
Abstract
Background
Recent studies have revealed that dysregulated expression of long non-coding RNA nicotinamide nucleotide transhydrogenase antisense RNA 1 (lncRNA NNT-AS1) is associated with cell tumorigenicity in non-small cell lung cancer. However, the exact molecular mechanisms of NNT-AS1 in lung squamous cell carcinoma (LUSC) remain largely unknown.
Methods
The expression of NNT-AS1, microRNA (miR)-22 and Forkhead box protein M1 (FOXM1) was measured using quantitative real-time polymerase chain reaction or western blot, respectively. The interaction between miR-22 and NNT-AS1 or FOXM1 was confirmed using a dual-luciferase reporter assay and RNA immunoprecipitation assay. Cell migration and invasion abilities were measured by Transwell assay. Flow cytometry was used to detect apoptotic cells.
Results
NNT-AS1 and FOXM1 were up-regulated but miR-22 was down-regulated in LUSC tissues and cell lines. NNT-AS1 was a sponge of miR-22, and NNT-AS1 deletion suppressed the migration and invasion but induced apoptosis in LUSC cells. FOXM1 was a target of miR-22, and overexpression of miR-22 inhibited cell carcinogenesis in LUSC by targeting FOXM1. Additionally, NNT-AS1 could directly regulate FOXM1 expression by binding to miR-22 in LUSC cells.
Conclusion
LncRNA NNT-AS1 contributes to cell carcinogenesis in LUSC by regulating the miR-22/FOXM1 axis, providing a novel insight into the pathogenesis of LUSC and a new potential therapeutic target for LUSC treatment.
Similar content being viewed by others


Background
Lung cancer remains the leading cause of cancer-associated mortality worldwide. Non-small cell lung cancer (NSCLC) is one of the main sub-types of lung cancer, accounting for around 80–85% of all lung cancers [1, 2]. NSCLC is histologically categorized into lung squamous cell carcinoma (LUSC), lung adenocarcinoma, large cell carcinoma and neuroendocrine cancer [3]. Among these, LUSC is a common type of NSCLC with a high risk of metastasis and relapse [4]. Thus, further investigations to identify therapeutic biomarkers and molecular mechanisms on the pathogenesis of LUSC are necessary.
Long non-coding RNAs (lncRNAs) is a kind of non-coding RNA of more than 200 nucleotides in length without protein-coding capacity [5]. Increasing evidence has identified that lncRNAs are novel master contributors in regulating malignant physiological or pathological cellular processes, such as carcinogenesis, metastasis, angiogenesis, metabolism, and so on [6, 7]. Also, lncRNAs have been identified to play important functions in the initiation and development of various malignancies, including lung cancer [8, 9]. LncRNA nicotinamide nucleotide transhydrogenase antisense RNA 1 (lncRNA NNT-AS1) is a newly detected cancer-related lncRNA. Recently, dysregulated expression of NNT-AS1 has been identified in several solid tumors, such as gastric, cervical, breast and ovarian cancer, and is associated with cancer tumorigenesis, metastasis, and progression of tumors [10,11,12,13]. Nevertheless, the role of NNT-AS1 in LUSC remains largely unknown.
MicroRNAs (miRNAs) are defined as small non-coding RNA molecules, which control gene expression via interacting with the mRNAs of target genes [14, 15]. To date, many miRNAs are known to be involved in the development of cancer through acting as anti-tumor or carcinogenic agents in LUSC, and miRNAs are potential biomarkers for diagnosis and prognosis and therapeutic targets for LUSC [16,17,18]. LncRNAs contain multiple miRNA-binding sites that have been investigated as competing endogenous RNAs (ceRNA) to indirectly modulate mRNAs through binding to miRNAs [19, 20]. The important roles of the lncRNA-miRNA-mRNA regulatory network and a protein-protein interaction network in lung cancer development have also been identified [21, 22]. Therefore, it is necessary to identify the important regulatory pathway or therapeutic biomarkers according to lncRNA-miRNA-mRNA networks.
In the present study, we aimed to investigate the biological effects of NNT-AS1 in LUSC cell carcinogenesis, and to explore the interaction network of NNT-AS1 in LUSC development. This study may provide novel therapeutic targets for LUSC treatment.
