
Abstract
The tryptic peptides from ice cold versus room temperature plasma were identified by C18 liquid chromatography and micro electrospray ionization tandem mass spectrometry (LC–ESI–MS/MS). Samples collected on ice showed low levels of endogenous tryptic peptides compared to the same samples incubated at room temperature. Plasma on ice contained peptides from albumin, complement, and apolipoproteins and others that were observed by the X!TANDEM and SEQUEST algorithms. In contrast to ice cold samples, after incubation at room temperature, greater numbers of tryptic peptides from well characterized plasma proteins, and from cellular proteins were observed. A total of 583,927 precursor ions and MS/MS spectra were correlated to 94,669 best fit peptides that reduced to 22,287 correlations to the best accession within a gene symbol and to 7174 correlations to at least 510 gene symbols with ≥ 5 independent MS/MS correlations (peptide counts) that showed FDR q-values ranging from E−9 (i.e. FDR = 0.000000001) to E−227. A set of 528 gene symbols identified by X!TANDEM and SEQUEST including C4B showed ≥ fivefold variation between ice cold versus room temperature incubation. STRING analysis of the protein gene symbols observed from endogenous peptides in normal plasma revealed an extensive protein-interaction network of cellular factors associated with cell signalling and regulation, the formation of membrane bound organelles, cellular exosomes and exocytosis network proteins. Taken together the results indicated that a pool of cellular proteins, or protein complexes, in plasma are apparently not stable and degrade soon after incubation at room temperature.
Similar content being viewed by others


Background
Many of the well-known proteins of human blood [1, 2] are cleaved by endogenous tryptic endopeptidases to release fully tryptic peptides that may be identified by collection over C18 followed by liquid chromatography, electrospray ionization and tandem mass spectrometry (LC–ESI–MS/MS) with a Paul ion trap or Qq-TOF [3,4,5]. Exopeptidases are active in blood fluid that give rise to non-tryptic peptides that are computationally challenging to identify [6,7,8]. However, incubating blood fluid samples at room temperature also releases tryptic peptides that have been confirmed by high mass accuracy measurements of the parent peptide masses and the use of tryptic protease inhibitors [5, 8]. The endogenous peptides of normal plasma may contain fragments of commonly known blood proteins released from tissues, cells and organs that might be cleaved by proteases ex vivo [3,4,5]. Thus, the representation of proteins by endogenous peptides may not be closely related to protein concentration but rather to the stability of the protein to ex vivo proteolytic attack. Human plasma contains many highly abundant proteins such as albumin, apolipoproteins, protease inhibitors and others that digest efficiently with the exogenous addition of trypsin which masks the detection of low abundance proteins by LC–ESI–MS/MS [3, 9]. The secretion or release of cellular proteins into extracellular space may result in the preferential cleavage of the cellular proteins upon exposure to extracellular protease activity that provides simple experimental access to the cellular factors of plasma. Incubating whole blood at room temperature is known to result in cell mediated degradation [10]. Sampling artefacts prior to freezing may be a major source of pre-analytical variation [8, 11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. To date there has been considerable variation in the peptides observed, and even the trends reported, in the degradation of blood proteins between groups, likely from the large variation in sample collection and processing times [8, 11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. Here sampling variation was addressed by collecting EDTA plasma directly onto ice and then purposefully degrading samples at room temperature under controlled conditions.
A comparison to random MS/MS spectra based on a computerized Random spectra generator (RSG) or instrument noise was used to estimate the type I error rate and showed that the peptides identified per protein by the X!TANDEM algorithm were well separated from the random expectation curve [28, 29]. The probability that the peptide-to-protein distribution of the authentic plasma proteins identified by X!TANDEM were the same as that of random expectation was estimated to be less than one chance in 10,000 (p < 0.0001) (< E−4) [28,29,30,31,32]. The peptide-to-protein counts and peptide intensity values may be compared by analysis with SQL and a generic statistical software system such as S, SAS or R between treatments and controls [30,31,32,33,34]. Comparing the Gene Ontology category distributions of the observed blood proteins to that of the entire protein library is an independent way to examine the likelihood that the proteins observed were simply a random assemblage of molecules [35, 36]. Since noise or near random mis-correlations may result in the identification of giant proteins such as Titin (TTN), Nebulin (NEB) and others at a low frequency in all experimental treatments, an additional way to filter out background noise or random correlations is to compare proteins in one treatment group against control treatments so that errors from noise or contamination that should be shared by all groups can be eliminated [37, 38]. Here, the endogenous tryptic peptides of normal human EDTA plasma collected on ice or incubated at room temperature were extracted by C18 and analyzed by liquid chromatography with micro electrospray ionization and a linear quadrupole ion trap, correlated by X!TANDEM and SEQUEST and analyzed by SQL, R and STRING.
Methods
The 1100 HPLC system was from Agilent (Santa Clara, CA, USA). The LTQ XL linear ion traps were obtained from Thermo Electron Corporation (Waltham, MA, USA). The random spectra generator was slightly modified from that provided by Zhu et al. [28]. The EDTA blood sample tubes were from Becton Dickinson (B367844 K2EDTA, 7.2 mg) (Franklin Lakes, NJ, USA). The C18 with 5 micron particle size and 300 Angstrom pore size was from Agilent Zorbax 300 SB-C18 5-micron (Agilent). The HPLC grade water and acetonitrile was from Caledon laboratories and the formic acid from Fluka (Georgetown, ON, Canada). Sequencing grade trypsin was from Roche (Basel, Switzerland). The ZipTip C18 micro preparative column (Millipore C18 ZipTips, cat # ZTC 18S 096, peptide capacity 5 µg) were from Millipore (Billerica, MA, USA).
Plasma sample collection
Human plasma from 30 normal controls both male and female were collected and aliquoted by JGM under a Comité National d’Ethique de Recherche (CNER) Protocol #201107 “Biospecimen Research” at the Centre Hospitalier de Luxembourg. The plasma was collected in EDTA tubes (B367844 K2EDTA, 7.2 mg, Becton Dickenson) that were rapidly inverted 10 times before packing in ice. The ice-cold plasma was then separated from blood cells at 12,000 RCF for 20 min in a centrifuge set at 4 °C prior to aliquoting the plasma to 225 µL and storing in a − 80 °C freezer. The plasma from one or a few patients per week was sampled by JGM over the course of several months and each plasma sample typically required about ~ 1 h of preparation time from blood collection on ice, and ice-cold centrifugation to obtain plasma, aliquoting and freezing or freeze drying on each of the many sampling days.
