
Abstract
Asthma is a complex heritable inflammatory disorder of the airways in which the development of clinical disease depends on environmental exposure. It has been well established that T helper type 2 (TH2) lymphocytes and their cytokines have an important role in allergic asthma. Interleukin (IL)-9, a member of the TH2 cytokine family, has recently been implicated as an essential factor in determining mucosal immunity and susceptibility to atopic asthma. In this review we examine the critical experiments and observations that support this hypothesis. We also discuss these results in comparison with the experiments supporting the involvement of other TH2 cytokines such as IL-4, IL-5 and IL-13.
Similar content being viewed by others

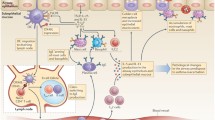

Introduction
Asthma is a complex heritable inflammatory disorder of the airways associated with airway obstruction as well as a variety of clinical signs [1,2]. Airway hyperresponsiveness (AHR), a risk factor and a cardinal feature of asthma, is characterized as an exaggerated airway response to an irritant stimulus, resulting in airway obstruction. Asthma is commonly associated with chronic elevation of serum IgE, another risk factor for the disorder, which is closely associated with allergy. Allergic asthma (atopic asthma) is the most common, accounting for 90% of cases under 30 years of age [3]. In addition, asthmatics frequently suffer from mucus overproduction that is thought to contribute to airway obstruction. Although allergic agents are numerous and the inflammatory responses produced are complex, clinical and experimental studies have strongly suggested the involvement of T helper type 2 (TH2) lymphocytes, and the cytokines that they produce, in the pathogenesis of allergic asthma [4,5]. Other forms of asthma include those induced by exercise, viruses, aspirin, and occupation. The mechanism underlying these forms of asthma might similarly involve TH2 lymphocytes and cytokines but might be triggered differentially by airway epithelium damage caused by viral infection, excess leukotriene production, or water loss that in turn stimulates the release of histamine by mast cells and basophils [6,7,8,9]. One rational approach to creating novel medications for asthma is to identify the underlying biological mechanisms to treat the root cause, not just the symptoms.
Genetic epidemiology confirms that there is a strong heritable component to asthma [2]. Genetic mapping studies reveal that chromosome 5q31-q33 is of particular interest because it contains the cytokine gene cluster including interleukin (IL)-4, IL-5, IL-9, IL-13, and numerous growth factors and receptors implicated in allergic inflammation [10]. Because of the enormous body of literature demonstrating the important roles of these cytokines in biological responses associated with asthma, emphasis has previously focused on strategies to inhibit the activity of IL-4 and IL-5 [11]. However, the crucial roles of these factors in supporting the normal biology of specific cell types suggest that blocking their activity might compromise normal host defense. Recently, IL-9 and IL-13 have been shown to mediate AHR in the allergic lung, and blockage of either cytokine by using neutralizing antibody or soluble receptor is sufficient to attenuate AHR in selective animal models [12,13,14,15,16,17]. In this review we examine recent evidence supporting a central role for IL-9 as a critical mediator of allergic asthma, comparing these data with similar studies on IL-4, IL-5, and IL-13.
