IM, and OmniPath. The resultant network contained the subset of proteins that form physical interactions with at least one other member in the list. The Molecular Complex Detection (MCODE) algorithm was applied to identify densely connected network components. Pathway and process enrichment analysis was applied to each MCODE component independently, and the three best-scoring terms by p-value have been retained as the functional description of the corresponding components.
Protein–protein association network analysis by STRING
The collection of proteins identified from this study were also analyzed with STRING, a database of known and predicted protein–protein interactions [14]. Specific and meaningful protein–protein associations represent proteins jointly contributing to a shared function, but not necessarily physically binding to each other. The database currently covers 24,584,628 proteins from 5090 organisms. Known interactions are sourced from curated databases (metabolic pathways, protein complexes, signal transduction pathways, etc.), experimental evidence (co-purification, co-crystallization, Yeast2Hybrid, genetic interactions, etc.), gene neighborhoods (groups of genes that are frequently observed in each other’s genomic neighborhood), gene fusions (genes that are sometimes fused into single open reading frames), gene co-occurrence (gene families whose occurrence patterns across genomes show similarities), gene text mining (automated, unsupervised searching for proteins that are frequently mentioned together), and co-expression (proteins whose genes are observed to be correlated in expression, across a large number of experiments).
Results
DS-affinity fractionation enriches certain HEp-2 proteins
Proteins were extracted from freshly cultured HEp-2 cells and fractionated with DS-affinity resins. Since DS molecules are poly-anionic, ionic interactions are expected to be a main contributor to DS affinity. We therefore developed an NaCl salt step-gradient method to sequentially dissociate and elute proteins with from DS resins. The identities of proteins in each DS-affinity enriched fraction were obtained by mass spectrometry sequencing.
Proteins with high DS-affinity
From fractions eluted with 1.0 M NaCl from DS-affinity resins, only 7 proteins were identified by mass spectrometry sequencing (Table 1). These include two histone proteins (H4 and H2BE), Scl-70, and Ro/SS-A, all of which are among the most classical nuclear autoantigens (Table 1). Three isoforms of ribosomal proteins are also identified, including 60S ribosomal proteins (L6 and L7) and 40S ribosomal protein S9. Ribosomal proteins are also a class of well-known autoantigens, and heterogenous forms have been reported to be recognize autoantibodies, however, it is not clear exactly which and how many isoforms are autoantigens. L6 and L7 have been reported as autoantigens, whereas S9 awaits further confirmation (Table 1).
Proteins with medium DS-affinity
From fractions eluted with 0.6 M NaCl from DS-affinity resins, 31 proteins were identified by mass spectrometry (Table 1). Histone H4 and H2B were redundantly identified in both 1.0 M and 0.6 M fractions and thus not counted again. Among these, there are 6 histone autoantigens (H1.0, H1.2, H1.5, H2A.V, H2A.1, and H3.2). Other known protein autoantigens include L5, hnRNP A3, nucleolin, nucleophosmin, lamin A/C, DHX9, PABP4, PABP3, YBX3, DNA-PKcs, PCNA, Ku80 and Ku70, lupus La antigen, filamin A and B, and HSPA8 (Table 1). Several proteins have not been found in literature as reported autoantigens, including SET, PRKCSH, ANP32A, ANP32B, L39, MYBBP1A, and hnRNP protein U and C-like-1.
Proteins with low DS-affinity
From fractions eluted with 0.4 M NaCl from DS-affinity resins, 69 proteins were identified (Table 1). The 8 proteins that were also identified in the 0.6 M fraction are not counted here. These proteins ranged from nuclear, cytosol, mitochondrial, cytoskeleton, to proteasome. Proteins related to cellular cytoskeleton include tubulin, tropomyosin, actin, alpha-actinin 1 and 4, spectrin, vimentin, calmodulin, calreticulin, and myosin, most of which have been previously reported as autoantigens. Several interesting groups of proteins are identified, including DNA replication 3 licensing factor proteins (MCM6, MCM2, and MCM3), 6 14-3-3 proteins (epsilon, zeta/delta, gamma, theta, beta/alpha, and sigma), 6 heat shock proteins (HSPA5, HSPA9, HSP90AA1, HSP90AA2, HSP90AB1, and HSP90B1), and 5 proteasomal proteins (PSMA2, PSMA5, PSMA7, PSMD6, and PSMD13). Several enzymes that function in the ER were identified, including P4HB, PDIA4, PDIA6, ERO1A, AHSG, and VCP. Other interesting proteins include Golgi proteins (COPG1 and COPB2), mitochondrial LRPPRC, growth factor HDGF, C1QBP, IQGAP1, BASP1, and clathrin.
DS affinity strongly enriches for autoantigenic proteins
Overall, this study identified a total of 107 proteins from DS-affinity enrichment of HEp-2 cellular extracts (Table 1). Based on current literature, 78 of 107 (72.9%) proteins are confirmed autoantigens. The rest (29 proteins) are potential autoantigens yet to be verified in future studies.