Experimental treatments
In an effort to sample the peptides that might degrade in normal human EDTA plasma in circulation, 82 random samples were collected and aliquoted on ice (ICE) and never warmed to room temperature before sampling to prevent the action of proteases ex vivo. In contrast, a set of 88 normal plasma samples were subsequently incubated at room temperature (RT) and sampled from 0 h over 72 h at room temperature to produce the complete range of peptides that might be cleaved in normal plasma under clinical conditions. In total, 170 peptide samples were prepared separately over C18 and then individually analyzed by LC–ESI–MS/MS. The results of the incubation and sampling from 0 to 72 h at room temperature, alongside preserved samples frozen in liquid nitrogen, frozen at − 80 °C and control samples incubated on ice from 0 to 72 h were combined for statistical analysis.
Manual C18 solid phase extraction and injection
A total of 25 μL of the plasma sample was diluted in 200 μL of 5% formic acid for peptide extraction over a 0.6 µL C18 preparative column (Millipore C18 ZipTips, cat # ZTC 18S 096, peptide capacity 5 ug) [7, 8]. The resin was wet 10× with 5% formic acid and 65% acetonitrile, the resin was equilibrated 5× with 5% formic acid, the sample was collected, concentrated and desalted over the resin and eluted in 2 µL (> 3 resin volumes) of 5% formic acid and 65% acetonitrile according to the manufacturer’s instructions. The 2 µL eluent was immediately diluted with 18 µL of 5% formic acid for injection via a 20 µL loop with a Rheodyne manual injector.
LC–ESI–MS/MS
The mass spectrometer was cleaned and calibrated with the manufacturer’s standard mixture, tuned to the Glu Fibrinogen and Angiotensin peptides, and tested for sensitivity by the infusion of GluFib and Angiotensin prior to each block of samples. Three linear ion trap LC–ESI–MS/MS systems were fitted with naive 300-micron ID silica C18 columns (Zorbax 300 SB-C18 5-micron, cat.no.899999-777, Agilent). The columns were conditioned and quality control tested with a digested mixture of alcohol dehydrogenase, cytochrome c and glycogen phosphorylase to confirm the system was working normally versus historical benchmarks [30]. The sensitivity of the system was tested with a BSA digest on the conditioned columns that showed a sensitivity for automatic identification by SEQUEST to ~ 1 fmol on column. The sample was introduced via a 20 µL sample loop and blank samples were injected into a 2 µL/min flow of 5% acetonitrile and 0.1% acetic acid. The gradient of acetonitrile was commenced after 12 min from 15 to 45% ACN over the course of 60 min and then to 65% ACN over 30 min, cleaning at 65% for 5 min before returning to 5% ACN. A roughly equal number of ice versus room temperature samples were analyzed on each of the C18 analytical columns in random order independently a few weeks apart. The column was washed extensively with 50% ACN until a clean background was obtained between samples. A total of ~ 5 µg of extracted and purified peptides from a 0.6 µL ZipTip was manually injected for each analytical HPLC separation at 2 µL/min over a 300 micron ID column (15 cm) with inline filter frits. The peptides were ionized at 4.5 kV via a micro electrospray ion source with 10 L/min of dry N2 gas with a transfer capillary temperature of 200 °C. The precursor ions were randomly and independently sampled (with up to 4 MS/MS spectra samples per precursor) without replacement as the peptides eluted from the HPLC from 350 to 2000 m/z with a Thermo Electron Corporation LTQ XL ion trap mass spectrometer [39]. There was no dynamic exclusion employed.
Peptide MS/MS spectra correlation analysis
A federated library of 154,208 human proteins that differed by at least one amino acid was assembled from NCBI, Ensembl and Swiss Prot and made non-redundant using Structured Query Language (SQL). About 74% of the accession numbers in the human FASTA library have defined gene symbols. A physical filter of at least one thousand (E3) intensity counts for precursor ions was used to limit type I error [28]. The MS and MS/MS spectra of peptides recorded were correlated to the federated library with fully tryptic enzyme specification, a charge state of 2+ or 3+ with up to three missed cleavages with ± 3 m/z and the fragments within 0.5 Da [28,29,30,31, 33] using the X!TANDEM [40] and SEQUEST [41] algorithms.
Computational analysis in SQL and statistical analysis with R
The random and independently sampled parent and fragment m/z and intensity values from MS and MS/MS spectra and the resulting peptide and protein identifications were parsed into an SQL database [33]. The SQL database utilized a complex key also known as an SHA1-HASH for each MS/MS peptide-protein identification to ensure that only the best fit of each MS/MS spectra at only one charge state was accepted, and thus no MS/MS spectra was assigned to more than one peptide sequence. The peptide-to-protein counts of serum samples were previously statistically analyzed using the generic statistical system S [8] or SAS [28, 30, 31, 33, 34] but in this study the data was analyzed using the generic open-source R statistical system [37]. The total peptide-to-protein fits (MS/MS correlation to peptides in proteins) of the authentic experimental data were compared to that of a computer generated set of random MS/MS spectra to compute the probability of type I error over the entire experiment using the goodness of fit test [28,29,30,31,32,33,34]. The distributions and quantile plots of the peptide-to-protein counts, log10 intensity, expectation values (E), peptide mass and delta mass values calculated by X!TANDEM were computed in R [30,31,32]. The peptide p-values from X!TANDEM were used to compute the cumulative p-values for peptide sequences and gene symbols [33]. The corresponding FDR q-values were computed by the method of Benjamini and Hochberg [42].
STRING analysis
Protein identifications made by LC–ESI–MS/MS of human blood may be tested by comparing the distribution of protein descriptive terms over categories using goodness of fit analysis [36] or looking for protein networks and interactions with respect to random expectation [43]. The STRING algorithm V10 (Search Tool for the Retrieval of Interacting Genes/Proteins) was used to create a network of plasma proteins from the set of proteins with at least 5 tryptic peptides [35]. The probability distribution of proteins over Gene Ontology (GO) terms, as well as their protein interactions from the Kyoto Encyclopedia of Genes and Genomes (KEGG) protein interactions were calculated with respect to random expectation using the corrections provided by Bonferroni, or Benjamini and Hochberg [42] for the number of hypotheses tested.