IL-9 is a TH2 cytokine that has pleiotropic effects on various cell types important in the pathogenesis of asthma
IL-9 was originally described as a mast cell growth factor for its ability to enhance the survival of primary mast cells and to induce their production of the inflammatory cytokine IL-6 [18,19]. IL-9 also stimulates the production of mast cell proteases and the high-affinity IgE receptor (FcεRIα), suggesting that IL-9 primes mast cells to respond to allergen via increased cell surface expression of the high-affinity IgE receptor and the production of inflammatory mediators including IL-6 and several proteases [20]. IL-9 was cloned as a T-cell growth factor for its ability to support antigen-independent growth of T helper clones [21,22]. Expression studies in vitro show that IL-9 could be produced by TH2-type lymphocytes, making it a member of the TH2 cytokine family [18]. In addition, studies of immunoglobulin production in vitro by B cells demonstrated that IL-9 enhances the IL-4-mediated production of IgE in human and murine B cells [23]. Recently, IL-9 has been shown to promote eosinophil maturation in synergy with IL-5 [24]. IL-9 modulates the expression of IL-5Rα in myeloid precursor cell lines [25,26]. IL-9 might therefore induce airway eosinophilia through the upregulation of IL-5 response and potentiating the IL-5-mediated maturation of eosinophil precursors [25,26]. Furthermore, IL-9 has been shown to activate airway epithelial cells by stimulating the production of several chemokines, proteases, ion channels and selective mucin genes [27,28]. Longphre et al [29] concluded, on the basis of studies in vitro and in vivo, that IL-9, but not IL-5 or IL-13, might account for most of the mucin-stimulatory activity of lung fluids in allergic airway disease. Taken together, these results indicate that IL-9 has pleiotropic activities on various cell types that are important in the pathogenesis of asthma. It is noteworthy that, unlike the IL-4-mediated production of IgE by B cells, or the IL-5-induced maturation of eosinophils, IL-9 seems to act primarily by enhancing the activities of many other cytokines and factors [23,28]. Blockage of its activity is therefore less likely to compromise normal host defense.
Genetic studies that identify the gene encoding IL-9 as a candidate gene for asthma
Asthma is an ecogenetic disorder in which the genetic background influences the environmental response [30]. Genetic mapping studies with asthma families identified several chromosomal regions that seem to harbor genes determining susceptibility to asthma and related risk factors including AHR and serum IgE [31,32,33,34,35,36]. Chromosome 5q31-q33 is one location in which asthma and its risk factors have been mapped by genetic linkage [10,34,35]. Chromosome 5q31-q33 contains numerous genes, such as those for cytokines, growth factors, and receptors, that are potentially involved in airway inflammation and atopic asthma [10]. Structural analyses of these candidate genes have identified polymorphisms in the promoter regions or coding regions of IL-4, IL-9, and IL-13 that are thought to alter gene regulation or function, and are potentially associated with atopy and asthma [37,38].
Significant biological variability in airway responsiveness in rodents has also been observed [39,40]. Linkage studies in mice associate AHR with a small region on chromosome 13 where the IL-9 gene is located [40]. This region is homologous with chromosome 5q31-q33 in humans [40]. In contrast, the cytokine genes encoding IL-4, IL-5, and IL-13 map to the syntenic region of mouse chromosome 11, excluding the linkage of these candidates in the mouse. Analyses of the murine IL-9 gene identified a genetic defect at the C57BL/6 (B6) IL-9 locus associated with no detectable lung IL-9 expression and reduced airway responsiveness in naive B6 mice. In contrast, robust expression of IL-9 gene and protein in the lung was observed in DBA/2J mice associated with AHR. Furthermore, (B6D2)F1 mice were intermediate in airway responsiveness and lung IL-9 levels, demonstrating a tight genotype-phenotype relation. These inbred strains showed no difference in IL-4 or IL-5 expression levels in the lung [40]. This naturally occurring variant, which lacks IL-9 (B6 mice), is healthy and does not seem to be immunocompromised. Thus, whereas IL-9 seems to act primarily by enhancing the activity of many other cytokines and chemokines, B6 mice suggest that it is dispensable.
Human studies corroborate these animal data. Elevated IL-9 expression, as determined by immunocytochemistry and in situ hybridization, demonstrated a highly significant and specific association between the expression of this cytokine with asthma and AHR ([41], and D Robinson, personal communication). Thus, IL-9 seems to be important in regulating the known risk factors for asthma, and represents an important target identified through genetic means for therapeutic intervention in this disorder.