The DS-affinity HEp-2 proteome shows protein network and functional enrichment
To understand the set of DS-affinity-associated proteins identified in this study, we performed various protein network analyses. From protein association network STRING analysis, 106 nodes (proteins, with HSP90AA2 not recognized) gave rise to 330 edges (protein–protein associations) vs. the expected 142 edges (with average node degree of 6.23, and average local clustering coefficient of 0.531, and PPI enrichment p-value of < 1.0e−16). The STRING analysis results indicate that the set of proteins identified from this study have significantly more interactions among themselves than what would be expected of a random set of proteins of similar size drawn from the genome. This insight suggests that DS-affinity proteins may share certain biological functions. While not definitive, it may also indicate that these proteins share functional or biochemical properties (i.e., forming macromolecular charge affinity complexes) and perhaps immunological properties (we have previously shown that DS-autoAg complexes stimulate autoreactive B cells and autoantibody production [1,2,3,4]).
The proteins identified from this study are not randomly distributed, but rather can be classified into 4 clusters, chromosome-binding, RNA-binding, vesicle, and cytoskeleton (Fig. 1a). Based on GO Molecular Function and Cellular Component analysis, nuclear proteins are the most significant group. Of the 107, 82 proteins can be found in the nucleus, including 36 DNA-binding, 7 histone-binding, and 28 RNA-binding proteins. In particular, 17 proteins are expressed in the M phase of cell cycle (Fig. 1b). According to Reactome Pathways, 24 proteins can be involved in cell cycle, with 20 potentially attributable to the mitotic phase and 17 protein attributable to the G2/M check points. In addition to nuclear proteins, another prominent group is related to vesicles/granulates. This group comprises of 48 proteins, including 35 vesicle components, 20 vesicle-mediated transporters, and 12 ribonucleoprotein granule proteins (Fig. 1c). Another prominent group are proteins associated with cytoskeleton organization (32/107).

Protein-protein association network of DS-affinity enriched proteins analyzed with STRING. a The protein network can be primarily clustered into four clusters: chromosome binding (Red: 36 DNA-binding proteins; Gold: seven histone-binding), 28 RNA-binding proteins (Blue), 35 vesicle components (Purple), and 32 cytoskeleton components (Green). b The same network highlighting 82 proteins found in the nucleus (Red, based on GO cellular component) and 17 associated with the mitotic cell cycle (Blue, based on Reactome Pathways). c 35 vesicle component proteins (Green) and 20 vesicle-mediated transport proteins (dark Green), and 12 ribonucleoprotein granule (Blue). These classifications are based on GO Molecular Function, GO Cellular Component, and Reactome Pathways. Minimum required interaction score was set to high confidence (0.700) in the network
To further capture the relationships among these proteins, Metascape pathway and process enrichment analyses were conducted. The three most prominent ontology clusters identified are mRNA metabolic process regulation, apoptosis, and DNA conformation change (Fig. 2). Other significant clusters include translocation of SLC2A4 to plasma membrane, protein processing in the ER, Nop56p-associated pre-rRNA complex, nucleocytoplasmic transport, Emerin complex 52, C complex spliceosome, DGCR8 multiprotein complex, H2AX complex, telomere maintenance, ACTB-ANP32A-C1QBP-PSMA1-PTMA-PSMA1 complex, DHX9-ADAR-vigilin-DNA-PK-Ku antigen complex, and systemic lupus erythematosus (Fig. 2).

a Heatmap of top 20 enriched pathways and processes identified by Metascape. b The top four protein–protein interaction networks and components identified by MCODE algorithm of Metascape analysis. Red network is most likely involved in cellular response to heat stress, vesicle-mediated transport, and kinase maturation complex 1. Blue network is most likely involved in PID BARD1 pathway, DHX9-ADAR-vigillin-DNA-PK-Ku antigen complex, and Nop56p-associated pre-rRNA complex. Green network is mostly likely involved in mRNA splicing and C complex spliceosome. Purple network is most likely involved in Nop56p-associated pre-rRNA complex, actin-mediated contraction, and actin filament-based movement
The MCODE (molecular complex detection) algorithm identified four significant networks (Fig. 2). The first MCODE network consists of 23 proteins (CLTC, YWHAB, HSPA9, PABPC3, KPNB1, ACTB, P4HB, ACTA2, UBA1, VCP, H2BC21, COPB2, CLTCL1, YWHAE, PDIA4, PABPC4, YWHAQ, TUBB4B, ENO1, HSPA5, HSPA8, HNRNPA1, and HSP90AA1). This network is likely associated with cellular response to heat stress, vesicle-mediated transport, or kinase maturation complex 1. The second MCODE network consists of 18 proteins (PCNA, MYH9, TPM2, ACTN4, TPM4, IQGAP1, SPTAN1, HSP90B1, XRCC6, PRKDC, PRPF8, DHX9, PSMD13, RPL5, RPS9, COPG1, RPL7, and NPM1). This network is likely associated with PID BARD1 pathway, DHX9-ADAR-vigilin-DNA-PK-Ku antigen complex, or Nop56p-associated pre-rRNA complex. The third MCODE network consists of 12 proteins (CALR, HSP90AB1, ACTN1, YWHAZ, HNRNPA2B1, SRSF7, HNRNPA3, EFTUD2, YWHAG, MCM3, MCM2, and HNRNPR), which are likely associated with mRNA splicing or C complex spliceosome. The fourth MCODE network also consists of 12 proteins (VIM, TPM3, PRMT1, TPM1, H1-2, HNRNPU, SF3B3, TOP1, XRCC5, FLNA, RPL6, and NCL), which are likely associated with Nop56p-associated pre-rRNA complex, actin-mediated cell contraction, or actin filament-based movement.