Results
Plasma versus random, noise and dust controls
The best fit peptides from X!TANDEM and SEQUEST were collected together in SQL Server. Plasma collected on ice showed tryptic peptides from many well-known proteins C4A, C4B, C3, KNG1, ITIH4, ALB, FGA, APOA4 from both the X!TANDEM and SEQUEST algorithms. The X!TANDEM and SEQUEST algorithms further identified many proteins, and protein complexes, of an apparent cellular origin. Comparison to random MS/MS spectra, source noise, and dust were used to control type I error [7, 28, 29, 44]. The SEQUEST results were apparently contaminated by giant proteins [33] such as TTN (34,350 aa), NEB (8560 aa), CCDC168 (7081 aa), SYNE1 (8797 aa), OBSCN (6620 aa), TROPH (6907), SYNE2 (6885), MACF1 (7388 aa), AHNAK (5890) and others such as DST, LRP1, XIRP2, MUC16, FSIP2, LRP1B, PLEC that were identified in random spectra controls and are likely random mis-correlations [28, 29]. In addition to TTN, the analysis of source noise from blank runs indicated that ZNF503, VWCE, API5, TMEM199, SYT7, TPSG1, USP9X, DACT3, SUGP2, CXorf31, MYO3A, KLRC4, CYCS, RGP1, SYN1, DSPP, IMMP1L CAMK1, ASPM, AHNAK, among others were correlated by SEQUEST from noise spectra collected from LC–ESI–MS/MS gradient runs with blank samples [28, 29]. Furthermore, a tryptic digest of laboratory dust may contain keratins, POTEI, POTEE, POTEF, ACTA2, ACTB, ACTG2, GFAP, POTEM, MUC5AC and others in addition to those found in solvent gradients alone [7]. In contrast to SEQUEST, relatively few peptides from TTN were observed by X!TANDEM. Furthermore, X!TANDEM identified many cellular proteins from endogenous peptides in EDTA plasma from the ion trap that showed good agreement with the proteins from exogenous tryptic digestion of blood fluids and in agreement with previous results of endogenous peptides from the Paul 3D ion trap [3]. After discounting apparently spurious proteins from the negative controls of random, noise (solvents alone) or typically digested dust controls, the many cellular proteins identified by X!TANDEM and SEQUEST show excellent agreement with the many cellular proteins identified from serum or plasma by independent chromatographic separation and exogenous tryptic digestion [3, 33]. In agreement with previous results from exogenous digest by trypsin [44], endogenous peptides were observed from proteins in the ~ 1 ng/ml range such as creatine kinase CKB that exists in 1–5 ng/mL, acid phosphatase ACP in the 1–5 ng/ml range and carcinoembryonic antigen CEACAM5 in the 0.5–5 ng/ml concentration range.
Statistical analysis of X!TANDEM results at the level of peptides
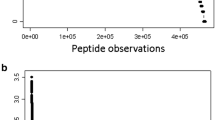
The X!TANDEM algorithm was selected to examine the proteins in plasma on ice or at room temperature since it directly computes a p-value that the protein detected was merely random chance and has been compared to that of random spectra or noise for fully tryptic peptides from exogenous digestion [28,29,30,31]. The X!TANDEM algorithm fit MS/MS spectra to peptides in proteins but there may be multiple protein accessions that contain the same peptide sequence. One method to remove these redundant correlations to more than one protein accession number is to select the accession number with the highest number of correlations per gene symbol and then analyze the data at the peptide and protein level to yield a list of distinct RANK1 peptides. Starting with 583,927 unique MS/MS Spectra of > 1000 counts, the X!TANDEM algorithm correlated to some 181,962 peptide sequences and after selecting only the best scoring charge state (+ 2 or + 3) only 181,032 correlations remained. Once only the lowest p-value peptide sequence was accepted 94,669 redundant peptides potentially observed in more than 1 protein collapsed to 22,287 distinct RANK1 peptide observations in 7030 protein Gene Symbols with a 1% FDR and 3979 with a 0.02% FDR. The delta mass distribution was Gaussian from − 2 to + 2 Da (Fig. 1). There were many more high-intensity peptides observed in the room temperature versus the preserved EDTA plasma (Fig. 2). The list of Gene Symbols identified with the cumulative p-value and the FDR corrected q-value are provided in the Additional file 1, Additional file 2, Additional file 3, Additional file 4.

Statistical analysis of the 22,287 RANK1 correlations by X!TANDEM to tryptic peptides and phosphopeptides. The redundant peptide correlations from X!TANDEM were filtered to only RANK1 peptides from the protein accession with the highest number of correlations in each Gene Symbol category. a Peptide counts per gene symbol; b the intensity distribution of the correlated peptides; c the intensity of the preserved (1) versus room temperature (2) samples; d the average p-value of the peptides for each gene symbol; e the cumulative p-value for each gene symbol (inset, the log10 cumulative p-value for each gene symbol); f the normal quantile plot of peptide delta mass. The Filtered (Filter 2) data can be found in Additional file 1, Additional file 2, Additional file 3, Additional file 4

The Log10 Intensity distribution of 22,287 Density RANK1 correlations to tryptic peptide sequences with and without phosphorylation by X!TANDEM for preserved versus degraded samples. The RANK1 peptides from X!TANDEM were sorted into log10 intensity bins to compare preserved and degraded samples. a The frequency versus log intensity distribution of 82 preserved samples; b the frequency versus log intensity distribution of 88 room temperature samples. The Filtered (Filter 2) data can be found in Additional file 1, Additional file 2, Additional file 3, Additional file 4
X!TANDEM filtering
The complex key system in the SQL Server database was used to filter out all but the best p-value MS/MS to peptide fit at either 2+ or 3+ such that no mass spectra was assigned to more than one peptide sequence at one charge state. Filtering out re-use of the MS/MS spectra at different charge states or peptides sequences to accept only the best fit of the data to Gene Symbols with 5 or more independent correlations to precursors of greater than 1000 detector counts to fully tryptic peptides by X!TANDEM provided robust statistical reliability in agreement with previous results [28,29,30,31,32]. Accepting only protein accessions with 5 or more independent peptides resulted in 28,580 correlations. However, many protein types are represented by more than one related protein accession sequence that share the same gene symbol and so filtering only the single accession with the most correlations per gene symbol results in 7174 remaining MS/MS correlations to peptides in 733 distinct Gene Symbols (protein types). The STRING algorithm further reduced the number of gene symbols to about 660 (Table 1). Manual annotation to discount contaminants from source noise, random MS/MS spectra, dust controls, and closely related proteins from large gene families such as collagens results in at least 510 protein types. The set of ≥ 510 ten gene symbols with at least 5 best-fit peptides by X!TANDEM showed an FDR q-value of E−9 (i.e. FDR = 0.000000001) to E−227.
