
Abstract
Background
Despite the pervasiveness of migraine, the underlying pathophysiological mechanisms initiating migraine attacks are far from well understood and are matter of scientific debate.
Objective
In this narrative review, we discuss key evidence for that suggest a peripheral origin or central origin and provide directions for future studies that may provide further clarification.
Discussion
Migraine pathogenesis is considered to involve the trigeminovascular system, a term that encompasses the trigeminal nerve and its axonal projections to the intracranial blood vessels. Beyond any doubt both peripheral and central mechanisms are involved in migraine pathogenesis, but an unresolved question is the how the initial activation occurs in a migraine attack. Evidence favoring a peripheral origin of migraine attacks, i.e., initial events occur outside of the blood–brain barrier, include the importance of sensitization of perivascular sensory afferents early on in a migraine attack. Evidence favoring a central origin include the occurrence of prodromal symptoms, migraine aura, and activation of structures within the central nervous system early in and during a migraine attack.
Conclusions
Both peripheral and central mechanisms are likely involved in a migraine attack, e.g., peripheral nociceptive input is necessary for pain transmission and cortical activity is necessary for pain perception. Yet, the debate of whether migraine attacks are initiated a peripheral or central site remains unresolved. The increased focus on prodromal symptoms and on the development of a human model of migraine aura will possibly provide key arguments needed to answer this question in the near future. Until then, we cannot draw firm conclusions and the debate goes on.
Video link
Video recording of the debate held at the 1st International Conference on Advances in Migraine Sciences (ICAMS 2022, Copenhagen, Denmark) is available at: https://www.youtube.com/watch?v=NC0nlcKohz0.
Graphical Abstract

Similar content being viewed by others
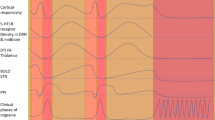


Introduction
Migraine is a common neurological disorder characterized by recurrent headache attacks of moderate-to-severe pain intensity accompanied by a range of symptoms including nausea, vomiting and hypersensitivity to light and sounds [1, 2]. Despite its pervasiveness, the underlying pathophysiological mechanisms initiating migraine attacks are far from well understood and are matter of scientific debate. On one side, evidence favoring a peripheral origin of migraine attacks, i.e., initial events occur outside of the blood–brain barrier, include the importance of sensitization of perivascular sensory afferents early on in a migraine attack [3, 4], and that migraine attacks can be triggered and attenuated using pharmacological compounds that do not appear to need to cross the blood–brain barrier to exert their effects [5]. In contrast, occurrence of prodromal symptoms, i.e., a symptoms before the onset of headache, migraine aura, and data supporting dysfunction of the diencephalon and the brainstem suggest a migraine attack generator localized within the central nervous system [3, 4]. In this narrative review, we will describe some of the key evidence that suggest a peripheral origin or a central origin of migraine attacks, respectively. In particular, we focus on the possible role of prodromal symptoms and central dysfunction, migraine aura, and migraine pain (Table 1).
Search strategy and selection criteria
We searched MEDLINE (in both cases from database inception to August 1, 2022) for original research articles, systematic reviews and meta-analyses. We used the search term “migraine” in combination with the terms “epidemiology”, “pathophysiology”, “premonitory”, “prodromal”, “aura”, “central dysfunction”, “nociception”, “diagnosis” and/or “treatment”. We preferentially selected publications from the past 10 years but did not exclude commonly referenced and highly regarded older publications. We also searched the reference lists of articles identified by this search strategy and selected those we judged relevant.
Migraine attacks are of peripheral origin
Prodromal symptoms and central dysfunction
Individuals with migraine may report a symptomatic phase before the onset of pain in migraine [6, 7], so-called prodromal symptoms (previously known as premonitory symptoms). Although not all patients report prodromal symptoms, understanding the underlying mechanisms of this proposed prodromal symptomatic phase in migraine may provide insights into the mechanisms of migraine attack initiation [1, 2, 7]. Yet, estimates of the relative frequency of prodromal symptoms fluctuate considerably between epidemiological studies [7], and it remains unclear if any specific individual symptoms are characteristic of this proposed phase [8]. Non-specific symptoms, e.g., fatigue, irritability, food cravings, yawning, are among the most frequently reported symptoms before onset of a migraine attack [7], which may suggest limbic dysfunction (e.g., dopaminergic and hypothalamic dysfunction). However, these are common symptoms amongst both individuals without and with migraine, and often have no association with a migraine attack [9]. These uncertainties question whether the available evidence can confirm the existence of a prodromal phase as a distinct component of a migraine attack. As patients can have migraine attacks without reporting prodromal symptoms, this suggests that the underlying mechanisms are not necessary to develop a migraine attack, but rather, they may co-occur as epiphenomena. A key feature of migraine is that attacks can be provoked, which provides a framework for investigating migraine pathophysiology by deliberately triggering migraine attacks in humans, i.e., a human provocation model [5]. Interestingly, this model has demonstrated that intravenous administration of calcitonin gene-related peptide (CGRP) and pituitary adenylate cyclase-activating peptide (PACAP) to susceptible individuals can induce migraine attacks [10, 11]. However, these migraine attacks occurred without prodromal symptoms in most cases following administration of CGRP and PACAP in a clinical trial [12]. Moreover, in those individuals who did report prodromal symptoms, these sometimes occur at or after onset of headache – or without any migraine attack developing at all [12].