Discussion
Autoantigens are the key molecules in autoimmunity and autoimmune diseases, as they are the tissue-resident targets of autoimmune responses and autoimmune diseases. The mechanism by which the normally non-antigenic self-molecules become auto-antigenic holds a key to the understanding of autoimmunity. We proposed that dermatan sulfate has particular affinity for autoAgs and that DS can convert self-molecules to autoAgs by forming DS-autoAg molecular complexes to induce immune responses, and hence any self-molecule with affinity to DS would have the potential to be an autoAg [1]. Using this unifying mechanism of autoantigenicity, we have developed a DS-affinity enrichment strategy to identify potential autoAgs in cell lines and animal organs [2,3,4]. In this study, we tested whether this strategy would enable us to identify a profile of known autoantigens and perhaps to uncover unknown potential autoantigens from HEp-2 cells.
HEp-2 cells are the classic substrate in clinical tests of autoantibodies for autoimmune diseases [12]. In the indirect immunofluorescence antibody test, microscope glass slides are coated with HEp-2 cells, and human serum is incubated with the HEp-2 cells to allow serum autoantibodies to react with autoantigens in HEp-2 cells. The binding of autoantibodies is detected by fluorescently tagged anti-human Igs and viewed under a microscope. HEp-2 cells give rise to ~ 30 nuclear and cytoplasmic staining patterns associated with various autoimmune conditions (HEp-2 Image Library of University of Birmingham and [12]).
Nuclear patterns from ANAs (antinuclear antibodies) are the most commonly found, ranging from homogeneous, peripheral and nuclear rim, centromere, nuclear pores, to speckled pattern. A few unique cell cycle specific patterns are only identifiable in dividing and mitotic cells, revealing autoantigens such as those expressed in metaphase centrioles and mitotic spindles. Cytoplasmic staining patterns range from Golgi apparatus, mitochondrial, rods and rings, to uncharacterized patterns. Cytoplasmic fiber staining patterns are typically found in association with actin, vimentin, tubulin, cytokeratin, and tropomyosin, all of which we have found in this study.
Despite its wide utility in clinical autoantibody screening for autoimmune diseases, HEp-2 indirect fluorescence test is significantly limited by the lack of molecular specificity. While some patterns are known to be associated with certain autoantibodies/autoantigens, it remains to be better characterized which and how many autoantigens are involved with each pattern. In clinical diagnosis, a particular staining pattern can appear from patients with different autoimmune diseases. For example, a nuclear homogeneous pattern can be from patients with systemic lupus erythematosus, chronic autoimmune hepatitis, or juvenile idiopathic arthritis, and a fine speckled pattern can be from patients with Sjogren’s syndrome, subcutaneous or neonatal lupus erythematosus, or congenital heart block, or other overlap syndrome [12]. For further differentiated diagnoses, samples displaying homogenous patterns are further analyzed by molecular assays such as ELISA for anti-dsDNA, anti-histone, and anti-ENA, whereas samples displaying speckled patterns are further analyzed for anti-ENA, anti-SSA, anti-SSB, and anti-dsDNA.
In this study, we identified various commonly found autoantigens with known associations to different HEp-2 cell staining patterns. For example, histones are mostly involved in homogeneous patterns, and lamin A, B, and C are involved in membranous nuclear rim patterns. Large, variable sized speckles in sponge-like patterns are usually associated with hnRNP, whereas fine speckles are mostly due to SSA/Ro, SSB/La, RNA polymerases and others. Cytoplasmic fiber staining patterns are in association with actin, vimentin, tubulin, cytokeratin, and tropomyosin.
In this study, we also identified a number of interesting potential autoantigens. For example, we identified the family of 14-3-3 proteins that have been reported as autoantigens elsewhere (Table 1), but their association with any particular HEp-2 staining pattern has not been described. As another example, mitotic cell cycle-related patterns have thus far remain poorly understood, whereas our study identified 14 proteins could potentially contribute to mitotic cell staining patterns (PCNA, XRCC5, XRCC6, PRKDC, MCM6, MCM3, MCM2, TUBB4B, IQGAP1, and others) (Fig. 1b). As a third example, COPA mutations impair ER-Golgi transport and cause hereditary autoimmune mediated lung disease and arthritis, and four deleterious variants in the COPA gene (encoding coatomer subunit alpha) were identified in families with an apparent Mendelian syndrome of autoimmunity characterized by high-titer autoantibodies [15]. Circulating anti-COPE (coatomer protein complex subunit epsilon) has been identified as a potential marker for cardiovascular and cerebrovascular events in patients with obstructive sleep apnea [16]. Our study identified two members, COPG1 and COPB2, as potential autoantigens (Table 1).
In addition to cellular staining location, this study also revealed possible functional association of autoAgs identified from DS-affinity. As derived from protein–protein interaction network and pathway analyses, the collection of proteins identified in this study are most likely involved in mRNA metabolism and apoptosis (Fig. 2a). Although the former has no clear role in autoimmunity, apoptosis has established connections to autoimmunity and autoimmune diseases. Apoptosis is well recognized as a source of autoAgs [17]. Our previous study also showed clear evidence that DS is particularly attracted to apoptotic cells and their autoAgs expressed in these cells [1]. In regard to the significant role of apoptosis in autoimmunity, our current findings have reached a consistent conclusion with previous reports by others and us.