Ice cold samples protein interactions
Collecting EDTA plasma samples on ice and aliquoting the samples on ice resulted in low amounts of endogenous peptides as analyzed by LC–ESI–MS/MS. Proteins with at least 5 peptides from X!TANDEM showed that peptides from albumin were prominent in plasma collected on ice and never incubated at room temperature along with peptides from PLC, PTK2, PELP1, CACNA1A Zinc Finger Proteins, TRRAP (Transformation/transcription domain-associated protein) and others (Fig. 3 and Additional file 1, Additional file 2, Additional file 3, Additional file 4).

The filtered (best charge state 2+ or 3+ and peptide sequence) and sorted ratio peptide-to-protein counts (MS/MS correlations to peptides in a protein accession). The ratio of peptide-to-protein counts for each protein was computed from all room temperature samples (ALLRT) over all preserved samples (ALLICE) from a roughly equal number of LC–ESI–MS/MS runs for each group treatment. The proteins with the highest number of correlations in Room Temperature plasma (ALLRT) relative to samples preserved on Ice (ALLICE) was complement C4B. The Filtered (Filter 2) data can be found in Additional file 1, Additional file 2, Additional file 3, Additional file 4
Room temperature incubation protein interactions
Plasma has been shown to express a net weak tryptic activity towards exogenous protein substrates [45]. New peptides, and larger numbers of peptides, were observed from plasma proteins incubated at room temperature including many cellular proteins such as G-proteins, dyneins, myosin and actin associated filament proteins, kininogen, signalling enzymes such as small G proteins and other factors, such as nucleic acid binding proteins among many others (Fig. 4 and Additional file 1, Additional file 2, Additional file 3, Additional file 4).

The filtered proteins of human plasma frozen or maintained on ice from at least 5 fully tryptic endogenously cleaved peptides as correlated by the X!TANDEM algorithm. The Network was produced using STRING confidence view. The Filtered (Filter 2) data can be found in Additional file 1, Additional file 2, Additional file 3, Additional file 4
Analysis of X!TANDEM results across gene ontology terms
The set of 7174 proteins sequences with 5 independent peptides was collapsed by STRING to a set of 660 Gene Symbols that after manual annotation were examined in detail using the R generic statistical analysis system. To date about 74% of the 154,208 accession numbers in the Federated Human library have defined gene symbols. Where gene symbols exist they may be used to trigger the automated bioinformatics analysis by STRING. The STRING algorithm looks for previously established relations between the set of identified proteins [35, 36, 43]. The STRING algorithm tests to determine if the data set is merely a random assemblage of proteins or a network of proteins that show structural and functional relationships and so are known to exist and function together. The comparison of the distribution of proteins across category distributions provides a means to estimate the type I error rate of the data set [35, 36, 43]. The STRING analysis shows a previously established relationship with many of the processed proteins of human plasma including myosins, kininogen, leucine-rich repeat proteins, adenylate cyclase, EH Domain proteins Wiscott Aldrich Syndrome interacting protein WAS/WASL and dynactin that in turn show relationships to other preferentially degraded proteins. Multiple peptides from hundreds of cellular factors that are associated with human disease were observed in normal human plasma including phosphoinositide kinase, FYVE-finger-containing protein (PIKFYVE), serum/glucocorticoid regulated kinase (SGK3), monomeric GTPase activating proteins such as ARHGAP31 or RHOJ, Williams-Beuren Syndrome Chromosomal Region 18 Protein (DNAJC30), Zinc Finger proteins (ZNF), homeobox proteins and many other signalling and nucleic acid binding factors. The complete network derived from at least three peptides from X!TANDEM is too large to illustrate here and so is in the Additional file 1, Additional file 2, Additional file 3, Additional file 4.
Biological process
The endogenous peptides of normal human plasma were highly enriched in regulatory factors involved in morphogenesis, differentiation, neuron projection/development, cell organization, cellular organization or biogenesis and others to name a few (Table 2).
Molecular function
The molecular functions of the proteins identified from the normal plasma peptides included a striking enrichment of proteins involved in ion and anion binding, adenyl ribonucleotide binding such as ATP/adenyl nucleotide binding, protein binding, carbohydrate binding, structural molecules and others (Table 3).
Cellular component
The normal human plasma was enriched in endogenous peptides from proteins of the extracellular matrix, cytoskeleton, basement membrane, microtubule, cytoplasm, organelles and others (Table 4). Proteins associated with membrane bound vesicles, secretion, exocytosis and exosomes were also observed.
Tissues
The peptides detected by LC–ESI–MS/MS showed a strongly non-random distribution and were enriched in proteins that are expressed pleiotropically in all tissues and organs that should be expected to appear in plasma. Proteins observed in cells as well as from specific tissues including the ovary and internal female genital organs, the colorectum and large intestine, adrenal gland, spinal cord, gall bladder, heart, urinary bladder, and blood plasma among others were enriched (not shown).
Disease
Blood plasma was found to be enriched in peptides from proteins previously associated with disease, including monogenic, anatomical, autosomal, nervous system, musculoskeletal, recessive and sensory system disease (not shown).
Protein–protein interaction and pathway analysis
The proteins of normal human plasma as identified by the degradation products from endogenous tryptic activities were closely associated with KEGG pathways (Kyoto Encyclopedia of Genes and Genomes) such as extracellular matrix, focal adhesions as well as cell and calcium signalling (Table 5). A total of 1165 protein interactions were observed (expected 590) from 461 gene symbols that showed a probability of much less than p < 0.0001 [35].
Room temperature-specific proteins
In contrast to samples maintained on ice over time, samples incubated at room temperature from 1 to 72 h showed some individual proteins such as C4B with 1 or 2 orders of magnitude more peptide correlations per protein (Fig. 5). Random mis-correlations from SEQUEST to giant proteins, noise and dust contamination are likely shared by all treatments to a roughly equal amount. Since false positive identifications should tend to be roughly equal in the room temperature versus ice cold samples, imposing a fivefold increase with temperature should tend to remove false positive identifications that should be common to both conditions. Thus, an alternative means to reduce the presence of random or noise mis-correlations to proteins was to compute the ratio of room temperature to ice cold samples that resulted in a list of 1171 gene symbols specific to room temperature including many cellular factors associated with membrane bound organelles, vesicle formation, exocytosis and vesicle targeting (Tables 6, 7, 8). The ratio of RT to ICE was computed for the proteins identified by SEQUEST where at least one peptide was also observed per gene symbol by X!TANDEM.