Furthermore, the hypothesis of a hypothalamic or brainstem generator of migraine attacks does not explain specificity to migraine headache in humans and exclusion of other nociceptive dorsal horn neurons [4]. Most likely, brainstem activation as observed on neuroimaging studies in humans is dependent on activation of meningeal or other intracranial nociceptors [4]. Of note, several neuroimaging studies reporting activation of central structures, e.g., brainstem, were reliant on peripheral noxious stimulation or human provocation model with a peripheral administration of a pharmacological trigger [13]. Interestingly, activation of the periaqueductal grey matter is a consequence of nociceptor activation anywhere in the body; it is highly unspecific and involve areas outside the trigeminal system and cannot be used as a marker for a pain phenomenon restricted exclusively to trigeminal regions [14,15,16,17]. In clinical case reports of headache secondary to brainstem lesions, it cannot be excluded that there was direct or indirect activation of intracranial nociceptors due to close proximity, and the phenotype of these headaches rarely resemble migraine-like pain [18,19,20].
Another line of reasoning for a central sensitization/dysfunction is the occurrence of allodynia in individuals with migraine [21, 22]. One would expect that allodynia would develop before the onset of a migraine attack if central sensitization/dysfunction is the etiology, but allodynia typically takes one hour or longer for development after onset of a spontaneous migraine attack [23]. Furthermore, one-third of individuals with migraine do not experience allodynia as an accompanying symptom of migraine attacks [21, 22]. More likely, central sensitization in migraine is driven by a nociceptive peripheral input [4].
Migraine aura
While migraine aura clearly has a cortical origin with cortical spreading depression (CSD) as the underlying neurobiological mechanism [24], this is not a prerequisite for migraine headache. Migraine aura is only experienced by one-third of individuals with migraine [25], and in some of these patients, migraine aura does not occur consistently through all attacks. In addition, headache, as well as other migraine-associated symptoms, are present early during the aura phase in most patients, and sometimes even before the aura phase [26]. More importantly, migraine aura does not provide an explanation for migraine attacks without aura. Occurrence of “silent” migraine aura, i.e., an event of CSD without focal neurological symptoms, has been proposed to occur prior to migraine attacks without aura. However, this has never been demonstrated in humans and remains speculative. Hemodynamic changes associated with CSD has been described on neuroimaging in individuals experiencing migraine aura [27, 28], but these findings cannot be reproduced in studies of individuals with migraine attacks without aura [29]. Furthermore, observations of individuals who experience migraine aura without headache suggests that cortical spreading depression is merely another potential trigger of a migraine headache [30,31,32,33]. This is emphasized through human provocation studies of patients with migraine with aura, who report a migraine attack without aura for the first time in their lives following peripheral administration of an experimental trigger compound, e.g., CGRP [5, 34]. As CGRP is not able to cross the blood–brain barrier [35], and current evidence suggest there is no blood–brain barrier disruption in migraine pathophysiology [36, 37], the site of action is likely outside of the brain. Interestingly, there are reports of migraine aura following peripheral administration of CGRP [5, 34], and migraine aura-like phenomena and CSD can be caused by vascular events, e.g., carotid dissection, arteriovenous malformations [38, 39]. Whether CSD in these cases is caused by reasons such as microembolization, focal ischemia through disruption in blood flow or disturbances in local homeostasis secondary to these events remains speculative, but fact is that symptomatic migraine with aura can occur due to an initial vascular circumstance [40].
In vivo studies of mice with knock-ins of two different familial hemiplegic migraine (FHM)-genes showed increased susceptibility to and propagation velocity of cortical spreading depression compared with wild-type animals [41,42,43,44]. However, these findings are not necessarily relevant for other migraine types [45,46,47,48], and many of the traits found in these monogenic subtypes of migraine (e.g., hemiplegia during aura, progressive ataxia, attacks triggered by mild head trauma; brain edema, mental retardation, and progressive ataxia) are certainly not found in common migraine subtypes.
Based on these observations, migraine aura and cortical spreading depression are likely another potential trigger of migraine headache and does not provide an explanation for the most common phenotype, migraine attacks without aura.