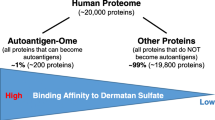
It is estimated that there are ~ 20,000 protein-coding genes in the human genome, and ~ 10,000 of these protein-coding genes are expressed in a typical cell. After DS-affinity fractionation, we identified only a small but specific subset 107 proteins (~ 1% of the expressed cellular proteome) from fractions with low to high DS affinity. With 73% of these DS-affinity proteins having verified autoantigenicity, this study demonstrates the powerful feasibility of DS-affinity enrichment for identifying potential autoantigens.
Conclusions
By DS-affinity enrichment of autoantigens from HEp-2 cellular protein extracts, this study identified 107 proteins, with 78 being verified and 29 being potential autoantigens. These proteins are not a random pool, but rather are clustered in the nucleus, vesicles, and cytoskeleton. This set of proteins shows significantly more interactions than random sets of proteins, revealing apoptosis as a prominent underlying process. Results from this study are consistent with our prior work on autoimmunity [1,2,3,4,5] and provide further support for a more general principle of autoantigenicity on how self-molecules become non-self autoAgs, which may help unravel the molecular etiology of autoimmunity and deepen our understanding of autoimmune diseases. The exact association of these proteins with HEp-2 staining patterns and associated diseases merits extensive investigation in future studies. This study also provides many interesting potential autoantigens for future studies.
Availability of data and materials
All data generated and analyzed during this study are included in this published article.
Abbreviations
- AutoAg:
-
Autoantigen
- DS:
-
Dermatan sulfate
References
Wang JY, Lee J, Yan M, Rho JH, Roehrl MH. Dermatan sulfate interacts with dead cells and regulates CD5(+) B-cell fate: implications for a key role in autoimmunity. Am J Pathol. 2011;178(5):2168–76.
Rho JH, Zhang W, Murali M, Roehrl MH, Wang JY. Human proteins with affinity for dermatan sulfate have the propensity to become autoantigens. Am J Pathol. 2011;178(5):2177–90.
Zhang W, Rho JH, Roehrl MH, Wang JY. A comprehensive autoantigen-ome of autoimmune liver diseases identified from dermatan sulfate affinity enrichment of liver tissue proteins. BMC Immunol. 2019;20(1):21.
Zhang W, Rho JH, Roehrl MW, Roehrl MH, Wang JY. A repertoire of 124 potential autoantigens for autoimmune kidney diseases identified by dermatan sulfate affinity enrichment of kidney tissue proteins. PLoS ONE. 2019;14(6):e0219018.
Wang JY, Roehrl MH. Glycosaminoglycans are a potential cause of rheumatoid arthritis. Proc Natl Acad Sci U S A. 2002;99(22):14362–7.
Mizumoto S, Kosho T, Yamada S, Sugahara K. Pathophysiological significance of dermatan sulfate proteoglycans revealed by human genetic disorders. Pharmaceuticals. 2017;10(2):34.
Islam S, Chuensirikulchai K, Khummuang S, Keratibumrungpong T, Kongtawelert P, Kasinrerk W, Hatano S, Nagamachi A, Honda H, Watanabe H. Accumulation of versican facilitates wound healing: implication of its initial ADAMTS-cleavage site. Matrix Biol. 2020;87:77–93.
Wisowski G, Kozma EM, Bielecki T, Pudelko A, Olczyk K. Dermatan sulfate is a player in the transglutaminase 2 interaction network. PLoS ONE. 2017;12(2):e0172263.
Belvedere R, Bizzarro V, Parente L, Petrella F, Petrella A. Effects of Prisma(R) Skin dermal regeneration device containing glycosaminoglycans on human keratinocytes and fibroblasts. Cell Adh Migr. 2018;12(2):168–83.
Aoyama E, Yoshihara R, Tai A, Yamamoto I, Gohda E. PKC- and PI3K-dependent but ERK-independent proliferation of murine splenic B cells stimulated by chondroitin sulfate B. Immunol Lett. 2005;99(1):80–4.
Yoshihara R, Aoyama E, Kadota Y, Kawai S, Goto T, Zhong M, Gohda E. Differentiation of murine B cells induced by chondroitin sulfate B. Cell Immunol. 2007;250(1–2):14–23.
Damoiseaux J, Andrade LEC, Carballo OG, Conrad K, Francescantonio PLC, Fritzler MJ, Garcia de la Torre I, Herold M, Klotz W, Cruvinel WM, et al. Clinical relevance of HEp-2 indirect immunofluorescent patterns: the International Consensus on ANA patterns (ICAP) perspective. Ann Rheum Dis. 2019;78(7):879–89.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13.
Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, Thamsen M, Santos-Cortez RL, Lee K, Gambin T, et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet. 2015;47(6):654–60.
Matsumura T, Terada J, Kinoshita T, Sakurai Y, Yahaba M, Ema R, Amata A, Sakao S, Nagashima K, Tatsumi K, et al. Circulating Anti-Coatomer Protein Complex Subunit Epsilon (COPE) autoantibodies as a potential biomarker for cardiovascular and cerebrovascular events in patients with obstructive sleep apnea. J Clin Sleep Med. 2017;13(3):393–400.
Lleo A, Selmi C, Invernizzi P, Podda M, Gershwin ME. The consequences of apoptosis in autoimmunity. J Autoimmun. 2008;31(3):257–62.