The protein interaction network of proteins from EDTA plasma that was incubated at room temperature detected from at least 5 fully tryptic endogenous peptides by the X!TANDEM algorithm. The Network was produced using STRING confidence view. The Filtered (Filter 2) data can be found in Additional file 1, Additional file 2, Additional file 3, Additional file 4
Discussion
Linear ion trap mass spectrometry
High resolution and low resolution mass spectrometers have been compared for the analysis of peptides from human blood fluids that showed good agreement but the ion trap is more sensitive and more robust [3,4,5, 7, 8]. High resolution mass spectrometers are required for the use of accurate mass strategy [46], for the use of isotopic [47, 48] or isobaric strategies [49] or carbohydrate analysis [50]. Simple ion traps are sufficient for the identification of tryptic peptides from Eukaryotes and so are appropriate for the sampling of peptides from human samples that have been fractioned by chromatography [51,52,53]. The low error rate of an ion trap mass spectrometer for identifying tryptic peptides does not result from a high resolution measurement of the precursor m/z value (± 3 m/z) but rather from fitting the MS/MS fragmentation spectra (± 0.5 Da) by algorithms such as SEQUEST [41] and X!TANDEM [40]. The low error rate of the sensitive ion trap for proteins with multiple peptides is well established and shows that confidence builds with the number of peptides identified such that proteins with three or more independent peptides show a low error rate [28,29,30,31, 54, 55]. The masses of most of the amino acid residues in MS/MS spectra can be effectively resolved by measurement to 0.5 Da by a simple ion trap but a high resolution mass spectrometer does not perform this task much better in practical terms. Moreover, proteins identified by low resolution mass spectrometers have been confirmed by Western blots, siRNA, drugs, immune cyto staining or GFP fusions [3, 37, 38]. Robust and sensitive ion traps [39, 56] may not always resolve the isotopic forms (R ≥ 1000–3000) leading to a concern that more than one peptide might be isolated resulting in mis-correlation of the MS/MS spectra. However, pre-separation of proteins or polypeptides by organic solvents, partition chromatography (including bio-specific or affinity separation), or differential centrifugation effectively enriches a sub-set of polypeptides that prevents co-elution of multiple tryptic peptides with similar m/z values [30, 31, 44]. Where polypeptides were efficiently pre-fractionated prior to analytical separation for LC–ESI–MS/MS the probability of tryptic peptides from more than one protein with similar m/z values eluting at the same time is dramatically reduced. The analysis of 300,000 synthetic peptides [57] agrees with the use of random and noise MS/MS control spectra that the fit of experimental MS/MS spectra to the predicted fragmentation patterns is statistically excellent and thus the reliable basis of peptide identification. The recent paper of Zolg using 300,000 synthetic peptides [57] confirms the observed MS/MS spectra closely matches the theoretical and thus MS/MS spectra matching with a simple ion trap is apparently sufficient to identify and quantify tryptic peptides that show low type I error by statistical analysis with respect to random expectation from null models of noise and random MS/MS spectra [28,29,30,31, 54, 55]. If there was a concern for false positive identification, the proteins p-values generated by the goodness of fit of the experimental MS/MS spectra might be corrected by the bona fide FDR method of Benjamini and Hochberg [42]. However, given the low p-values presented here, the corrected q-values from the FDR calculated by the method of Benjamini and Hochberg [42] were highly significant.
Agreement between algorithms
The aim of this study was to determine the nature of the proteins and protein complexes that are susceptible to degradation by comparing endogenous tryptic peptides from normal human EDTA plasma on ice versus plasma incubated at room temperature. There are multiple sources of experimental and algorithm error that must be computed efficiently using SQL Server and a classical statistical analysis system such as R to interpret the results of the LC–ESI–MS/MS experiments with excellence. Plasma has a net mild tryptic activity [45] but also shows both C and N terminal exopeptidase activity [8, 58] that might confound the results of tryptic correlations. It is well established that C and N terminal exopeptidases are active in blood plasma and these enzymes cleave amino acids from both ends of tryptic peptides [8]. The SEQUEST algorithm has been shown to provide a type I error rate of about 1% where three independent peptides correlate to the same protein in tryptic digests [55]. In agreement with previous statistical analysis, the results of X!TANDEM were found to be highly reliable. Thus, to provide a more complete list of plasma peptides for the Additional file 1, Additional file 2, Additional file 3, Additional file 4 that also limits error, we have selected the proteins identified by SEQUEST with at least 5 peptides where the proteins were also detected by X!TANDEM at least once (see Additional file 5: Figure S1, Additional file 6: Figure S2).
Multiple correlations from the same MS/MS spectra
A large source of error in LC–ESI–MS/MS is the re-use of the same MS/MS spectra that is correlated to multiple peptide sequences. The multiple peptide sequences correlated to the same MS/MS spectra may be eliminated by using a complex key also known as an SHA1-HASH for each MS/MS peptide-protein identification to retain only the highest scoring charge state and peptide sequence in SQL Server. Hence, the use of the automated functions of the SQL Server Database has the profound effect of avoiding the re-use of MS/MS spectra in proteomics to lower type I errors.
Reporting results per Gene Symbol
There is significant redundancy in the LC–ESI–MS/MS of peptides since the same peptide sequence might be observed in multiple isoforms of the same protein and multiple predicted protein sequences. Collapsing the identical peptides observed in homologous proteins to one representative gene symbol is a convenient way to represent proteomic data that reduces the redundancy from proteins that share overlapping amino acid sequences. The analysis of the results stored in SQL Server can be computed on a per Gene Symbol basis using R to eliminate redundancy from similar protein sequences. Previously we collapsed these multiple proteins into protein types by manual annotation [7], or mapping short sequences into longer representative sequences using SQL or BLAST [7, 44]. The Gene symbols associated with the protein variants may be used to efficiently summarize the LC–ESI–MS/MS data using the SQL Server/R data storage and statistical analysis system.
Random mis-correlation, noise and contamination
The SEQUEST algorithm may result in the false positive identification of large proteins [33] such as Titin (TTN), Nebulin (NEB), Spectrin protein (SYNE1) and others from random or noise spectra [28] at a low frequency. However, when the results of many LC–ESI–MS/MS experiments are analyzed together the number of correlations to these contaminants may become a concern. Another concern is the presence of keratins, mucins like MUC5AC, and POTE that may be detected in laboratory dust. A direct experimental and practical means to limit type I error from these sources is to compare experimental results to random spectra, noise from blank LC–ESI–MS/MS runs, and laboratory dust samples as negative controls. Alternatively making a ratio of treatments to look for proteins that differ between treatments also appears to be an effective means to limit the false positive identification of large proteins that tend to show a ratio of ~ 1.0 between sample sets. We conclude that caution must be exercised when analyzing proteins on these known contaminant lists using SEQUEST to ensure the frequency of detection is much higher than background.