Migraine pain
The most convincing arguments for a peripheral origin of migraine attacks is the fact that migraine attacks can be produced and attenuated entirely at peripheral sites of action. A key feature of migraine is that various trigger factors are able to provoke migraine attacks. Human provocation models draw advantage of this feature, wherein endogenous molecules or other putative triggers are administered in humans to induce migraine, to identify signaling pathways that are involved in migraine pathophysiology. Series of randomized trials with human provocation models have consistently demonstrated that intravenous administration of various neuropeptides, e.g., CGRP and PACAP [49,50,51], is able to induce migraine attacks in susceptible individuals [5]. The site of action of these neuropeptides (e.g., CGRP and PACAP) includes the cranial arteries as neuroimaging studies consistently demonstrate a marked extracerebral vasodilation but not of the large cerebral arteries following administration [2, 5, 11]. Interestingly, animal models demonstrate that administration of these n CGRP and PACAP within the central nervous system can induce antinociception rather than nociception [52,53,54,55]. Observations during neurosurgical procedures in awake patients suggest that the pain-sensitive structures are limited to the surrounding structures of the brain, e.g., dura mater, pia mater and their feeding vessels, whereas stimulation of the brain parenchyma itself does not evoke pain [56]. The trigeminovascular system provides a framework wherein peripheral input can lead to migraine attacks through sensitization and activation of trigeminal primary afferents, mediated through vasodilation of intracranial arteries [2, 3]. A proposed mechanism of how this vascular signaling contributes to pain perception involves sensitization through increased extracellular potassium [2]. Arterial vasodilation is caused by opening of cation channels, mainly potassium channels, in vascular smooth muscle cells and results in accumulation of positively charged ions in the extracellular space [2]. In turn, this electrical gradient drives positively charged ions into and activate neighboring trigeminal pain fibers [2]. This is supported by the observation that activation of downstream targets of CGRP and PACAP, e.g., KATP channels and BKCa channels, induces migraine attacks at a much higher rate in parallel with a marked vasodilation, which suggests a vascular site of action [57, 58]. Interestingly, while these channels are also expressed in C- and Aδ-fibers, intradermal and intramuscular injections of levcromakalim, a KATP channel opener, does not evoke cutaneous or muscle pain [59]; in turn, a direct activation of these channels in peripheral neurons is an unlikely site of action for migraine attacks.
In other paroxysmal pain disorders, e.g., familial episodic pain syndrome, ion channels have been demonstrated to exhibit modulatory activity and provides a context for the episodic nature of migraine attacks [60, 61]. In line with these observations, a genome wide association meta-analysis identified 123 susceptibility loci that showed enrichment for genes expressed in vascular and smooth muscle tissues in individuals with migraine without or with aura [62]. These findings suggest vascular dysfunction, and possibly also smooth muscle dysfunction (consistent with a shared polygenic risk scores of migraine, stroke, and cardiovascular diseases) [63,64,65,66,67], are crucial in migraine pathogenesis and strongly implicates a vascular etiology of migraine. Interestingly, rare vasculopathies have an overrepresentation of migraine. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a hereditary small artery disease and one of its characteristic presentations include migraine [68]. Patients with mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS) are especially known to suffer from repeated episodes of migraine [69]. Morphological observations suggest MELAS involves mitochondrial angiopathy as autopsies of patients show abnormally proliferated mitochondria in the smooth muscle cells and endothelial cells of the small cerebral blood vessels [70].
Migraine drugs that purely exhibit their effects at a peripheral site of action exist, i.e., monoclonal antibodies targeting CGRP or its receptor, as they are very unlikely to cross the blood brain barrier [35, 71]. Interestingly, these monoclonal antibodies are also effective in patients with migraine with aura [72, 73]. Other compounds with a predominant peripheral site of action include onabotulinumtoxinA where the mechanism of action is likely to involve attenuation of peripheral pain transmission [74]. In individuals with chronic headache, the clinical effect of onabotulinumtoxinA has been suggested to involve reduction in periosteal inflammation [75]. These findings are consistent with the observation that onabotulinumtoxinA is able to reduce release of inflammatory and excitatory neurotransmitters and neuropeptides from primary nociceptors [76, 77]. Of note, adverse events related to the central nervous system are not reported for either monoclonal antibodies nor onabotulinumtoxinA [71]. These observations are also true for commonly used acute migraine medications. Dihydroergotamine (acute migraine drug) is not able to cross the blood–brain barrier in humans [78], and while sumatriptan (acute migraine drug) is able to cross the blood–brain barrier and may present with adverse events related to the central nervous system, preclinical models suggest the site of action for its migraine-attenuating effects is likely mediated through modulation of the first-order neuron localized outside of the blood–brain barrier [79]. This is supported by the observation that sumatriptan constricts the superficial temporal artery and middle meningeal artery, but not the middle cerebral artery, in migraine attacks in humans, which favors a perivascular site of action outside of the blood–brain barrier [80].
Migraine attacks are of central origin
Prodromal symptoms and central dysfunction
From a clinical perspective, the site of origin of a migraine attack is reflected by the earliest symptoms of the attack. Many migraine patients report prodromal symptoms, which include mood changes, excessive yawning, thirst, and cravings for certain foods [81]. Such symptoms would be expected to arise from brain regions such as the hypothalamus or other parts of the limbic system, and not from peripheral nerves or from blood vessels. The symptoms may not be specific for migraine but, importantly, a prospective study demonstrated that some patients can accurately predict their migraine attacks based on these early symptoms indicating that prodromal symptoms are truly linked to migraine at least in a subgroup of patients [82].
The notion that this early phase of the migraine attack originates in the brain is supported by a line of evidence based on advanced neuroimaging studies. One study investigated migraine patients during glyceryl trinitrate-induced prodromal symptoms and subsequent migraine headache and found increased activity of the hypothalamus, brainstem, and various cortical areas specifically during the prodromal phase [83]. Another study using BOLD functional MRI and painful trigeminal stimulation to study a migraine patient every day for 30 days, and during three spontaneous migraine attacks, found increased activation of the hypothalamus, and increased functional connectivity between the hypothalamus and the pons within 24 h before headache onset [13]. The authors later reproduced these findings of preictal hypothalamic activation in seven migraine patients scanned every day for at least 30 consecutive days [84].