IL-9 transgenic mice and recombinant IL-9 instillation in B6 mice provide further evidence for a role in asthma development
Two independent IL-9 transgenic models have been developed that indicate that the overexpression of IL-9 has a profound disturbance on hematopoietic cell lineages [15,42]. Systemic expression of an IL-9 transgene has been associated with lymphomagenesis, expansion of B-1 lymphocytes, enhanced IgE production, mastocytosis, and parasitic worm expulsion [42,43,44,45]. Both transgenic models, one with systemic expression, the other with expression of an IL-9 transgene controlled by a lung-specific promoter (CC-10 promoter), led to a phenotype consistent with asthma. This phenotype included mucus overproduction, sub-epithelial fibrosis, increased intra-epithelial mast cells, lung eosinophilia, elevated IgE levels, AHR, and increased responsiveness to antigen stimulation [12,15]. In contrast, overexpression of IL-4 with the CC-10 promoter resulted in baseline eosinophilia without AHR [46]. Overexpression of IL-13 in a similar model led to baseline AHR, eosinophilia, and mucus overproduction. However, bronchoalveolar lavage (BAL) fluid from IL-13 transgenic mice also contained mononuclear cells and enlarged multinucleate cells (a cell type not usually associated with human asthma), suggesting biological effects on granulocytes [47]. Mast cells are also important effector cells in asthma, and increased numbers of intra-epithelial lung mast cells were a noteworthy, apparently unique, finding in both strains of IL-9 transgenic mice [15,42]. Moreover, mast cells isolated from IL-13-/- transgenic knockouts were able to secrete levels of IL-4 and IL-5 that were equivalent to wild-type mast cells [48]. These data therefore suggest that IL-13 is not important in mast cell biology.
To elucidate further the temporal and spatial requirement of IL-9 in vivo on the pathophysiology of asthma, studies were made on recombinant mouse IL-9 (rIL-9) instilled into the airways of B6 mice, which had previously been shown to be genetically deficient in lung IL-9 protein [26]. Lung instillation of rIL-9 for up to 10 days produced all the histopathological features of asthma, including a time-dependent and dose-dependent increase in AHR, BAL eosinophils, total serum IgE, and lung proteases, along with submucosal membrane thickening [26]. Consistent with observations in IL-9 transgenic animals, increased mucus production in the rIL-9 instilled lung was associated with the specific upregulation of MUC2 and MUC5AC mucin gene products [28]. Collectively, these results suggest that IL-9 is sufficient to produce a classical TH2 response in vivo, and regulates AHR, lung eosinophilia, and serum total IgE.
Comparisons with mice instilled intratracheally with IL-13 demonstrated both similarities and some notable differences [49]. Mice similarly treated with IL-13 or IL-9 showed increased allergic inflammation, AHR, and mucous cell metaplasia, and induction of genes encoding lung chemokines, proteases, adhesion molecules, and mucins. Compared with mice receiving IL-9, mice treated with IL-13 had a greater cellular influx and inflammation-related gene upregulation at lower IL-13 doses and after fewer intratracheal instillations. Although IL-13 instillation caused eosinophil and lymphocyte influx into the lung, it was unique in its ability to recruit neutrophils as the dominant cell type. IL-9 administration resulted in eosinophil and lymphocyte infiltration without neutrophilia. At similar doses, both IL-9 and IL-13 produced comparable mucous metaplasia after only 3 days of instillations.
IL-9-neutralizing antibody ablates the asthmatic response without compromising the normal host defense system
Neutralizing antibodies or soluble receptors have been successful strategies for demonstrating the involvement of IL-4, IL-5, and IL-13 in asthmatic responses [13,14,50]. Recently, two studies using IL-9 neutralizing antibody confirmed that IL-9 is a necessary mediator of asthma [16,17]. In one of the models, IL-9-neutralizing antibody was instilled directly into the lungs of mice exposed to either Aspergillus fumigatus or dust mite antigen. In these asthma models of mucosal TH2 immunity, A. fumigatus or dust mite antigen produced a marked allergic inflammatory response in (B6D2)F1 mice, including significant increases in BAL eosinophils, elevated serum total IgE, increased mucin production, and AHR in comparison with control or naive animals. Intratracheal administration of IL-9-neutralizing antibody reduced the constellation of allergic inflammation in these models including AHR, serum total IgE elevations, and increases in BAL eosinophils in comparison with treatments of isotype-specific control antibody or untreated sensitized mice. Alcian Blue/PAS stains for mucus and overall histopathologic grading confirmed that the antibody treatment effectively abrogated the allergic asthmatic response in the antigen-challenged animals [16]. An independent study showed that similar blockage results from systematic administration of an IL-9 neutralization antibody in ovalbumin-treated BALB/c mice [17]. Taken together, these results demonstrate that IL-9 is both necessary and sufficient to produce the asthmatic response.