Materials and methods
Patients and specimens
This study was permitted by the Ethics Committee of Huaihe Hospital of Henan University and informed consent was obtained from all patients. Tumor specimens and paratumor samples were obtained from 46 paired LUSC patients who underwent surgical resections at Huaihe Hospital of Henan University and were immediately stored at − 80 °C until further analysis. None of the patients received any preoperative treatment and all underwent histopathological examination. Moreover, all procedures carried out in studies involving human participants were in accordance with the ethical standards of the Ethics Committee of Huaihe Hospital of Henan University, and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Cell culture
Human bronchial epithelial cell line 16HBE and LUSC cell lines (H1703, SW900, H2170 and U1752) were obtained from Shanghai Academy of Life Science (Shanghai, China), and were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Carlsbad, CA, USA) harboring 10% fetal bovine serum and 1% penicillin-streptomycin solution (Gibco) at 37 °C with 5% CO2.
Cell transfection
The small interfering RNA (siRNA) sequences targeting NNT-AS1 (si-NNT-AS1), siRNA negative control (si-con), pcDNA3.1-NNT-AS1 overexpression vector (NNT-AS1), pcDNA3.1-Forkhead box protein M1 (FOXM1) overexpression vector (FOXM1), and pcDNA3.1 empty vector (vector) were synthesized by Invitrogen (Carlsbad, CA, USA). The miR-22 mimic (miR-22), miR-22 inhibitor (in-miR-22) and their corresponding negative control (miR-con or in-miR-con) were obtained from RIBOBIO (Guangzhou, China). All oligonucleotides or vectors were respectively transfected into H1703 and H2170 cells using Lipofectamine 3000 (Invitrogen) according to the standard procedure.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted using a TRIzol kit (Invitrogen) according to the standard protocol. For NNT-AS1 and FOXM1 detection, total RNA was reversely transcribed into complementary DNA (cDNA) using a high-capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA), and then qPCR was carried out with SYBR Premix Ex Taq (Qiagen, Valencia, CA, USA). As to miR-22 detection, cDNA was synthesized using a TaqMan Reverse Transcription Reagents Kit (Applied Biosystems) and amplified with TransScript Green One-Step qRT-PCR SuperMix (Qiagen). The relative expression was analyzed by the 2-△△Ct method and normalized byglyceraldehyde-3-phosphate dehydrogenase (GADPH) or U6 small nuclear RNA (snRNA). The specific primer sequences used were as follows: NNT-AS1, F: 5′-TCTCCTAAGTCGAGGACTAGC-3′, R: 5′-AGGCACTCACTAGCATCACGCT-3′;FOXM1, F: 5′-GGAGCAGCGACAGGTTAAGG-3′, R: 5′-GTTGATGGCGAATTG TATCATGG-3′; miR-22, F: 5′-GGGGGATCCCTGGGGCAGGACCCT-3′, R: 5′-GGGGAATTCAACGTATCATCCACCC-3′; U6: F:5′-GCTTCGGCAGCACATATACTAAAAT-3′, R: 5′-CGCTTCACGAATTTGCGTGTCAT-3′; GAPDH, F: 5′-AACTTTGGCATTGTGGAAGG-3′, R: 5′-ACACATTGGGGGTAGGAACA-3′.
Dual-luciferase reporter assay
The WT or MUT NNT-AS1/FOXM1 3′UTR containing a miR-22 binding site was amplified and cloned into the pmirGLO Vector (Promega, Shanghai, China). Then H1703 and H2170 cells were seeded in 24-well plates and co-transfected with miR-22 or miR-con and the corresponding luciferase reporter vectors using Lipofectamine 2000 for 48 h. Subsequently, a dual-luciferase reporter assay kit (Promega) was used to measure the changes in relative luciferase activity.
RNA immunoprecipitation (RIP) assay
Transfected cells were lysed, and the lysate was incubated with magnetic beads coated with anti-Ago2 (Millipore, Billerica, MA, USA) or IgG antibody (Abcam, Cambridge, MA, USA). Finally, the enrichment of NNT-AS1 or FOXM1 was examined using qRT-PCR.