Migraine aura
Approximately one-third of migraine patients experience aura symptoms. The aura symptoms begin before the onset of pain in nearly all cases [85] and based on their clinical presentation they clearly originate from the cerebral cortex [86]. The underlying mechanism of migraine aura is widely accepted to be the electrophysiological phenomenon of CSD, involving a wave of neuronal and glial depolarization spreading across the cerebral cortex at an approximate rate of 3 mm/min [87]. Although electrophysiological recordings from the cortical surface have not been performed during attacks of migraine with aura, gradually spreading changes of cerebral blood flow consistently shown in functional neuroimaging studies support that the aura phase of migraine is indeed due to CSD [28, 88]. Interestingly, a recent PET-MRI study in migraine aura patients, applying a radioactive marker of inflammation, indicated that CSD may directly induce meningeal inflammation and thereby potentially head pain and associated symptoms of migraine [89]. Thus, CSD appears to be a primary event that precedes, and causes, the pain phase of migraine. In support of this, animal studies have demonstrated that experimentally induced CSD leads to activation of meningeal nociceptors and central trigeminovascular neurons [90]. In addition, CSD leads to pain and anxiety behavior in animals even when elicited in a minimally invasive manner using optogenetics [91].
Collectively, migraine attacks with aura clearly originate from the cerebral cortex. In the subset of patients with familial hemiplegic migraine (FHM), the site of origin can even be specified at the molecular level. Three genes are known to be involved in familial hemiplegic migraine. In FHM type 1, mutations in CACNA1A, encoding the α1 subunit of the voltage-gated channel CaV2.1 calcium leads to gain of channel function. FHM type 2 mutations in the ATP1A2 gene encoding the α2 subunit of Na/K-ATPases result in a loss of function in glial cells. In FHM type 3, mutations in the SCN1A gene lead to a gain of function of NaV1.1 sodium channels. These mutations facilitate neuronal depolarization. Knock-in mouse models have been developed for all three types of familial hemiplegic migraine and in these an enhanced susceptibility to experimental CSD elicitation has been demonstrated [92]. Interestingly, a recent genome-wide analysis of 102,084 migraine cases indicated that the CACNA1A gene is also involved in migraine with typical aura [62]. Likely, migraine with aura patients in general are susceptible to CSD initiation due to ion channel dysfunction leading to occasional neuronal depolarization.
Attacks of symptoms that are clinically indistinguishable from migraine with aura may occur due to e.g., carotid dissection or carotid aneurysms [40]. The likely mechanism behind this observation is that carotid pathology may lead to hypoperfusion or microembolization, which is known to be able to trigger CSD [93]. Clear-cut cases of “symptomatic migraine aura”, although apparently rare, have been reported and are most often caused by lesions to the cerebral cortex including brain arteriovenous malformations or brain tumors [40]. Rare vasculopathies including CADASIL, MELAS, Sneddon syndrome, and Moyamoya disease may present with migraine with aura as well as stroke [94]. In these cases, attacks of migraine with aura are likely secondary to cortical lesions caused by the cerebrovascular pathology.
Migraine pain
It is possible that the pain of migraine originates from peripheral structures although there is no firm evidence to support this. Vasodilation seems not be the cause of pain in migraine. Even strong vasodilation of cephalic arteries causes only mild headache [95] and there is no correlation between the degree of vasodilation and pharmacologically induced headache in healthy volunteers [96]. An MR angiography study of spontaneous migraine attacks without aura reported slight dilation of intracranial, but not extracranial, arteries during attacks [97]. Administration of subcutaneous sumatriptan resulted in pain relief but not constriction of the dilated intracerebral arteries. Triptans, 5-HT1B/1D agonists cross the blood–brain barrier [98] and their anti-migraine effects may depend on binding to central serotonin receptors. Likewise, ditans, i.e., 5-HT1F agonists, cause relief of migraine headache, cross the blood–brain barrier, and do not appear to cause vasoconstriction [99, 100]. Even a central action of monoclonal antibodies cannot be excluded with certainty since these may cross the blood–brain barrier although at a small rate of 1:1000 [101].
Migraine-inducing neuropeptides like CGRP may exert their effects in the periphery but this does not provide evidence of a peripheral origin of spontaneous migraine attacks. Interestingly, drugs that provoke migraine attacks clinically generally do not cause migraine aura symptoms, even in patients who always experience aura during their spontaneous migraine attacks [5]. Thus, these substances likely exert their migraine-provoking effects peripherally and downstream as opposed to spontaneous migraine attacks that originate centrally and subsequently lead to peripheral effects.
Lessons learned and future directions
Prodromal symptoms and central dysfunction
Methodological uncertainties limits inferences from reports on prodromal symptoms in migraine, which otherwise may assist our understanding of migraine attack initiation. Heterogeneity between investigations, including application of different definitions and matters of enquiry, allows for large discrepancies. Furthermore, non-specific symptoms such as fatigue and mood change are commonly reported, and strict criteria need to be applied to allow discrimination between spontaneous occurrence or migraine-associated occurrence. These shortcomings can be addressed through harmonization of studies by standardized methodology and data reporting. The fact there are no internationally acknowledged guidelines yet warrants an investment.