Most recently, the first IL-9-deficient mice were reported as being healthy with no overall development abnormalities or phenotype [51]. Naive cytokine profiles, TH1 and TH2 cell development, and generation of naive or antigen-specific antibody responses were all normal in IL-9-deficient mice. Using a primary pulmonary granulomatous model induced by Schistosoma mansoni eggs, the authors demonstrated a transient reduction in mucus production and lung mast cell numbers in the IL-9-deficient mice, indicating a fundamental role of IL-9 in the rapid and robust pulmonary mucus production and mastocytosis in response to lung challenge. Otherwise, IL-9-deficient mice developed normal granulomas with eosinophilia. Lymph nodes from IL-9-deficient mice expressed normal levels of IL-4, IL-5, and IL-13. In addition, the requirement for IL-9 in the expulsion of the intestinal parasitic nematode Nippostrongylus brasiliensis, as previously suggested in the IL-9 transgenic mice, was completely compensated for by increased expression of IL-4 and IL-5 [51]. However, in marked contrast to the specific reduction in goblet cell and mast cell responses observed in IL-9-deficient mice in the pulmonary granulomatous model, IL-13-deficient mice developed a global downregulation of the TH2 response with smaller granulomas around S. mansoni eggs, a reduction in lung eosinophilia, and reduced IL-4, IL-5, and IL-9 productions from cultured lymph nodes [51], indicating a role for IL-13 as a general enhancing factor on TH2 response.
Conclusion
IL-9 was first suggested as an asthma candidate gene by unbiased genetic mapping studies on AHR and linkage homology in both humans and mice. Cell biology in vitro has shown that IL-9 is an important growth factor and stimulator of numerous cell types important in the pathogenesis of asthma. Mice overexpressing IL-9 and neutralizing antibodies further demonstrate a crucial role for IL-9 in the development of the allergic asthmatic response. Importantly, increased lung IL-9 expression in mice and humans is tightly and specifically associated with asthma and its risk factors; decreased IL-9 is protective from antigen challenge. IL-9-deficient animals, including naturally occurring mutants and transgenic knockout mice, are healthy, suggesting that IL-9 therapeutic intervention will be well tolerated. Thus, IL-9 seems to be both necessary and sufficient to produce the TH2 response in vivo and is an important therapeutic target for allergic asthma.
Abbreviations
- AHR:
-
airway hyperresponsiveness
- BAL:
-
bronchoalveolar lavage
- IL:
-
interleukin
- rIL-9:
-
recombinant mouse IL-9
- TH2:
-
T helper type 2.
References
McFadden ER, Gilbert IA: Asthma. N Engl J Med. 1992, 327: 1928-1937.
Holgate ST, Church MK, Howarth PH, Morton EN, Frew AJ, Djukanovic R: Genetic and environmental influences on airway inflammation in asthma. Int Arch Allergy Immunol. 1995, 107: 29-33.
Sporik R, Ingram JM, Price W, Sussman JH, Honsinger RW, Platts-Mills TA: Association of asthma with serum IgE and skin test reactivity to allergens among children living at high altitude. Tickling the dragon's breath. Am J Respir Crit Care Med. 1995, 151: 1388-1392.
Kon OM, Kay AB: T cells and chronic asthma. Int Arch Allergy Immunol. 1999, 118: 133-135. 10.1159/000024049.
Yssel H, Groux H: Characterization of T cell subpopulations involved in the pathogenesis of asthma and allergic diseases. Int Arch Allergy Immunol. 2000, 121: 10-18. 10.1159/000024292.
Minor TE, Dick EC, DeMeo AN, Ouellette JJ, Cohen M, Reed CE: Viruses as precipitants of asthmatic attacks in children. J A M A. 1974, 227: 292-298. 10.1001/jama.227.3.292.