Cell migration and invasion
For detection of migrated cells, top chambers were loaded with cell suspension (5 × 104 cells). Then 500 μL of DMEM mixed with 10% FBS was added to the bottom chambers. After incubation for 48 h at 37 °C, migrated cells were fixed with methanol and stained with 0.1% crystal violet. For detection of invaded cells, the upper chamber membranes were pre-coated with Matrigel (Becton Dickinson, Franklin Lakes, NJ, USA) and the measurement method was similar to the steps of cell migration. Finally, cells in five randomly selected fields were counted with a microscope.
Cell apoptosis assay
Cell apoptosis was analyzed using an Annexin V-FITC/PI apoptosis detection kit (BD Biosciences, San Jose, CA, USA) according to the supplier’s direction. Transfected cells were collected and re-suspended with binding buffer, followed by staining with 10 μL of fluorescein isothiocyanate (FITC) annexin V and propidium iodide (PI). Finally, apoptotic cells were measured by flow cytometry.
Western blot
Proteins were isolated using RIPA lysis buffer (Beyotime, Shanghai, China) and then were quantified by a bicinchoninic acid (BCA) method following the standard protocol. An equal amount of the extracts was treated with 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride membranes (Millipore). Subsequently, the membranes were incubated with specific primary antibodies against FOXM1 (1:1000, 20,459, Cell Signaling Technology, Boston, MA, USA) and β-actin (1:1000, 4967, Cell Signaling Technology), followed by incubation with HRP-conjugated secondary antibody (1:1000; ab9482; Abcam). Finally, protein signals were visualized using an ECL method.
Statistical analysis
Experimental data from three independent experiments were expressed as the mean ± standard deviation (SD) and analyzed using GraphPad Prism 7 software (GraphPad Inc., San Diego, CA, USA). The differences between two groups were analyzed by Student’s t test and multiple comparisons were performed by one-way analysis of variance (ANOVA). The correlation analysis was performed using Spearman’s correlation test. P < 0.05 was regarded as statistically significant.
Results
NNT-AS1 and FOXM1 are up-regulated but miR-22 is down-regulated in LUSC tissues and cell lines
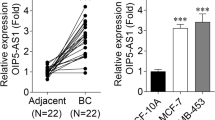
To explore the potential biological functions of NNT-AS1, miR-22 and FOXM1 involved in LUSC carcinogenesis, we detected the levels of NNT-AS1, miR-22 and FOXM1 in 46 paired LUSC tissues and paratumor tissues. Subsequently, qRT-PCR results showed that, compared with the non-tumor tissues, NNT-AS1 and FOXM1 were up-regulated, while miR-22 was down-regulated in LUSC tissues (Fig. 1a-c). The same expression changes were also found in the LUSC cell lines compared with the human bronchial epithelial cell line 16HBE (Fig. 1d-f). After that, expression association was analyzed and a negative correlation between miR-22 and NNT-AS1 (R = -0.3807, P = 0.0091) or FOXM1 (R = -0.4157, P = 0.0041), and a positive correlation between NNT-AS1 and FOXM1 were observed (R = 0.5750, P < 0.0001) (Fig. 1g-i). Therefore, we suspected that NNT-AS1, miR-22 and FOXM1 might be associated with the progression of LUSC.

NNT-AS1 and FOXM1 are up-regulated but miR-22 is down-regulated in LUSC tissues and cell lines. a-c Levels of NNT-AS1, miR-22 and FOXM1 were detected in LUSC tissues and non-tumor tissues using qRT-PCR. d-f Levels of NNT-AS1, miR-22 and FOXM1 in human bronchial epithelial cell line 16HBE and LUSC cell lines (H1703, SW900, H2170 and U1752) were detected using qRT-PCR. g-i The expression association among NNT-AS1, miR-22 and FOXM1 was analyzed using Spearman’s correlation test. *P < 0.05, **P < 0.01, ***P < 0.001.