Vordenbaumen S, Bohmer P, Brinks R, Fischer-Betz R, Richter J, Bleck E, Rengers P, Gohler H, Zucht HD, Budde P, et al. High diagnostic accuracy of histone H4-IgG autoantibodies in systemic lupus erythematosus. Rheumatology. 2018;57(3):533–7.
van Bavel CC, Dieker J, Muller S, Briand JP, Monestier M, Berden JH, van der Vlag J. Apoptosis-associated acetylation on histone H2B is an epitope for lupus autoantibodies. Mol Immunol. 2009;47(2–3):511–6.
Wesierska-Gadek J, Penner E, Lindner H, Hitchman E, Sauermann G. Autoantibodies against different histone H1 subtypes in systemic lupus erythematosus sera. Arthritis Rheum. 1990;33(8):1273–8.
Burlingame RW, Boey ML, Starkebaum G, Rubin RL. The central role of chromatin in autoimmune responses to histones and DNA in systemic lupus erythematosus. J Clin Invest. 1994;94(1):184–92.
Vila JL, Juarez C, Illa I, Agusti M, Gelpi C, Amengual MJ, Rodriguez JL. Autoantibodies against the H1(0) subtype of histone H1. Clin Immunol Immunopathol. 1987;45(3):499–503.
Rubin RL, Bell SA, Burlingame RW. Autoantibodies associated with lupus induced by diverse drugs target a similar epitope in the (H2A-H2B)-DNA complex. J Clin Invest. 1992;90(1):165–73.
Dieker J, Berden JH, Bakker M, Briand JP, Muller S, Voll R, Sjowall C, Herrmann M, Hilbrands LB, van der Vlag J. Autoantibodies against modified histone peptides in SLE patients are associated with disease activity and lupus nephritis. PLoS ONE. 2016;11(10):e0165373.
Scholz J, Grossmann K, Knutter I, Hiemann R, Sowa M, Rober N, Rodiger S, Schierack P, Reinhold D, Bogdanos DP, et al. Second generation analysis of antinuclear antibody (ANA) by combination of screening and confirmatory testing. Clin Chem Lab Med. 2015;53(12):1991–2002.
Mofors J, Holmqvist M, Westermark L, Bjork A, Kvarnstrom M, Forsblad-d’Elia H, Magnusson Bucher S, Eriksson P, Theander E, Mandl T, et al. Concomitant Ro/SSA and La/SSB antibodies are biomarkers for the risk of venous thromboembolism and cerebral infarction in primary Sjogren’s syndrome. J Intern Med. 2019;286(4):458–68.
Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526–33.
Brankin B, Skaar TC, Brotzman M, Trock B, Clarke R. Autoantibodies to the nuclear phosphoprotein nucleophosmin in breast cancer patients. Cancer Epidemiol Biomarkers Prev. 1998;7(12):1109–15.
Ola TO, Biro PA, Hawa MI, Ludvigsson J, Locatelli M, Puglisi MA, Bottazzo GF, Fierabracci A. Importin beta: a novel autoantigen in human autoimmunity identified by screening random peptide libraries on phage. J Autoimmun. 2006;26(3):197–207.
Vlachoyiannopoulos PG, Frillingos S, Tzioufas AG, Seferiadis K, Moutsopoulos HM, Tsolas O. Circulating antibodies to prothymosin alpha in systemic lupus erythematosus. Clin Immunol Immunopathol. 1989;53(2 Pt 1):151–60.
Nahamura H, Yoshida K, Kishima Y, Enomoto H, Uyama H, Kuroda T, Okuda Y, Hirotani T, Ito H, Kawase I. Circulating auto-antibody against hepatoma-derived growth factor (HDGF) in patients with ulcerative colitis. Hepatogastroenterology. 2004;51(56):470–5.
Moscato S, Pratesi F, Sabbatini A, Chimenti D, Scavuzzo M, Passatino R, Bombardieri S, Giallongo A, Migliorini P. Surface expression of a glycolytic enzyme, alpha-enolase, recognized by autoantibodies in connective tissue disorders. Eur J Immunol. 2000;30(12):3575–84.
Braunschweig D, Krakowiak P, Duncanson P, Boyce R, Hansen RL, Ashwood P, Hertz-Picciotto I, Pessah IN. Autism-specific maternal autoantibodies recognize critical proteins in developing brain. Transl Psychiatry. 2013;3:e277.
Kistner A, Bigler MB, Glatz K, Egli SB, Baldin FS, Marquardsen FA, Mehling M, Rentsch KM, Staub D, Aschwanden M, et al. Characteristics of autoantibodies targeting 14-3-3 proteins and their association with clinical features in newly diagnosed giant cell arteritis. Rheumatology (Oxford). 2017;56(5):829–34.
Chakravarti R, Gupta K, Swain M, Willard B, Scholtz J, Svensson LG, Roselli EE, Pettersson G, Johnston DR, Soltesz EG, et al. 14-3-3 in Thoracic Aortic aneurysms: identification of a novel autoantigen in large vessel vasculitis. Arthritis Rheumatol. 2015;67(7):1913–21.
Qiu J, Choi G, Li L, Wang H, Pitteri SJ, Pereira-Faca SR, Krasnoselsky AL, Randolph TW, Omenn GS, Edelstein C, et al. Occurrence of autoantibodies to annexin I, 14-3-3 theta and LAMR1 in prediagnostic lung cancer sera. J Clin Oncol. 2008;26(31):5060–6.