X!TANDEM
X!TANDEM is known to be a rigorous MS/MS correlation algorithm that relies on the fragment pattern from the precursor peptide to match endogenous tryptic peptides to amino acid sequences [40, 59]. In agreement with previous results, the measured type I error rate of fully tryptic peptides from X!TANDEM was low and so is one means to assign identity to low abundance endogenous tryptic peptides from plasma with confidence [28, 29]. Here, the use of a physical noise filter of E3 counts, together with the significant fit of at least three peptides [55] by X!TANDEM led to the apparent identification of many cellular proteins in plasma that are provided in the Additional file 1, Additional file 2, Additional file 3, Additional file 4. The X!TANDEM algorithm indicates that a pool of cellular proteins, with some major plasma proteins, are degraded in plasma with incubation at room temperature. The results indicate that the cellular proteins released into plasma are not as stable as the major plasma proteins. The cumulative p-values and FDR corrected q-values from X!TANDEM indicate that peptides from 510 types of functionally or structurally related proteins [7, 44] can be accurately identified by a one-step C18 chromatography preparation.
STRING analysis
The STRING algorithm revealed a complex set of relationships at the level of homology, transcriptional regulation, protein–protein interactions, protein families and signal pathways. In agreement with the significant p-values assigned by the X!TANDEM correlation algorithms to MS/MS sequence matches, the STRING algorithm indicated that the results are not a random assemblage of proteins. It has been previously established that the distribution of proteins across categories may be used to estimate the probability that the data set is no different than random expectation that may serve as an estimate of type I error of identification [35, 36]. Structural, functional and protein-interactions were revealed in the relationships between many of the degraded proteins of human plasma and a variety of cellular factors were observed that may have great biological significance. Myosins, kininogen, leucine-rich repeat proteins, adenylate cyclase, EH Domain proteins Wiscott Aldrich Syndrome interacting protein WAS/WASL and dynactin and their interactors as well as cellular signaling proteins such as PYK2 including regulatory proteins such as receptors, GTPases such as RHO, SH2 and SH3 domain containing proteins, signalling enzymes such as phospholipases or MAPK may be released into the plasma as protein complexes. Cytoskeleton associated Ankyrin itself from red blood cells has been previously shown to encode an activity that results in the cleavage of complement [60]. The endogenous peptides of normal human serum, including kininogen (KNG1), and Ankyrin Domain Family proteins such as POTEJ have been previously detected by DEAE [7]. The significant distributions across networks, GO terms, the significant protein-interactions, and the expectation of the MS/MS spectra to the predicted peptide sequences using goodness of fit by X!TANDEM all agree the data obtained was highly significant [3, 33, 34]. A significantly large number of reciprocal protein–protein interactions were observed between the set of proteins identified that essentially precludes high rates of false positive identification, and clearly indicates that some circulating protein complexes are susceptible to degradation by endogenous tryptic peptide activities soon after incubation at room temperature. The simplest explanation is that functionally or structurally related proteins and/or protein complexes were preferentially cleaved by endopeptidases within the plasma.
Enrichment of tryptic peptides at room temperature
One simply strategy to avoid potential false positive correlations is to take the ratio of the room temperature (where tryptic proteases may act) versus ice cold samples (where tryptic protease activity is significantly reduced) thus largely removing the results of random mis-correlations and noise shared by both. Accepting only tryptic peptides that show a fivefold increase in correlation after incubation at room temperature using the SEQUEST and X!TANDEM algorithms results in a list of cellular proteins that are cleaved in plasma at room temperature. Since the apparently spurious mis-correlations to titin (TTN) or other giant proteins from SEQUEST are shared by all treatments, one strategy to avoid noise and mis-correlation from the sensitive SEQUEST algorithm is to select tryptic peptides that are specific to room temperature incubation thereby largely eliminating the bias towards erroneous identifications to giant proteins. Quantifying the endogenous peptides may represent a direct approach to monitor levels of cellular protein in the plasma between experimental treatments or physiological states while avoiding the interfering peptides from albumin, apolipoproteins and other common plasma molecules [7]. Peptides from KNG1 were observed at room temperature and it is known to play a role in the regulation of the complement cascade [61]. Alternative splicing of the KNG1 transcript generates the cysteine protease inhibitor, high-molecular-weight kininogen (HMWK), that functions in the regulation of plasminogen that activates the complement system and blood coagulation, and in turn is cleaved by the enzyme kallikrein to produce bradykinin that regulates inflammatory responses [62]. Kininogen and Ankyrin have been previously observed from tryptic peptides from serum [7] and in degraded plasma samples [27].
Conclusion
The results here showed agreement that LC–ESI–MS/MS of high intensity blood peptides via an electrospray source with a linear ion trap may confidently identify peptides from blood plasma that are associated with extracellular matrix, cellular factors, specific tissues and organs as well as known diseases. The Type I error from correlating experimental MS/MS spectra to those predicted for human tryptic peptides was limited by X!TANDEM or by taking the ratio of room temperature versus preserved samples that was supported by the comparison to the null model of random peptides and the non-random distribution across GO terms. The agreement between independent algorithms, the p-value of the peptides generated in X!TANDEM and the FDR q-value of the proteins generated by SQL Server/R, the ratio between experimental treatments, and the statistically significant structural and functional relationships between the proteins identified were all consistent with the veracity of the set of peptides and proteins identified from endogenous tryptic peptides by LC–ESI–MS/MS. The simplest model that explains all of the data collected here and elsewhere is that the major proteins of plasma such as albumin and apolipoproteins are relatively stable in plasma but that complexes of cellular proteins in circulation are more susceptible to degradation. A pool of cellular proteins and/or protein complexes were apparently degraded by a tryptic like protease activity soon after incubation of plasma at room temperature. The ex vivo artefacts of tryptic peptides released by endogenous endopeptidases may be used to identify and quantify plasma proteins. More than 500 proteins including cellular regulatory factors such as phospholipases, GTPases, Zinc finger proteins, lipases, and others may be directly monitored in human plasma by a one step C18 solid phase extraction followed by micro electrospray LC–ESI–MS/MS. It will apparently be possible to analyze the low abundance blood proteins using the artefacts of endogenous tryptic peptides by simple extraction with C18 followed by micro electrospray LC–ESI–MS/MS using the precursor m/z and resulting fragment ions from ≥ 22,000 endogenous tryptic peptides fit by X!TANDEM to ≥ 500 gene symbols presented in the Additional file 1, Additional file 2, Additional file 3, Additional file 4. Understanding and documenting the set of peptides cleaved from proteins that appear over the baseline immediately after sample collection is a pre-condition for the rational exploration of blood peptides across disease or other physiological states. Collecting EDTA plasma directly on ice essentially prevents the degradation of blood proteins.