Migraine aura
Although migraine aura and migraine headache are temporally associated in many cases, it may be counter-productive to discuss these disorders as a single entity as their mechanistic relationship is not clarified. Experimental research in humans has been limited by the lack of a potent human model of migraine aura. Recent findings suggest that opening of potassium channels may be a potent trigger of migraine aura in humans [102]. If these observations are confirmed, it would allow for investigations into the mechanisms that link migraine aura and migraine headache using neuroimaging, electrophysiology, and biochemistry.
Migraine pain
Beyond any doubt, migraine pain is modulated through activity of CGRP and other neuropeptides within the trigeminovascular system [3]. Release of CGRP and other neuropeptides have been demonstrated to be released at a peripheral site at the level of the trigeminal ganglion [103], but we cannot at the present time confirm or reject a downstream regulation through central dysfunction as there is an absence of evidence, not evidence of absence. While cortical spreading depression is able to depolarize meningeal nociceptors [24], thereby causing pain, this does not provide an explanation for the majority of migraine attacks experienced in the world: migraine attacks without aura. Experimental investigations need to address whether it is possible to induce a peripheral release of these neuropeptides through a central mechanism in humans with migraine without aura.
Conclusions
Both peripheral and central mechanisms are likely involved in a migraine attack, e.g., peripheral nociceptive input is necessary for pain transmission and cortical activity is necessary for pain perception. Yet, the debate of whether migraine attacks are initiated a peripheral or central site remains unresolved. The increased focus on prodromal symptoms and on the development of a human model of migraine aura will possibly provide key arguments needed to answer this question in the near future. Until then, we cannot draw firm conclusions and the debate goes on.
Availability of data and materials
No data were generated for this manuscript.
References
Ashina M, Terwindt GM, Al-Karagholi MA-M et al (2021) Migraine: disease characterisation, biomarkers, and precision medicine. Lancet 397:1496–1504
Ashina M (2020) Migraine. N Engl J Med 383:1866–1876
Ashina M, Hansen JM, Do TP et al (2019) Migraine and the trigeminovascular system—40 years and counting. Lancet Neurol 18:795–804
Olesen J, Burstein R, Ashina M et al (2009) Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol 8:679–690
Ashina M, Hansen JM, Á Dunga BO et al (2017) Human models of migraine-short-Term pain for long-Term gain. Nat Rev Neurol 13:713–724
Headache Classification Committee of the International Headache Society (IHS) (2018) The International Classification of Headache Disorders, 3rd edition. Cephalalgia 38(1):1–211. https://doi.org/10.1177/0333102417738202
Karsan N, Goadsby PJ (2018) Biological insights from the premonitory symptoms of migraine. Nat Rev Neurol 14:699–710
Eigenbrodt AK, Christensen RH, Ashina H et al (2022) Premonitory symptoms in migraine: a systematic review and meta-analysis of observational studies reporting prevalence or relative frequency. J Headache Pain 23:140
Ricci JA, Chee E, Lorandeau AL et al (2007) Fatigue in the U.S. Workforce: Prevalence and Implications for Lost Productive Work Time. J Occup Environ Med 49:1–10
Lassen L, Haderslev P, Jacobsen V et al (2002) Cgrp May Play A Causative Role in Migraine. Cephalalgia 22:54–61
Schytz HW, Birk S, Wienecke T et al (2009) PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain 132:16–25
Guo S, Vollesen ALH, Olesen J et al (2016) Premonitory and nonheadache symptoms induced by CGRP and PACAP38 in patients with migraine. Pain 157:2773–2781
Schulte LH, May A (2016) The migraine generator revisited: continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain 139:1987–1993
Derbyshire SWG, Jones AKP, Creed F et al (2002) Cerebral Responses to Noxious Thermal Stimulation in Chronic Low Back Pain Patients and Normal Controls. Neuroimage 16:158–168
Hsieh J-C, Ståhle-Bäckdahl M, Hägermark Ö et al (1996) Traumatic nociceptive pain activates the hypothalamus and the periaqueductal gray: a positron emission tomography study. Pain 64:303–314
Iadarola M (1998) Neural activation during acute capsaicin-evoked pain and allodynia assessed with PET. Brain 121:931–947
Petrovic P, Ingvar M, Stone-Elander S et al (1999) A PET activation study of dynamic mechanical allodynia in patients with mononeuropathy. Pain 83:459–470
Haas DC, Kent PF, Friedman DI (1993) Headache Caused by a Single Lesion of Multiple Sclerosis in the Periaqueductal Gray Area. Headache J Head Face Pain 33:452–454
Afridi S (2003) New onset migraine with a brain stem cavernous angioma. J Neurol Neurosurg Psychiatry 74:680–681
Goadsby P (2002) Neurovascular Headache and A Midbrain Vascular Malformation: Evidence for A Role of the Brainstem in Chronic Migraine. Cephalalgia 22:107–111
Burstein R, Yarnitsky D, Goor-Aryeh I et al (2000) An association between migraine and cutaneous allodynia. Ann Neurol 47:614–624
Lipton RB, Bigal ME, Ashina S et al (2008) Cutaneous allodynia in the migraine population. Ann Neurol 63:148–158
Burstein R, Collins B, Jakubowski M (2004) Defeating migraine pain with triptans: A race against the development of cutaneous allodynia. Ann Neurol 55:19–26
Pietrobon D, Moskowitz MA (2014) Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci 15:379–393