NNT-AS1 is a sponge of miR-22
To detect the relationship between NNT-AS1 and miR-22, StarBase software was used. As shown in Fig. 2a, miR-22 was identified to have a binding site with NNT-AS1. Moreover, the transfection efficiency of miR-22 overexpression, NNT-AS1 overexpression, and NNT-AS1 deletion is shown and examined in Sup. Fig. 1. To verify this prediction, the dual-luciferase reporter assay was performed. The results indicated that miR-22 overexpression reduced the luciferase activities of the NNT-AS1-WT reporter vector compared to the control group, while there was no obvious change in NNT-AS1-MUT reporter after miR-22 transfection in H1703 and H2170 cells (Fig. 2b, c). Furthermore, RIP assay also suggested that NNT-AS1 pull-down by Ago2 was significantly enriched in miR-22-transfected H1703 and H2170 cells (Fig. 2d, e). We also found that overexpression of NNT-AS1 inhibited miR-22 expression, while NNT-AS1 deletion promoted miR-22 expression in H1703 and H2170 cells (Fig. 2f, g). These data indicated that NNT-AS1 was a sponge of miR-22 and negatively regulated miR-22 expression in LUSC cells.

NNT-AS1 is a sponge of miR-22. a The binding site between miR-22 and NNT-AS1 is shown. b, c Luciferase activity was examined in H1703 and H2170 cells co-transfected with NNT-AS1-WT, or NNT-AS1-MUT and miR-22, or miR-con. d, e Enrichment of NNT-AS1 was analyzed in H1703 and H2170 cells transfected with miR-con or miR-22 after RIP. f, g The level of miR-22 was detected in H1703 and H2170 cells transfected with vector, NNT-AS1, si-con, or si-NNT-AS1 using qRT-PCR. *P < 0.05, **P < 0.01, ***P < 0.001
NNT-AS1 deletion suppresses cell migration and invasion but induces cell apoptosis in LUSC by sponging miR-22
Based on the relationship between NNT-AS1 and miR-22, we further wanted to explore the roles of the NNT-AS1/miR-22 axis in LUSC cells. Then H1703 and H2170 cells were transfected with si-con, si-NNT-AS1, si-NNT-AS1 + in-miR-con, or si-NNT-AS1 + in-miR-22. Meanwhile, the knockdown efficiency of in-miR-22 was detected and presented in Sup Fig. 1. Then, qRT-PCR results revealed that miR-22 expression was significantly elevated when NNT-AS1 was silenced, but this increase was attenuated after miR-22 inhibition in H1703 and H2170 cells (Fig. 3a, b). After that, transwell assay indicated that NNT-AS1 deletion inhibited the migration and invasion of H1703 and H2170 cells, while this inhibition could be reversed by the inhibition of miR-22 (Fig. 3c-f). Meanwhile, we also found that apoptotic cells were significantly increased by NNT-AS1 silence but were decreased by following miR-22 inhibition in H1703 and H2170 cells (Fig. 3g, h). Taken together, the evidence showed that NNT-AS1 deletion suppressed cell migration and invasion but induced cell apoptosis in LUSC by interacting with miR-22.

NNT-AS1 deletion suppresses the migration and invasion but induces apoptosis in LUSC cells by sponging miR-22. H1703 and H2170cells were transfected with si-con, si-NNT-AS1, si-NNT-AS1 + in-miR-con, or si-NNT-AS1 + in-miR-22. a, b Expression of miR-22 was measured using qRT-PCR. c-f Cell migration and invasion were determined by transwell assay. g, h Cell apoptosis was analyzed by flow cytometry. *P < 0.05, **P < 0.01, ***P < 0.001
FOXM1 is a target of miR-22
We further explored the molecular mechanisms by which the NNT-AS1/miR-22 axis regulated LUSC cell carcinogenesis, the potential target genes of miR-22 were screened using microT-CDS program and FOXM1 was identified to contain the putative binding site of miR-22 (Fig. 4a). Subsequently, a dual-luciferase reporter assay analysis showed that overexpression of miR-22 reduced the luciferase activities of the FOXM1-WT reporter vector, but not the FOXM1-MUT reporter vector inH1703 and H2170 cells (Fig. 4b, c). Moreover, RIP assay also proved the interaction between miR-22 and FOXM1 because of the obvious enrichment of FOXM1 in H1703 and H2170 cells (Fig. 4d, e). Additionally, we observed that the expression of FOXM1 was inhibited by overexpression of miR-22 but was promoted by miR-22 inhibition (Fig. 4f, g). Thus, these results showed that miR-22 interacted with FOXM1 to suppress its expression in LUSC cells.