Kaneda K, Takasaki Y, Takeuchi K, Yamada H, Nawata M, Matsushita M, Matsudaira R, Ikeda K, Yamanaka K, Hashimoto H. Autoimmune response to proteins of proliferating cell nuclear antigen multiprotein complexes in patients with connective tissue diseases. J Rheumatol. 2004;31(11):2142–50.
Hoa S, Hudson M, Troyanov Y, Proudman S, Walker J, Stevens W, Nikpour M, Assassi S, Mayes MD, Wang M, et al. Single-specificity anti-Ku antibodies in an international cohort of 2140 systemic sclerosis subjects: clinical associations. Medicine. 2016;95(35):e4713.
Mimori T, Ohosone Y, Hama N, Suwa A, Akizuki M, Homma M, Griffith AJ, Hardin JA. Isolation and characterization of cDNA encoding the 80-kDa subunit protein of the human autoantigen Ku (p70/p80) recognized by autoantibodies from patients with scleroderma-polymyositis overlap syndrome. Proc Natl Acad Sci USA. 1990;87(5):1777–81.
Schild-Poulter C, Su A, Shih A, Kelly OP, Fritzler MJ, Goldstein R, Hache RJ. Association of autoantibodies with Ku and DNA repair proteins in connective tissue diseases. Rheumatology. 2008;47(2):165–71.
Frampton G, Moriya S, Pearson JD, Isenberg DA, Ward FJ, Smith TA, Panayiotou A, Staines NA, Murphy JJ. Identification of candidate endothelial cell autoantigens in systemic lupus erythematosus using a molecular cloning strategy: a role for ribosomal P protein P0 as an endothelial cell autoantigen. Rheumatology. 2000;39(10):1114–20.
Absi M, La Vergne JP, Marzouki A, Giraud F, Rigal D, Reboud AM, Reboud JP, Monier JC. Heterogeneity of ribosomal autoantibodies from human, murine and canine connective tissue diseases. Immunol Lett. 1989;23(1):35–41.
Neu E, von Mikecz AH, Hemmerich PH, Peter HH, Fricke M, Deicher H, Genth E, Krawinkel U. Autoantibodies against eukaryotic protein L7 in patients suffering from systemic lupus erythematosus and progressive systemic sclerosis: frequency and correlation with clinical, serological and genetic parameters. The SLE Study Group. Clin Exp Immunol. 1995;100(2):198–204.
Guialis A, Patrinou-Georgoula M, Tsifetaki N, Aidinis V, Sekeris CE, Moutsopoulos HM. Anti-5S RNA/protein (RNP) antibody levels correlate with disease activity in a patient with systemic lupus erythematosus (SLE) nephritis. Clin Exp Immunol. 1994;95(3):385–9.
Qin Z, Lavingia B, Zou Y, Stastny P. Antibodies against nucleolin in recipients of organ transplants. Transplantation. 2011;92(7):829–35.
Scofield RH. Do we need new autoantibodies in lupus? Arthritis Res Ther. 2010;12(3):120.
Siapka S, Patrinou-Georgoula M, Vlachoyiannopoulos PG, Guialis A. Multiple specificities of autoantibodies against hnRNP A/B proteins in systemic rheumatic diseases and hnRNP L as an associated novel autoantigen. Autoimmunity. 2007;40(3):223–33.
Guarneri C, Aguennouz M, Guarneri F, Polito F, Benvenga S, Cannavo SP. Autoimmunity to heterogeneous nuclear ribonucleoprotein A1 in psoriatic patients and correlation with disease severity. J Dtsch Dermatol Ges. 2018;16(9):1103–7.
Hassfeld W, Chan EK, Mathison DA, Portman D, Dreyfuss G, Steiner G, Tan EM. Molecular definition of heterogeneous nuclear ribonucleoprotein R (hnRNP R) using autoimmune antibody: immunological relationship with hnRNP P. Nucleic Acids Res. 1998;26(2):439–45.
Konig MF, Giles JT, Nigrovic PA, Andrade F. Antibodies to native and citrullinated RA33 (hnRNP A2/B1) challenge citrullination as the inciting principle underlying loss of tolerance in rheumatoid arthritis. Ann Rheum Dis. 2016;75(11):2022–8.
Beutgen VM, Schmelter C, Pfeiffer N, Grus FH. Autoantigens in the trabecular meshwork and glaucoma-specific alterations in the natural autoantibody repertoire. Clin Transl Immunology. 2020;9(3):e01101.
Bach M, Winkelmann G, Luhrmann R. 20S small nuclear ribonucleoprotein U5 shows a surprisingly complex protein composition. Proc Natl Acad Sci USA. 1989;86(16):6038–42.
Hwang HM, Heo CK, Lee HJ, Kwak SS, Lim WH, Yoo JS, Yu DY, Lim KJ, Kim JY, Cho EW. Identification of anti-SF3B1 autoantibody as a diagnostic marker in patients with hepatocellular carcinoma. J Transl Med. 2018;16(1):177.
Overzet K, Gensler TJ, Kim SJ, Geiger ME, van Venrooij WJ, Pollard KM, Anderson P, Utz PJ. Small nucleolar RNP scleroderma autoantigens associate with phosphorylated serine/arginine splicing factors during apoptosis. Arthritis Rheum. 2000;43(6):1327–36.