Rasmussen BK, Olesen J (1992) Migraine With Aura and Migraine Without Aura: An Epidemiological Study. Cephalalgia 12:221–228
Hansen JM, Lipton RB, Dodick DW et al (2012) Migraine headache is present in the aura phase: A prospective study. Neurology 79:2044–2049
Olesen J, Larsen B, Lauritzen M (1981) Focal hyperemia followed by spreading oligemia and impaired activation of rcbf in classic migraine. Ann Neurol 9:344–352
Hadjikhani N, Sanchez del Rio M, Wu O et al (2001) Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci 98:4687–4692
Olesen J, Lauritzen M, Tfelt-Hansen P et al (1982) Spreading Cerebral Oligemia in Classical- and Normal Cerebral Blood Flow in Common Migraine. Headache J Head Face Pain 22:242–248
Aiba S, Tatsumoto M, Saisu A et al (2010) Prevalence of typical migraine aura without headache in Japanese ophthalmology clinics. Cephalalgia 30:962–967
Amos JF, Fleming JB (2000) Clinical description and review of migraine aura without headache. Optometry 71:372–380
Fisher CM (1986) Late-life migraine accompaniments–further experience. Stroke 17:1033–1042
Wijman CAC, Wolf PA, Kase CS et al (1998) Migrainous Visual Accompaniments Are Not Rare in Late Life. Stroke 29:1539–1543
Hansen JM, Hauge AW, Olesen J et al (2010) Calcitonin gene-related peptide triggers migraine-like attacks in patients with migraine with aura. Cephalalgia 30:1179–1186
Wiggers A, Ashina H, Hadjikhani N et al (2022) Brain barriers and their potential role in migraine pathophysiology. J Headache Pain 23:16
Amin FM, Hougaard A, Cramer SP et al (2017) Intact blood−brain barrier during spontaneous attacks of migraine without aura: a 3T DCE-MRI study. Eur J Neurol 24:1116–1124
Hougaard A, Amin FM, Christensen CE et al (2017) Increased brainstem perfusion, but no blood-brain barrier disruption, during attacks of migraine with aura. Brain 140:1633–1642
Donnelly A, Sinnott B, Boyle R, et al. Beware the middle-aged migraine: internal carotid artery dissection mimicking migraine in the emergency department. BMJ Case Rep. 2017;bcr-2017–221774.
Ramadan NM, Tietjen GE, Levine SR et al (1991) Scintillating scotomata associated with internal carotid artery dissection: Report of three cases. Neurology 41:1084–1084
Thomsen AV, Sørensen MT, Ashina M et al (2021) Symptomatic migraine: a systematic review to establish a clinically important diagnostic entity. Headache J Head Face Pain 61:1180–1193
van den Maagdenberg AMJ, Pietrobon D, Pizzorusso T et al (2004) A cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 41:701–710
Eikermann-Haerter K, Dileköz E, Kudo C, et al. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest. Epub ahead of print 22 December 2008. https://doi.org/10.1172/JCI36059.
van den Maagdenberg AMJM, Pizzorusso T, Kaja S et al (2010) High cortical spreading depression susceptibility and migraine-associated symptoms in Ca v 2.1 S218L mice. Ann Neurol 67:85–98
Leo L, Gherardini L, Barone V et al (2011) Increased susceptibility to cortical spreading depression in the mouse model of familial hemiplegic migraine type 2. PLoS Genet 7:e1002129
Nyholt DR, LaForge KS, Kallela M et al (2008) A high-density association screen of 155 ion transport genes for involvement with common migraine. Hum Mol Genet 17:3318–3331
Kirchmann M, Thomsen LL, Olesen J (2006) The CACNA1A and ATP1A2 genes are not involved in dominantly inherited migraine with aura. Am J Med Genet Part B Neuropsychiatr Genet 141B:250–256
Jen JC, Kim GW, Dudding KA et al (2004) No Mutations in CACNA1A and ATP1A2 in Probands With Common Types of Migraine. Arch Neurol 61:926
Wieser T, Mueller C, Evers S, et al. Absence of Known Familial Hemiplegic Migraine (FHM) Mutations in the CACNA1A Gene in Patients with common Migraine: Implications for Genetic Testing. Clin Chem Lab Med; 41. Epub ahead of print 24 January 2003. https://doi.org/10.1515/CCLM.2003.042.
Younis S, Do TP, Ashina M (2021) Human Models. In: Maassen van den Brink, A., Martelletti P. (eds) Monoclonal Antibodies in Headache . Headache. Springer, Cham. https://doi.org/10.1007/978-3-030-69032-8_5
Ghanizada H, Al-Karagholi MA-M, Arngrim N et al (2020) PACAP27 induces migraine-like attacks in migraine patients. Cephalalgia 40:57–67
Amin FM, Hougaard A, Schytz HW et al (2014) Investigation of the pathophysiological mechanisms of migraine attacks induced by pituitary adenylate cyclase-activating polypeptide-38. Brain 137:779–794
Pecile A, Guidobono F, Netti C et al (1987) Calcitonin gene-related peptide: antinociceptive activity in rats, comparison with calcitonin. Regul Pept 18:189–199
Candeletti S, Ferri S (1990) Antinociceptive profile of intracerebroventricular salmon calcitonin and calcitonin gene-related peptide in the mouse formalin test. Neuropeptides 17:93–98
Zhang YZ, Sjőlund B, Moller K et al (1993) Pituitary adenylate cyclase activating peptide produces a marked and long-lasting depression of a C-fibre-evoked flexion reflex. Neuroscience 57:733–737
Yamamoto T, Tatsuno I (1995) Antinociceptive effect of intrathecally administered pituitary adenylate cyclase activating polypeptide (PACAP) on the rat formalin test. Neurosci Lett 184:32–35
Fontaine D, Almairac F, Santucci S et al (2018) Dural and pial pain-sensitive structures in humans: new inputs from awake craniotomies. Brain 141:1040–1048
Al-Karagholi MA-M, Ghanizada H, Nielsen CAW, et al. Opening of ATP sensitive potassium channels causes migraine attacks with aura. Brain. Epub ahead of print March 2021. https://doi.org/10.1093/brain/awab136.