FOXM1 is a target of miR-22. a The binding site between miR-22 and FOXM1 is presented. b, c Luciferase activity was examined in H1703 and H2170 cells co-transfected with FOXM1-WT, or FOXM1-MUT and miR-22, or miR-con. d, e The interaction between miR-22 and FOXM1 was analyzed using RIP assay. f, g The level of FOXM1 was examined in H1703 and H2170 cells transfected with miR-con, miR-22, in-miR-con, in-miR-22 using western blot. *P < 0.05, **P < 0.01, ***P < 0.001
Overexpression of miR-22 inhibits cell mobility in LUSC by targeting FOXM1
According to the interaction between miR-22 and FOXM1, we further investigated whether the miR-22/FOXM1 axis was responsible for LUSC cell mobility. Synchronously, transfection efficiency of FOXM1 overexpression was detected and exhibited in Sup Fig. 1. Then H1703 and H2170 cells were transfected with miR-con, miR-22, miR-22 + vector, or miR-22 + FOXM1. After transfection, we found that the level of FOXM1 was inhibited by overexpression of miR-22, but was rescued by following FOXM1 restoration in H1703 and H2170 cells (Fig. 5a, b). Immediately, rescue assay was performed and the results indicated that overexpression of FOXM1 could reverse miR-22 restoration induced inhibition on cell migration and invasion (Fig. 5c-f), as well as promotion on cell apoptosis in LUSC (Fig. 5g, h). In all, we demonstrated that overexpressed miR-22 inhibited cell mobility in LUSC by directly interacting with FOXM1.

Overexpression of miR-22 inhibits cell carcinogenesis in LUSC by targeting FOXM1. H1703 and H2170 cells were transfected with miR-con, miR-22, miR-22 + vector, or miR-22 + FOXM1. a, b Western blot was used to detect the level of FOXM1 in H1703 and H2170 cells. c-f Migrated and invaded cells were detected by transwell assay. g, h Apoptotic cells were measured using flow cytometry. *P < 0.05, **P < 0.01, ***P < 0.001
NNT-AS1 indirectly regulates FOXM1 expression by sponging miR-22 in LUSC cells
Based on the miR-22/FOXM1 axis, we further explored whether NNT-AS1 could regulate the FOXM1 expression by binding to miR-22. Subsequently, western blot analysis suggested that the level of FOXM1 was promoted by overexpression of NNT-AS1, while it was inhibited by following miR-22 restoration in H1703 cells (Fig. 6a). Meanwhile, we found that inhibition of miR-22 could rescue NNT-AS1 deletion induced inhibition of the expression of FOXM1 in H2170 cells (Fig. 6b). Altogether, these data suggested that NNT-AS1 functioned as a molecular sponge of miR-22 to regulate FOXM1 expression.

NNT-AS1 indirectly regulates FOXM1 expression by sponging miR-22 in LUSC cells. a Expression of FOXM1 was examined using western blot in H1703 cells transfected with vector, NNT-AS1, NNT-AS1 + miR-con, or NNT-AS1 + miR-22. b Expression of FOXM1 was determined by western blot in H2170 cells transfected with si-con, si-NNT-AS1, si-NNT-AS1 + in-miR-con, or si-NNT-AS1 + in-miR-22. *P < 0.05, **P < 0.01, ***P < 0.001
Discussion
LUSC is the second most common type and accounts for roughly 30% of NSCLC. To date, molecular-targeted therapies have remarkably improved the overall survival of lung adenocarcinoma, but have little effect on the treatment of LUSC [23, 24]. Thus, there is an unmet need to develop novel therapeutics based on current genomic approaches for LUSC. LncRNAs are the principal elements of the mammalian transcriptome, which are emerging as master regulators in a wide variety of cellular processes, such as the cell cycle, differentiation, growth, and apoptosis, mediating the pathogenesis of many malignancies, including LUSC [25]. For examples, lncRNA HULC contributed to LUSC development by regulating the PTPRO/NF-κB signaling pathway [26]. LncRNA SNHG1 was over-expressed in LUSC tissues and promoted cell tumorigenesis in LUSC through regulation of ZEB1 by TAp63 [27]. Thus, lncRNAs may be useful therapeutic targeting in LUSC due to their multiple functions.