Abbreviations
- ACN:
-
Acetonitrile
- RSG:
-
Random spectra generator
References
Putnam F. The plasma proteins: structure function, and genetic control. 2nd ed. New York: Academic Press; 1975.
Ashwood ER, Burtis CA, editors. Tietz fundamentals of clinical chemistry. 5th ed. Philadelphia: Saunders; 2001. p. 1091.
Tucholska M, et al. The endogenous peptides of normal human serum extracted from the acetonitrile-insoluble precipitate using modified aqueous buffer with analysis by LC–ESI–Paul ion trap and Qq-TOF. J Proteomics. 2010;73(6):1254–69.
Tucholska M, et al. Endogenous peptides from biophysical and biochemical fractionation of serum analyzed by matrix-assisted laser desorption/ionization and electrospray ionization hybrid quadrupole time-of-flight. Anal Biochem. 2007;370:228–45.
Williams D, et al. Precipitation and selective extraction of human serum endogenous peptides with analysis by quadrupole time-of-flight mass spectrometry reveals posttranslational modifications and low-abundance peptides. Anal Bioanal Chem. 2010;396:1223–47.
Adkins JN, et al. Toward a human blood serum proteome: analysis by multidimensional separation coupled with mass spectrometry. Mol Cell Proteomics. 2002;1(12):947–55.
Marshall J, et al. Human serum proteins preseparated by electrophoresis or chromatography followed by tandem mass spectrometry. J Proteome Res. 2004;3(3):364–82.
Marshall J, et al. Processing of serum proteins underlies the mass spectral fingerprinting of myocardial infarction. J Proteome Res. 2003;2:361–72.
Zhu P, et al. Mass spectrometry of peptides and proteins from human blood. Mass Spectrom Rev. 2011;30(5):685–732.
Kaisar M, et al. Plasma degradome affected by variable storage of human blood. Clin Proteomics. 2016;13:26.
Banks RE. Preanalytical influences in clinical proteomic studies: raising awareness of fundamental issues in sample banking. Clin Chem. 2008;54(1):6–7.
Tuck MK, et al. Standard operating procedures for serum and plasma collection: early detection research network consensus statement standard operating procedure integration working group. J Proteome Res. 2009;8(1):113–7.
Karsan A, et al. Analytical and preanalytical biases in serum proteomic pattern analysis for breast cancer diagnosis. Clin Chem. 2005;51(8):1525–8.
Semmes OJ, et al. Evaluation of serum protein profiling by surface-enhanced laser desorption/ionization time-of-flight mass spectrometry for the detection of prostate cancer: I. Assessment of platform reproducibility. Clin Chem. 2005;51(1):102–12.
de Noo ME, et al. Reliability of human serum protein profiles generated with C8 magnetic beads assisted MALDI-TOF mass spectrometry. Anal Chem. 2005;77(22):7232–41.
Li J, et al. Independent validation of candidate breast cancer serum biomarkers identified by mass spectrometry. Clin Chem. 2005;51(12):2229–35.
Pilny R, et al. Surface-enhanced laser desorption ionization/time-of-flight mass spectrometry reveals significant artifacts in serum obtained from clot activator-containing collection devices. Clin Chem. 2006;52(11):2115–6.
Albrethsen J, et al. Preanalytical and analytical variation of surface-enhanced laser desorption-ionization time-of-flight mass spectrometry of human serum. Clin Chem Lab Med. 2006;44(10):1243–52.
Koomen JM, et al. Direct tandem mass spectrometry reveals limitations in protein profiling experiments for plasma biomarker discovery. J Proteome Res. 2005;4(3):972–81.
Rai AJ, et al. HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics. 2005;5(13):3262–77.
Callesen AK, et al. Reproducibility of mass spectrometry based protein profiles for diagnosis of breast cancer across clinical studies: a systematic review. J Proteome Res. 2008;7(4):1395–402.
van Winden AW, et al. Validation of previously identified serum biomarkers for breast cancer with SELDI-TOF MS: a case control study. BMC Med Genomics. 2009;2(1):4.
Spencer K. The influence of different sample collection types on the levels of markers used for Down’s syndrome screening as measured by the Kryptor Immunosassay system. Ann Clin Biochem. 2003;40(Pt 2):166–8.
Govorukhina NI, et al. Influence of clotting time on the protein composition of serum samples based on LC–MS data. J Chromatogr B Anal Technol Biomed Life Sci. 2008;877:1281–91.
Gast MC, et al. Influence of sample storage duration on serum protein profiles assessed by surface-enhanced laser desorption/ionisation time-of-flight mass spectrometry (SELDI-TOF MS). Clin Chem Lab Med. 2009;47(6):694–705.
West-Nielsen M, et al. Sample handling for mass spectrometric proteomic investigations of human sera. Anal Chem. 2005;77(16):5114–23.
West-Norager M, et al. Unravelling in vitro variables of major importance for the outcome of mass spectrometry-based serum proteomics. J Chromatogr, B: Anal Technol Biomed Life Sci. 2007;847(1):30–7.
Zhu P, et al. Chi square comparison of tryptic peptide-to-protein distributions of tandem mass spectrometry from blood with those of random expectation. Anal Biochem. 2011;409(2):189–94.
Zhu P, et al. Peptide-to-protein distribution versus a competition for significance to estimate error rate in blood protein identification. Anal Biochem. 2011;411:241–53.
Bowden P, et al. Quantitative statistical analysis of standard and human blood proteins from liquid chromatography, electrospray ionization, and tandem mass spectrometry. J Proteome Res. 2012;11:2032–47.
Florentinus AK, et al. Identification and quantification of peptides and proteins secreted from prostate epithelial cells by unbiased liquid chromatography tandem mass spectrometry using goodness of fit and analysis of variance. J Proteomics. 2012;75:1303–17.
Florentinus AK, et al. The Fc receptor-cytoskeleton complex from human neutrophils. J Proteomics. 2011;75:450–68.
Bowden P, Beavis R, Marshall J. Tandem mass spectrometry of human tryptic blood peptides calculated by a statistical algorithm and captured by a relational database with exploration by a general statistical analysis system. J Proteomics. 2009;73:103–11.
Bowden P, et al. Meta sequence analysis of human blood peptides and their parent proteins. J Proteomics. 2010;73:1163–75.
von Mering C, et al. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005;33(Database issue):D433-7.
Zhu P, et al. Comparison of protein expression lists from mass spectrometry of human blood fluids using exact peptide sequences versus BLAST. Clin Proteomics. 2007;2(3–4):185–203.