Li FJ, Surolia R, Li H, Wang Z, Kulkarni T, Liu G, de Andrade JA, Kass DJ, Thannickal VJ, Duncan SR, et al. Autoimmunity to vimentin is associated with outcomes of patients with idiopathic pulmonary fibrosis. J Immunol. 2017;199(5):1596–605.
Presslauer S, Hinterhuber G, Cauza K, Horvat R, Rappersberger K, Wolff K, Foedinger D. RasGAP-like protein IQGAP1 is expressed by human keratinocytes and recognized by autoantibodies in association with bullous skin disease. J Invest Dermatol. 2003;120(3):365–71.
Iannaccone A, Giorgianni F, New DD, Hollingsworth TJ, Umfress A, Alhatem AH, Neeli I, Lenchik NI, Jennings BJ, Calzada JI, et al. Circulating autoantibodies in age-related macular degeneration recognize human macular tissue antigens implicated in autophagy, immunomodulation, and protection from oxidative stress and apoptosis. PLoS ONE. 2015;10(12):e0145323.
Shimizu F, Schaller KL, Owens GP, Cotleur AC, Kellner D, Takeshita Y, Obermeier B, Kryzer TJ, Sano Y, Kanda T, et al. Glucose-regulated protein 78 autoantibody associates with blood-brain barrier disruption in neuromyelitis optica. Sci Transl Med. 2017;9(397):eaai9111.
Harlow L, Rosas IO, Gochuico BR, Mikuls TR, Dellaripa PF, Oddis CV, Ascherman DP. Identification of citrullinated hsp90 isoforms as novel autoantigens in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheum. 2013;65(4):869–79.
Cid C, Regidor I, Alcazar A. Anti-heat shock protein 90beta antibodies are detected in patients with multiple sclerosis during remission. J Neuroimmunol. 2007;184(1–2):223–6.
Suzuki S, Utsugisawa K, Iwasa K, Satoh T, Nagane Y, Yoshikawa H, Kuwana M, Suzuki N. Autoimmunity to endoplasmic reticulum chaperone GRP94 in myasthenia gravis. J Neuroimmunol. 2011;237(1–2):87–92.
Kenderov A, Minkova V, Mihailova D, Giltiay N, Kyurkchiev S, Kehayov I, Kazatchkine M, Kaveri S, Pashov A. Lupus-specific kidney deposits of HSP90 are associated with altered IgG idiotypic interactions of anti-HSP90 autoantibodies. Clin Exp Immunol. 2002;129(1):169–76.
Kishore U, Sontheimer RD, Sastry KN, Zappi EG, Hughes GR, Khamashta MA, Reid KB, Eggleton P. The systemic lupus erythematosus (SLE) disease autoantigen-calreticulin can inhibit C1q association with immune complexes. Clin Exp Immunol. 1997;108(2):181–90.
Nagayama S, Yokoi T, Tanaka H, Kawaguchi Y, Shirasaka T, Kamataki T. Occurrence of autoantibody to protein disulfide isomerase in patients with hepatic disorder. J Toxicol Sci. 1994;19(3):163–9.
Miyachi K, Hosaka H, Nakamura N, Miyakawa H, Mimori T, Shibata M, Matsushima S, Chinoh H, Horigome T, Hankins RW, et al. Anti-p97/VCP antibodies: an autoantibody marker for a subset of primary biliary cirrhosis patients with milder disease? Scand J Immunol. 2006;63(5):376–82.
Ikeda Y, Toda G, Hashimoto N, Aotsuka S, Yokohari R, Maruyama T, Oka H. Anticalmodulin autoantibody in liver diseases: a new antibody against a cytoskeleton-related protein. Hepatology. 1987;7(2):285–93.
Tanaka M, Kishimura M, Ozaki S, Osakada F, Hashimoto H, Okubo M, Murakami M, Nakao K. Cloning of novel soluble gp130 and detection of its neutralizing autoantibodies in rheumatoid arthritis. J Clin Invest. 2000;106(1):137–44.
Weber CK, Haslbeck M, Englbrecht M, Sehnert B, Mielenz D, Graef D, Distler JH, Mueller RB, Burkhardt H, Schett G, et al. Antibodies to the endoplasmic reticulum-resident chaperones calnexin, BiP and Grp94 in patients with rheumatoid arthritis and systemic lupus erythematosus. Rheumatology. 2010;49(12):2255–63.
Yi JK, Chang JW, Han W, Lee JW, Ko E, Kim DH, Bae JY, Yu J, Lee C, Yu MH, et al. Autoantibody to tumor antigen, alpha 2-HS glycoprotein: a novel biomarker of breast cancer screening and diagnosis. Cancer Epidemiol Biomarkers Prev. 2009;18(5):1357–64.
Hong HS, Chung WH, Hung SI, Chen MJ, Lee SH, Yang LC. Clinical association of anti-golgi autoantibodies and their autoantigens. Scand J Immunol. 2004;59(1):79–87.
Minami S, Matsumoto K, Nagashio R, Hagiuda D, Fukuda E, Goshima N, Hattori M, Tsuchiya B, Hachimura K, Jiang SX, et al. Analysis of Autoantibodies Related to Tumor Progression in Sera from Patients with High-grade Non-muscle-invasive Bladder Cancer. Anticancer Res. 2017;37(12):6705–14.
Betteridge ZE, Gunawardena H, Chinoy H, North J, Ollier WE, Cooper RG, McHugh NJ. Collaboration UKAOMI: clinical and human leucocyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann Rheum Dis. 2009;68(10):1621–5.