Ferreri NR, Howland WC, Stevenson DD, Spiegelberg HL: Release of leukotrienes, prostaglandins, histamine into nasal secretions of aspirin-sensitive asthmatics during reaction to aspirin. Am Rev Respir Dis. 1988, 137: 847-854.
Finnerty JP, Wood-Baker R, Thomson H, Holgate ST: Role of leukotrienes in exercise-induced asthma. Inhibitory effect of ICI 20 a potent leukotriene D4 receptor antagonist. Am Rev Respir Dis. 4219, 145: 746-749.
Venables KM, Chan-Yeung M: Occupational asthma. Lancet. 1997, 349: 1465-1469. 10.1016/S0140-6736(96)07219-4.
Postma DS, Bleecker ER, Amelung PJ, Holroyd KJ, Xu J, Panhuysen CI, Meyers DA, Levitt RC: Genetic susceptibility to asthma - bronchial hyperresponsiveness coinherited with a major gene for atopy. N Engl J Med. 1995, 333: 894-900. 10.1056/NEJM199510053331402.
Drazen JM, Arm JP, Austen KF: Sorting out the cytokines of asthma. J Exp Med. 1996, 183: 1-5.
McLane MP, Haczku A, van de Rijn M, Weiss C, Ferrante V, MacDonald D, Renauld JC, Nicolaides NC, Holroyd KJ, Levitt RC: Interleukin-9 promotes allergen-induced eosinophilic inflammation and airway hyperresponsiveness in transgenic mice. Am J Respir Cell Mol Biol. 1998, 19: 713-720.
Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD: Interleukin-13: central mediator of allergic asthma. Science. 1998, 282: 2258-2261. 10.1126/science.282.5397.2258.
Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksley RM, Corry DB: Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998, 282: 2261-2263. 10.1126/science.282.5397.2261.
Temann UA, Geba GP, Rankin JA, Flavell RA: Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, bronchial hyperresponsiveness. J Exp Med. 1998, 188: 1307-1320. 10.1084/jem.188.7.1307.
McLane MP, Tepper J, Weiss C, Tomer Y, Tobin P, Tumas D, Zhou Y, Haczku A, Nicolaides NC, Levitt RC: Lung delivery of blocking IL9 antibody inhibits airway hyperresponsiveness, Bal eosinophilia, mucin production, serum IgE elevation to natural antigens in a murine model of asthma [abstract]. Am J Respir Crit Care Med. 2000, 161: A593-
Kung TT, Crawley Y, Luo B, Minnicozzi M, Jones H, Egan RW, Kreutner W, Chapman RW: Effect of anti-mIL-9 antibody on the development of pulmonary eosinophilia and airway hyperresponsiveness in allergic mice [abstract]. Am J Respir Crit Care Med. 2000, 161: A844-
Hultner L, Druez C, Moeller J, Uyttenhove C, Schmitt E, Rude E, Dormer P, Van Snick J: Mast cell growth-enhancing activity (MEA) is structurally related and functionally identical to the novel mouse T cell growth factor P40/TCGFIII (interleukin 9). Eur JImmunol. 1990, 20: 1413-1416.
Renauld JC, Vink A, Louahed J, Van Snick J: Interleukin-9 is a major anti-apoptotic factor for thymic lymphomas. Blood. 1995, 85: 1300-1305.
Louahed J, Kermouni A, Van Snick J, Renauld JC: IL-9 induces expression of granzymes and high-affinity IgE receptor in murine T helper clones. J Immunol. 1995, 154: 5061-5070.
Van Snick J, Goethals A, Renauld JC, Van Roost E, Uyttenhove C, Rubira MR, Moritz RL, Simpson RJ: Cloning and characterization of a cDNA for a new mouse T cell growth factor (P40). J Exp Med. 1989, 169: 363-368.
Uyttenhove C, Simpson RJ, Van Snick J: Functional and structural characterization of P40, a mouse glycoprotein with T-cell growth factor activity. Proc Natl Acad Sci USA. 1988, 85: 6934-6938.