Howard, J.C., et al., OxLDL receptor chromatography from live human U937 cells identifies SYK(L) that regulates phagocytosis of oxLDL. Anal Biochem, 2016.
Jankowski A, Zhu P, Marshall JG. Capture of an activated receptor complex from the surface of live cells by affinity receptor chromatography. Anal Biochem. 2008;380:235–48.
Schwartz JC, Senko MW, Syka JE. A two-dimensional quadrupole ion trap mass spectrometer. J Am Soc Mass Spectrom. 2002;13(6):659–69.
Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20(9):1466–7.
Yates JR 3rd, et al. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67(8):1426–36.
Benjamini Y, Hochberg Y. Controlling false discovery rate: A practical approach to multiple testing. J R Stat Soc. 1995;57(1):289–300.
Marshall J, et al. Creation of a federated database of blood proteins: a powerful new tool for finding and characterizing biomarkers in serum. Clin Proteomics. 2014;11(1):3.
Tucholska M, et al. Human serum proteins fractionated by preparative partition chromatography prior to LC–ESI–MS/MS. J Proteome Res. 2009;8:1143–55.
Sardesai VM, Provido HS. A fluorometric method for determining the tame esterase (tryptic) activity of plasma. J Lab Clin Med. 1965;65:1023–9.
Smith RD, et al. An accurate mass tag strategy for quantitative and high-throughput proteome measurements. Proteomics. 2002;2(5):513–23.
Shevchenko A, et al. Rapid ‘de novo’ peptide sequencing by a combination of nanoelectrospray, isotopic labeling and a quadrupole/time-of-flight mass spectrometer. Rapid Commun Mass Spectrom. 1997;11(9):1015–24.
Ong SE, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–86.
DeSouza L, et al. Search for cancer markers from endometrial tissues using differentially labeled tags iTRAQ and cICAT with multidimensional liquid chromatography and tandem mass spectrometry. J Proteome Res. 2005;4(2):377–86.
An HJ, et al. Profiling of glycans in serum for the discovery of potential biomarkers for ovarian cancer. J Proteome Res. 2006;5(7):1626–35.
Washburn MP, Wolters D, Yates JR 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19(3):242–7.
McCormack AL, et al. Direct analysis and identification of proteins in mixtures by LC/MS/MS and database searching at the low-femtomole level. Anal Chem. 1997;69(4):767–76.
Link AJ, et al. Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17(7):676–82.
Moore RE, Young MK, Lee TD. Qscore: an algorithm for evaluating SEQUEST database search results. J Am Soc Mass Spectrom. 2002;13(4):378–86.
Cargile BJ, Bundy JL, Stephenson JL Jr. Potential for false positive identifications from large databases through tandem mass spectrometry. J Proteome Res. 2004;3(5):1082–5.
Stafford GC. Instrumental aspects of positive and negative ion chemical ionization mass spectrometry. Environ Health Perspect. 1980;36:85–8.
Zolg DP, et al. Building ProteomeTools based on a complete synthetic human proteome. Nat Methods. 2017;14(3):259–62.
Li Y, et al. Circulating proteolytic products of carboxypeptidase N for early detection of breast cancer. Clin Chem. 2014;60(1):233–42.
Craig R, Beavis RC. A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun Mass Spectrom. 2003;17(20):2310–6.
Hermann J, Barel M, Frade R. Human erythrocyte ankyrin, a cytoskeleton component, generates the p57 membrane proteinase which cleaves C3, the third component of complement. Biochem Biophys Res Commun. 1994;204(2):453–60.
Dobo J, et al. Cleavage of kininogen and subsequent bradykinin release by the complement component: mannose-binding lectin-associated serine protease (MASP)-1. PLoS ONE. 2011;6(5):e20036.
Michael IP, et al. Biochemical and enzymatic characterization of human kallikrein 5 (hK5), a novel serine protease potentially involved in cancer progression. J Biol Chem. 2005;280(15):14628–35.
Authors’ contributions
JD calibrated, tuned and quality controlled the instruments and performed LC–ESI–MS/MS analysis and edited the manuscript. AF-M performed the sample aliquoting and performed LC–ESI–MS/MS analysis and edited the manuscript. JA performed LC–ESI–MS/MS analysis and edited the manuscript. AF performed LC–ESI–MS/MS analysis. PB collected the data into an SQL database for statistical analysis in R. JM planned the experiment, and performed the clinical plasma collection and sample treatments and storage experiments. All authors read and approved the final manuscript.
Acknowledgements
This research was paid for by the Fonds National de la Recherche, Mobility of Researcher award through Luxembourg Institute of Health LIH (formerly CRP Sante), Luxembourg to JGM and a Grant from the Integrated Biobank of Luxembourg. We gratefully acknowledge the review and critique of this manuscript by Dr. Fay Betsou and Dr. R.A. Phillips of IBBL.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The raw data is provided in the additional files.
Consent for publication
No material from any other publication was used in this publication.
Ethics approval and consent to participate
Ethical approval and informed consent was obtained through the Comité National d’Ethique de Recherche (CNER) Protocol #201107 “Biospecimen Research” at the Centre Hospitalier de Luxembourg.
Funding
This research was paid for by the Fonds National de la Recherche, Mobility of Researcher award through Luxembourg Institute of Health LIH (formerly CRP Sante) and the Integrated Biobank of Luxembourg (IBBL) to JGM.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
12014_2017_9174_MOESM5_ESM.bmp
Additional file 5: Figure S1. The proteins of human plasma from samples on ice plus samples at room temperature from at least 5 endogenously cleaved peptides as correlated by the X!TANDEM algorithm. The network was produced using STRING confidence view. The Filtered (Filter 2) data can be found in Additional file 1, Additional file 2, Additional file 3, Additional file 4.
12014_2017_9174_MOESM6_ESM.bmp
Additional file 6: Figure S2. The proteins of human plasma from samples on ice plus samples at room temperature from at least one fully tryptic peptides as correlated by the X!TANDEM algorithm. The network was produced using STRING confidence view. The probability that so many protein–protein interactions could be obtained by random chance was estimated by STRING to be p ≤ 0.0001. The Filtered (Filter 2) data can be found in Additional file 1, Additional file 2, Additional file 3, Additional file 4.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Dufresne, J., Florentinus-Mefailoski, A., Ajambo, J. et al. The proteins cleaved by endogenous tryptic proteases in normal EDTA plasma by C18 collection of peptides for liquid chromatography micro electrospray ionization and tandem mass spectrometry. Clin Proteom 14, 39 (2017). https://doi.org/10.1186/s12014-017-9174-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12014-017-9174-9