Al-Karagholi MA-M, Ghanizada H, Waldorff Nielsen CA, et al. Opening of BKCa channels causes migraine attacks. Pain; Publish Ah. Epub ahead of print 16 March 2021. https://doi.org/10.1097/j.pain.0000000000002238.
Al-Karagholi MA-M, Ghanizada H, Hansen JM et al (2019) Extracranial activation of ATP-sensitive potassium channels induces vasodilation without nociceptive effects. Cephalalgia 39:1789–1797
Kullmann DM, Waxman SG (2010) Neurological channelopathies: new insights into disease mechanisms and ion channel function. J Physiol 588:1823–1827
Kremeyer B, Lopera F, Cox JJ et al (2010) A Gain-of-Function Mutation in TRPA1 Causes Familial Episodic Pain Syndrome. Neuron 66:671–680
Hautakangas H, Winsvold BS, Ruotsalainen SE et al (2022) Genome-wide analysis of 102,084 migraine cases identifies 123 risk loci and subtype-specific risk alleles. Nat Genet 54:152–160
Winsvold BS, Nelson CP, Malik R et al (2015) Genetic analysis for a shared biological basis between migraine and coronary artery disease. Neurol Genet 1:e10
Malik R, Freilinger T, Winsvold BS et al (2015) Shared genetic basis for migraine and ischemic stroke: a genome-wide analysis of common variants. Neurology 84:2132–2145
Sacco S, Ornello R, Ripa P et al (2015) Migraine and risk of ischaemic heart disease: a systematic review and meta-analysis of observational studies. Eur J Neurol 22:1001–1011
Schurks M, Rist PM, Bigal ME et al (2009) Migraine and cardiovascular disease: systematic review and meta-analysis. BMJ 339:b3914–b3914
Mahmoud AN, Mentias A, Elgendy AY et al (2018) Migraine and the risk of cardiovascular and cerebrovascular events: a meta-analysis of 16 cohort studies including 1 152 407 subjects. BMJ Open 8:e020498
Dichgans M, Mayer M, Uttner I et al (1998) The phenotypic spectrum of CADASIL: Clinical findings in 102 cases. Ann Neurol 44:731–739
Montagna P, Gallassi R, Medori R et al (1988) MELAS syndrome: Characteristic migrainous and epileptic features and maternal transmission. Neurology 38:751–751
Ohama E, Ohara S, Ikuta F et al (1987) Mitochondrial angiopathy in cerebral blood vessels of mitochondrial eneephalomyopathy. Acta Neuropathol 74:226–233
Ashina M, Buse DC, Ashina H et al (2021) Migraine: integrated approaches to clinical management and emerging treatments. Lancet 397:1505–1518
Ashina M, Goadsby PJ, Dodick DW et al (2022) Assessment of Erenumab Safety and Efficacy in Patients With Migraine With and Without Aura. JAMA Neurol 79:159
Ashina M, McAllister P, Cady R, Hirman J, Ettrup A (2022) Efficacy and safety of eptinezumab in patients with migraine and self-reported aura: Post hoc analysis of PROMISE-1 and PROMISE-2. Cephalalgia 42(8):696–704. https://doi.org/10.1177/03331024221077646. Epub 2022 Mar 18
Burstein R, Blumenfeld AM, Silberstein SD et al (2020) Mechanism of Action of OnabotulinumtoxinA in Chronic Migraine: A Narrative Review. Headache J Head Face Pain 60:1259–1272
Gfrerer L, Xu W, Austen W et al (2022) OnabotulinumtoxinA alters inflammatory gene expression and immune cells in chronic headache patients. Brain 145:2436–2449
Lucioni A, Bales GT, Lotan TL et al (2008) Botulinum toxin type A inhibits sensory neuropeptide release in rat bladder models of acute injury and chronic inflammation. BJU Int 101:366–370
Durham PL, Cady R, Cady R (2004) Regulation of Calcitonin Gene-Related Peptide Secretion From Trigeminal Nerve Cells by Botulinum Toxin Type A: Implications for Migraine Therapy. Headache J Head Face Pain 44:35–43
Schankin CJ, Maniyar FH, Seo Y et al (2016) Ictal lack of binding to brain parenchyma suggests integrity of the blood–brain barrier for 11 C-dihydroergotamine during glyceryl trinitrate-induced migraine. Brain 139:1994–2001
Levy D, Jakubowski M, Burstein R (2004) Disruption of communication between peripheral and central trigeminovascular neurons mediates the antimigraine action of 5HT 1B/1D receptor agonists. Proc Natl Acad Sci 101:4274–4279
Khan S, Mohammad Amin F, Emil Christensen C et al (2019) Meningeal contribution to migraine pain: a magnetic resonance angiography study. Brain 142:93–102
Laurell K, Artto V, Bendtsen L et al (2016) Premonitory symptoms in migraine: A cross-sectional study in 2714 persons. Cephalalgia 36:951–959
Giffin NJ, Ruggiero L, Lipton RB et al (2003) Premonitory symptoms in migraine: An electronic diary study. Neurology 60:935–940
Maniyar FH, Sprenger T, Monteith T et al (2014) Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain 137:232–241