Dugas B, Renauld JC, Pene J, Bonnefoy JY, Peti-Frere C, Braquet P, Bousquet J, Van Snick J, Mencia-Huerta JM: Interleukin-9 potentiates the interleukin-4-induced immunoglobulin (IgG, IgM and IgE) production by normal human B lymphocytes. Eur J Immunol. 1993, 23: 1687-1692.
Louahed J, Zhou Y, Maloy WL, Rani PU, Weiss C, Tomer Y, Vink A, Renauld JC, Van Snick J, Nicolaides NC, Levitt RC, Haczku A: IL-9 promotes influx and local maturation of eosinophils. Blood. 2001.
Gounni AS, Gregory B, Nutku E, Aris F, Latifa K, Minshall E, North J, Tavernier J, Levitt R, Nicolaides N, Robinson D, Hamid Q: Interleukin-9 enhances interleukin-5 receptor expression, differentiation, survival of human eosinophils. Blood. 2000, 96: 2163-2171.
Levitt RC, McLane MP, MacDonald D, Ferrante V, Weiss C, Zhou T, Holroyd KJ, Nicolaides NC: IL-9 pathway in asthma: new therapeutic targets for allergic inflammatory disorders. J Allergy Clin Immunol. 1999, 103: S485-491.
Dong Q, Louahed J, Vink A, Sullivan CD, Messler CJ, Zhou Y, Haczku A, Huaux F, Arras M, Holroyd KJ, Renauld JC, Levitt RC, Nicolaides NC: IL-9 induces chemokine expression in lung epithelial cells and baseline airway eosinophilia in transgenic mice. Eur J Immunol. 1999, 29: 2130-2139. 10.1002/(SICI)1521-4141(199907)29:07<2130::AID-IMMU2130>3.0.CO;2-S.
Louahed J, Toda M, Jen J, Hamid Q, Renauld JC, Levitt RC, Nicolaides NC: Interleukin-9 upregulates mucus expression in the airways. Am JRespir Cell Mol Biol. 2000, 22: 649-656.
Longphre M, Li D, Gallup M, Drori E, Ordonez CL, Redman T, Wenzel S, Bice DE, Fahy JV, Basbaum C: Allergen-induced IL-9 directly stimulates mucin transcription in respiratory epithelial cells. J Clin Invest. 1999, 104: 1375-1382.
Marsh DG, Meyers DA, Bias WB: The epidemiology and genetics of atopic allergy. N Engl J Med. 1981, 305: 1551-1559.
Daniels SE, Bhattacharrya S, James A, Leaves NI, Young A, Hill MR, Faux JA, Ryan GF, le Souef PN, Lathrop GM, Musk AW, Cookson WO: A genome-wide search for quantitative trait loci underlying asthma. Nature. 1996, 383: 247-250. 10.1038/383247a0.
Anonymous: A genome-wide search for asthma susceptibility loci in ethnically diverse populations. Nat Genet. 1997, 15: 389-392.
Ober C, Cox NJ, Abney M, Di Rienzo A, Lander ES, Changyaleket B, Gidley H, Kurtz B, Lee J, Nance M, Pettersson A, Prescott J, Richardson A, Schlenker E, Summerhill E, Willadsen S, Parry R: Genome-wide search for asthma susceptibility loci in a founder population. The Collaborative Study on the Genetics of Asthma. Hum Mol Genet. 1998, 7: 1393-1398. 10.1093/hmg/7.9.1393.
Marsh DG, Neely JD, Breazeale DR, Ghosh B, Freidhoff LR, Ehrlich-Kautzky E, Schou C, Krishnaswamy G, Beaty TH: Linkage analysis of IL4 and other chromosome 5q31.1 markers and total serum immunoglobulin E concentrations. Science. 1994, 264: 1152-1156.
Meyers DA, Postma DS, Panhuysen CI, Xu J, Amelung PJ, Levitt RC, Bleecker ER: Evidence for a locus regulating total serum IgE levels mapping to chromosome 5. Genomics. 1994, 23: 464-470. 10.1006/geno.1994.1524.