Sugimoto K, Hiwasa T, Shibuya K, Hirano S, Beppu M, Isose S, Arai K, Takiguchi M, Kuwabara S, Mori M. Novel autoantibodies against the proteasome subunit PSMA7 in amyotrophic lateral sclerosis. J Neuroimmunol. 2018;325:54–60.
Mojtahedi Z, Safaei A, Yousefi Z, Ghaderi A. Immunoproteomics of HER2-positive and HER2-negative breast cancer patients with positive lymph nodes. OMICS. 2011;15(6):409–18.
Kamhieh-Milz J, Sterzer V, Celik H, Khorramshahi O. Fadl Hassan Moftah R, Salama A: identification of novel autoantigens via mass spectroscopy-based antibody-mediated identification of autoantigens (MS-AMIDA) using immune thrombocytopenic purpura (ITP) as a model disease. J Proteomics. 2017;157:59–70.
Konstantinov KN, Galcheva-Gargova Z, Hoier-Madsen M, Wiik A, Ullman S, Halberg P, Vejlsgaard GL. Autoantibodies to lamins A and C in sera of patients showing peripheral fluorescent antinuclear antibody pattern on HEP-2 cells. J Invest Dermatol. 1990;95(3):304–8.
Hanrotel-Saliou C, Segalen I, Le Meur Y, Youinou P, Renaudineau Y. Glomerular antibodies in lupus nephritis. Clin Rev Allergy Immunol. 2011;40(3):151–8.
Mande PV, Parikh FR, Hinduja I, Zaveri K, Vaidya R, Gajbhiye R, Khole VV. Identification and validation of candidate biomarkers involved in human ovarian autoimmunity. Reprod Biomed Online. 2011;23(4):471–83.
van Beers JJ, Schwarte CM, Stammen-Vogelzangs J, Oosterink E, Bozic B, Pruijn GJ. The rheumatoid arthritis synovial fluid citrullinome reveals novel citrullinated epitopes in apolipoprotein E, myeloid nuclear differentiation antigen, and beta-actin. Arthritis Rheum. 2013;65(1):69–80.
Garbarz M, Dhermy D, Bournier O, Bezeaud A, Boivin P. Anti-spectrin in sera containing smooth muscle autoantibodies from patients with chronic active hepatitis. Clin Exp Immunol. 1981;43(1):87–93.
von Muhlen CA, Chan EK, Peebles CL, Imai H, Kiyosawa K, Tan EM. Non-muscle myosin as target antigen for human autoantibodies in patients with hepatitis C virus-associated chronic liver diseases. Clin Exp Immunol. 1995;100(1):67–74.
Kimura A, Sakurai T, Yamada M, Koumura A, Hayashi Y, Tanaka Y, Hozumi I, Ohtaki H, Chousa M, Takemura M, et al. Anti-endothelial cell antibodies in patients with cerebral small vessel disease. Curr Neurovasc Res. 2012;9(4):296–301.
Geng X, Biancone L, Dai HH, Lin JJ, Yoshizaki N, Dasgupta A, Pallone F, Das KM. Tropomyosin isoforms in intestinal mucosa: production of autoantibodies to tropomyosin isoforms in ulcerative colitis. Gastroenterology. 1998;114(5):912–22.
Gajbhiye R, Sonawani A, Khan S, Suryawanshi A, Kadam S, Warty N, Raut V, Khole V. Identification and validation of novel serum markers for early diagnosis of endometriosis. Hum Reprod. 2012;27(2):408–17.
Zhao X, Cheng Y, Gan Y, Jia R, Zhu L, Sun X. Anti-tubulin-alpha-1C autoantibody in systemic lupus erythematosus: a novel indicator of disease activity and vasculitis manifestations. Clin Rheumatol. 2018;37(5):1229–37.
Prasannan L, Misek DE, Hinderer R, Michon J, Geiger JD, Hanash SM. Identification of beta-tubulin isoforms as tumor antigens in neuroblastoma. Clin Cancer Res. 2000;6(10):3949–56.
Acknowledgements
We are grateful to Ross Tomaino and the Taplin Biological Mass Spectrometry facility of Harvard Medical School for expert service with protein sequencing.
Funding
This work was partially funded by the NIH (R01 AI068826 to JYW) and Curandis. MHR acknowledges the NIH/NCI MSKCC Cancer Center Support Grant P30 CA008748. The funding bodies were not involved in the design of the study and the collection, analysis, and interpretation of data.
Author information
Authors and Affiliations
Contributions
JYW directed the study, analyzed data, and prepared the manuscript. WZ and JHR carried out experiments and reviewed the manuscript. MWR assisted in data analysis and manuscript preparation. MHR consulted on study design and data analysis and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
JYW is the founder and Chief Scientific Officer of Curandis. WZ and JHR were supported by the NIH during the course of this study and declare no competing interests. MWR is an employee of Curandis and declares no further competing interests. MHR is member of the Scientific Advisory Boards of Trans-Hit, Proscia, and Universal DX and declares no other competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, J.Y., Zhang, W., Rho, Jh. et al. A proteomic repertoire of autoantigens identified from the classic autoantibody clinical test substrate HEp-2 cells. Clin Proteom 17, 35 (2020). https://doi.org/10.1186/s12014-020-09298-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12014-020-09298-3