Schulte LH, Mehnert J, May A (2020) Longitudinal Neuroimaging over 30 Days: Temporal Characteristics of Migraine. Ann Neurol 87:646–651
Russell MB, Olesen J (1996) A nosographic analysis of the migraine aura in a general population. Brain 119:355–361
Lashley KS (1941) Patterns, of cerebral integration indicated by the scotomas of migraine. Arch Neurol Psychiatry 46:331
Ayata C, Lauritzen M (2015) Spreading Depression, Spreading Depolarizations, and the Cerebral Vasculature. Physiol Rev 95:953–993
Olesen J, Friberg L, Olsen TS et al (1990) Timing and topography of cerebral blood flow, aura, and headache during migraine attacks. Ann Neurol 28:791–798
Hadjikhani N, Albrecht DS, Mainero C et al (2020) Extra-Axial Inflammatory Signal in Parameninges in Migraine with Visual Aura. Ann Neurol 87:939–949
Zhang X, Levy D, Noseda R et al (2010) Activation of Meningeal Nociceptors by Cortical Spreading Depression: Implications for Migraine with Aura. J Neurosci 30:8807–8814
Harriott AM, Chung DY, Uner A et al (2021) Optogenetic Spreading Depression Elicits Trigeminal Pain and Anxiety Behavior. Ann Neurol 89:99–110
de Boer I, Terwindt GM, van den Maagdenberg AMJM (2020) Genetics of migraine aura: an update. J Headache Pain 21:64
Nozari A, Dilekoz E, Sukhotinsky I et al (2010) Microemboli may link spreading depression, migraine aura, and patent foramen ovale. Ann Neurol 67:221–229
Kurth T, Chabriat H, Bousser M-G (2012) Migraine and stroke: a complex association with clinical implications. Lancet Neurol 11:92–100
Wienecke T, Olesen J, Ashina M (2011) Discrepancy between strong cephalic arterial dilatation and mild headache caused by prostaglandin D 2 (PGD 2). Cephalalgia 31:65–76
Ashina M, Tfelt-Hansen P, Dalgaard P et al (2011) Lack of correlation between vasodilatation and pharmacologically induced immediate headache in healthy subjects. Cephalalgia 31:683–690
Amin FM, Asghar MS, Hougaard A et al (2013) Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: a cross-sectional study. Lancet Neurol 12:454–461
Deen M, Hougaard A, Hansen HD et al (2019) Association Between Sumatriptan Treatment During a Migraine Attack and Central 5-HT 1B Receptor Binding. JAMA Neurol 76:834
Rubio-Beltrán E, Labastida-Ramírez A, Villalón CM et al (2018) Is selective 5-HT1F receptor agonism an entity apart from that of the triptans in antimigraine therapy? Pharmacol Ther 186:88–97
Labastida-Ramírez A, Rubio-Beltrán E, Haanes KA et al (2020) Lasmiditan inhibits calcitonin gene-related peptide release in the rodent trigeminovascular system. Pain 161:1092–1099
Felgenhauer K (1974) Protein size and cerebrospinal fluid composition. Klin Wochenschr 52:1158–1164
Al-Karagholi MA-M, Ghanizada H, Nielsen CAW, et al. Opening of ATP sensitive potassium channels causes migraine attacks with aura. Brain. Epub ahead of print 26 March 2021. https://doi.org/10.1093/brain/awab136.
Goadsby PJ, Edvinsson L, Ekman R (1988) Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann Neurol 23:193–196
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors contributed to conception, design and critical revision of the work for important intellectual content. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
Thien Phu Do has received honoraria for delivering lectures for Teva.
Anders Hougaard reports no conflicts of interest.
Greg Dussor has received grant funding from Alder Biopharmaceuticals and Teva Pharmaceuticals.
KC Brennan reports consulting activities unrelated to this paper or topic of research from Eli Lilly and Allergan.
Faisal Mohammad Amin has received honoraria for delivering lectures and/or participation in advisory boards for Pfizer, Teva, Lundbeck, Novartis and Eli Lilly. Faisal Mohammad Amin serves as associate editor for the journals Headache Medicine, Acta Neurologica Scandinavica, Frontiers In Neurology and Frontiers In Pain Research. Faisal Mohammad Amin serves as president of the Danish Headache Society and a member of the Board of Directors in European Headache Federation. Faisal Mohammad Amin has no ownership interest and does not own stocks of any pharmaceutical company.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Do, T.P., Hougaard, A., Dussor, G. et al. Migraine attacks are of peripheral origin: the debate goes on. J Headache Pain 24, 3 (2023). https://doi.org/10.1186/s10194-022-01538-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-022-01538-1