NNT-AS1, a novel cancer-related lncRNA, has been identified to function as an oncogene to participate in tumor cell development in many cancers [28, 29]. Recent studies have also revealed that NNT-AS1 was elevated in NSCLC, and dysregulated NNT-AS1expression was involved in the regulation of cell proliferation, invasion and cisplatin resistance in NSCLC [30, 31]. Here, we found that NNT-AS1 might also be involved in the development of LUSC. In this study, StarBase databases and results proved that NNT-AS1 was increased in LUSC tissues, indicating the potential regulatory roles of NNT-AS1 in LUSC. Functionally, NNT-AS1 deletion was identified to suppress cell migration and invasion, but induce cell apoptosis in LUSC. Taken together, the evidence indicated that NNT-AS1 functioned as an oncogene to promote cell tumorigenesis in LUSC.
In our study of the StarBase databases, we also found that miR-22 was up-regulated and FOXM1 was down-regulated in LUSC tissues compared to normal tissues. Moreover, the same changes of their expression were also observed in LUSC tissues and cell lines. Therefore, miR-22 and FOXM1 might be associated with the progression of LUSC. MiR-22 has been suggested to be frequently decreased, and exert an anti-tumor function in several cancers, including lung cancer [32]. Furthermore, miR-22 is a useful predictive biomarker and therapeutic target for lung cancer [33, 34]. In this study, miR-22 was also down-regulated in LUSC, and functional experiments showed that miR-22 overexpression inhibited cell migration and invasion, but induced cell apoptosis in LUSC. Moreover, our results confirmed that miR-22 was a target of NNT-AS1, and was negatively regulated by NNT-AS1 in LUSC tissues. The rescue assay also indicated that NNT-AS1 regulated cell carcinogenesis by binding to miR-22 in LUSC. Thus, an NNT-AS1/miR-22 regulatory axis was identified in the regulation of LUSC development.
FOXM1 belongs to the Forkhead box (Fox) family of transcription factors. FOXM1 is often expressed in actively dividing cells and plays important roles in cell cycle progression [35]. FOXM1 has been identified to be upregulated in lung cancer and is critical for the tumorigenicity of malignant lung cells, making it a new target for cancer diagnosis and therapies [36, 37]. Furthermore, FOXM1, as the downstream target, is frequently involved in the regulation of cancer progression [38]. This study verified that FOXM1 was a target of miR-22. Thus the miR-22/FOXM1 axis was investigated in LUSC. Subsequently, the rescue assay showed that overexpression of miR-22 inhibited cell mobility in LUSC by targeting FOXM1. Moreover, co-expression analysis suggested that NNT-AS1 could regulate FOXM1 expression by binding to miR-22 in LUSC cells. Therefore, an NNT-AS1/miR-22/FOXM1 regulatory network was identified in the LUSC.
In conclusion, our results demonstrated that FOXM1 was up-regulated in LUSC and overexpression of FOXM1 contributed to the tumorigenicity of LUSC cells. Additionally, molecular analysis showed that an NNT-AS1/miR-22/FOXM1 regulatory network in LUSC and NNT-AS1 silence exerted anti-tumor effects through the miR-22/FOXM1 axis in LUSC, providing a novel insight into the pathogenesis of LUSC.
Availability of data and materials
The data from this study are available in this published article.
Abbreviations
- LUSC:
-
Lung squamous cell carcinoma
- FOXM1:
-
Forkhead box protein M1
- NSCLC:
-
Non-small cell lung cancer
- FITC:
-
Fluorescein isothiocyanate
- PI:
-
Propidium iodide
- BCA:
-
Bicinchoninic acid
- ANOVA:
-
analysis of variance
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30.
Goldstraw P, Ball D, Jett JR, Le Chevalier T, Lim E, Nicholson AG, et al. Non-small-cell lung cancer. Lancet. 2011;378(9804):1727–40.
Chansky K, Sculier JP, Crowley JJ, Giroux D, Van Meerbeeck J, Goldstraw P. The International Association for the Study of Lung Cancer staging project: prognostic factors and pathologic TNM stage in surgically managed non-small cell lung cancer. J Thorac Oncol. 2009;4(7):792–801.
Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22(6):725–36.
Zhang J, Zhang P, Wang L, Piao HL, Ma L. Long non-coding RNA HOTAIR in carcinogenesis and metastasis. Acta Biochim Biophys Sin Shanghai. 2014;46(1):1–5.
Peng WX, Koirala P, Mo YY. LncRNA-mediated regulation of cell signaling in cancer. Oncogene. 2017;36(41):5661–7.
Kumar MM, Goyal R. LncRNA as a therapeutic target for angiogenesis. Curr Top Med Chem. 2017;17(15):1750–7.
Zhang H, Chen Z, Wang X, Huang Z, He Z, Chen Y. Long non-coding RNA: a new player in cancer. J Hematol Oncol. 2013;6(37).
Liu L, Zhang Y, Cao W. Highly expressed lncRNA LOC730101 promotes lung cancer cell growth through Wnt canonical pathway. Biochem Biophys Res Commun. 2017;493(2):992–7.
Chen B, Zhao Q, Guan L, Lv H, Bie L, Huang J, et al. Long non-coding RNA NNT-AS1 sponges miR-424/E2F1 to promote the tumorigenesis and cell cycle progression of gastric cancer. J Cell Mol Med. 2018;22(10):4751–9.
Hua F, Liu S, Zhu L, Ma N, Jiang S, Yang J. Highly expressed long non-coding RNA NNT-AS1 promotes cell proliferation and invasion through Wnt/beta-catenin signaling pathway in cervical cancer. Biomed Pharmacother. 2017;92:1128–34.
Huang Y, Shi J, Xu Y. Long non-coding RNA NNT-AS1 contributes to cell proliferation, metastasis and apoptosis in human ovarian cancer. Oncol Lett. 2018;15(6):9264–70.
Li Y, Lv M, Song Z, Lou Z, Wang R, Zhuang M. Long non-coding RNA NNT-AS1 affects progression of breast cancer through miR-142-3p/ZEB1 axis. Biomed Pharmacother. 2018;103:939–46.
Jiang C, Hu X, Alattar M, Zhao H. miRNA expression profiles associated with diagnosis and prognosis in lung cancer. Expert Rev Anticancer Ther. 2014;14(4):453–61.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–8.
Wang Q, Liu S, Zhao X, Wang Y, Tian D, Jiang W. MiR-372-3p promotes cell growth and metastasis by targeting FGF9 in lung squamous cell carcinoma. Cancer Med. 2017;6(6):1323–30.
Hou Y, Li L, Ju Y, Lu Y, Chang L, Xiang X. MiR-101-3p regulates the viability of lung squamous carcinoma cells via targeting EZH2. J Cell Biochem. 2017;118(10):3142–9.
Gao X, Wang Y, Zhao H, Wei F, Zhang X, Su Y, et al. Plasma miR-324-3p and miR-1285 as diagnostic and prognostic biomarkers for early stage lung squamous cell carcinoma. Oncotarget. 2016;7(37):59664–75.
Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–52.
Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21(11):1253–61.
Wang X, Yin H, Zhang L, Zheng D, Yang Y, Zhang J, et al. The construction and analysis of the aberrant lncRNA-miRNA-mRNA network in non-small cell lung cancer. J Thorac Dis. 2019;11(5):1772–8.
Zhao J, Cheng W, He X, Liu Y, Li J, Sun J, et al. Construction of a specific SVM classifier and identification of molecular markers for lung adenocarcinoma based on lncRNA-miRNA-mRNA network. Onco Targets Ther. 2018;11:3129–40.
Raponi M, Dossey L, Jatkoe T, Wu X, Chen G, Fan H, et al. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res. 2009;69(14):5776–83.
Filipits M. New developments in the treatment of squamous cell lung cancer. Curr Opin Oncol. 2014;26(2):152–8.
Li R, Yang YE, Jin J, Zhang MY, Liu X, Liu XX, et al. Identification of lncRNA biomarkers in lung squamous cell carcinoma using comprehensive analysis of lncRNA mediated ceRNA network. Artif Cells Nanomed Biotechnol. 2019;47(1):3246–58.
Xu Y, Li J, Wang P, Zhang Z, Wang X. LncRNA HULC promotes lung squamous cell carcinoma by regulating PTPRO via NF-kappaB. J Cell Biochem. 2019;120(12):19415–421.
Zhang HY, Yang W, Zheng FS, Wang YB, Lu JB. Long non-coding RNA SNHG1 regulates zinc finger E-box binding homeobox 1 expression by interacting with TAp63 and promotes cell metastasis and invasion in Lung squamous cell carcinoma. Biomed Pharmacother. 2017;90:650–8.
Lu YB, Jiang Q, Yang MY, Zhou JX, Zhang Q. Long noncoding RNA NNT-AS1 promotes hepatocellular carcinoma progression and metastasis through miR-363/CDK6 axis. Oncotarget. 2017;8(51):88804–14.
Wang Q, Yang L, Hu X, Jiang Y, Hu Y, Liu Z, et al. Upregulated NNT-AS1, a long noncoding RNA, contributes to proliferation and migration of colorectal cancer cells in vitro and in vivo. Oncotarget. 2017;8(2):3441–53.
Cai Y, Dong ZY, Wang JY. LncRNA NNT-AS1 is a major mediator of cisplatin chemoresistance in non-small cell lung cancer through MAPK/slug pathway. Eur Rev Med Pharmacol Sci. 2018;22(15):4879–87.
Shen Q, Jiang Y. LncRNA NNT-AS1 promotes the proliferation, and invasion of lung cancer cells via regulating miR-129-5p expression. Biomed Pharmacother. 2018;105:176–81.
Xin M, Qiao Z, Li J, Liu J, Song S, Zhao X, et al. miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase: evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer. Oncotarget. 2016;7(28):44252–65.
Jiang W, Han X, Wang J, Wang L, Xu Z, Wei Q, et al. miR-22 enhances the radiosensitivity of small-cell lung cancer by targeting the WRNIP1. J Cell Biochem. 2019;120(10):17650–61.
Franchina T, Amodeo V, Bronte G, Savio G, Ricciardi GR, Picciotto M, et al. Circulating miR-22, miR-24 and miR-34a as novel predictive biomarkers to pemetrexed-based chemotherapy in advanced non-small cell lung cancer. J Cell Physiol. 2014;229(1):97–9.
Zhang J, Zhang J, Cui X, Yang Y, Li M, Qu J, et al. FoxM1: a novel tumor biomarker of lung cancer. Int J Clin Exp Med. 2015;8(3):3136–40.
Sun Q, Dong M, Chen Y, Zhang J, Qiao J, Guo X. Prognostic significance of FoxM1 expression in non-small cell lung cancer. J Thorac Dis. 2016;8(6):1269–73.
Koo CY, Muir KW, Lam EW. FOXM1: from cancer initiation to progression and treatment. Biochim Biophys Acta. 2012;1819(1):28–37.
Zhao L, Liu L, Dong Z, Xiong J. miR-149 suppresses human non-small cell lung cancer growth and metastasis by inhibiting the FOXM1/cyclin D1/MMP2 axis. Oncol Rep. 2017;38(6):3522–30.
Acknowledgements
None.
Funding
This work was approved by Major Science and Technology Projects of Kaifeng Science and Technology Bureau (19ZD011 and 18ZD008 ) and Kaifeng Science and Technology Bureau Project (No. 110342).
Author information
Authors and Affiliations
Contributions
GQ and LL were the main designers of this study. GQ and LL performed the experiments and analyzed the data. GQ drafted the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All the tumor specimens and paratumor samples were collected with written informed consent in accordance with the Declaration of Helsinki and with the approval of the Ethical Committee of Henan University (No. 2017055, Date: 2017/05/10, Henan, China).
Consent for publication
Not applicable.
Competing interests
All the authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1
Sup Fig. 1 Transfection efficiency of NNT-AS1 overexpression, si-NNT-AS1, miR-22 overexpression, in-miR-22, and FOXM1 overexpression was detected in LUSC cells.***P < 0.001.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, J., Qi, G. & Li, L. LncRNA NNT-AS1 promotes lung squamous cell carcinoma progression by regulating the miR-22/FOXM1 axis. Cell Mol Biol Lett 25, 34 (2020). https://doi.org/10.1186/s11658-020-00227-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11658-020-00227-8