Barnes KC, Neely JD, Duffy DL, Freidhoff LR, Breazeale DR, Schou C, Naidu RP, Levett PN, Renault B, Kucherlapati R, Iozzino S, Ehrlich E, Beaty TH, Marsh DG: Linkage of asthma and total serum IgE concentration to markers on chromosome 12q: evidence from Afro-Caribbean and Caucasian populations. Genomics. 1996, 37: 41-50. 10.1006/geno.1996.0518.
Rosenwasser LJ, Klemm DJ, Dresback JK, Inamura H, Mascali JJ, Klinnert M, Borish L: Promoter polymorphisms in the chromosome 5 gene cluster in asthma and atopy. Clin Exp Allergy. 1995, 25: 74-78.
Heinzmann A, Mao XQ, Akaiwa M, Kreomer RT, Gao PS, Ohshima K, Umeshita R, Abe Y, Braun S, Yamashita T, Roberts MH, Sugimoto R, Arima K, Arinobu Y, Yu B, Kruse S, Enomoto T, Dake Y, Kawai M, Shimazu S, Sasaki S, Adra CN, Kitaichi M, Inoue H, Yamauchi K, Tomichi N, Kurimoto F, Hamasaki N, Hopkin JM, Izuhara K, Shirakawa T, Deichmann KA: Genetic variants of IL-13 signalling and human asthma and atopy. Hum Mol Genet. 2000, 9: 549-559. 10.1093/hmg/9.4.549.
Pauwels R: The relationship between airway inflammation and bronchial hyperresponsiveness. Clin Exp Allergy. 1989, 19: 395-398.
Nicolaides NC, Holroyd KJ, Ewart SL, Eleff SM, Kiser MB, Dragwa CR, Sullivan CD, Grasso L, Zhang LY, Messler CJ, Zhou T, Kleeberger SR, Buetow KH, Levitt RC: Interleukin 9: a candidate gene for asthma. Proc Natl Acad Sci USA. 1997, 94: 13175-13180. 10.1073/pnas.94.24.13175.
Shimbara A, Christodoulopoulos P, Soussi-Gounni A, Olivenstein R, Nakamura Y, Levitt RC, Nicolaides NC, Holroyd KJ, Tsicopoulos A, Lafitte JJ, Wallaert B, Hamid QA: IL-9 and its receptor in allergic and nonallergic lung disease: increased expression in asthma. J Allergy Clin Immunol. 2000, 105: 108-115.
Renauld JC, van der Lugt N, Vink A, van Roon M, Godfraind C, Warnier G, Merz H, Feller A, Berns A, Van Snick J: Thymic lymphomas in interleukin 9 transgenic mice. Oncogene. 1994, 9: 1327-1332.
Vink A, Warnier G, Brombacher F, Renauld JC: Interleukin 9-induced in vivo expansion of the B-1 lymphocyte population. J Exp Med. 1999, 189: 1413-1423. 10.1084/jem.189.9.1413.
Faulkner H, Humphreys N, Renauld JC, Van Snick J, Grencis R: Interleukin-9 is involved in host protective immunity to intestinal nematode infection. Eur JImmunol. 1997, 27: 2536-2540.
Faulkner H, Renauld JC, Van Snick J, Grencis RK: Interleukin-9 enhances resistance to the intestinal nematode Trichuris muris. Infect Immun. 1998, 66: 3832-3840.
Rankin JA, Picarella DE, Geba GP, Temann UA, Prasad B, DiCosmo B, Tarallo A, Stripp B, Whitsett J, Flavell RA: Phenotypic and physiologic characterization of transgenic mice expressing interleukin 4 in the lung: lymphocytic and eosinophilic inflammation without airway hyperreactivity. Proc Natl Acad Sci USA. 1996, 93: 7821-7825. 10.1073/pnas.93.15.7821.
Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA: Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, eotaxin production. J Clin Invest. 1999, 103: 779-788.
McKenzie GJ, Emson CL, Bell SE, Anderson S, Fallon P, Zurawski G, Murray R, Grencis R, McKenzie AN: Impaired development of Th2 cells in IL-13-deficient mice. Immunity. 1998, 9: 423-432.
Reader R, Aldrich M, Li C, Stoddard A, Schelegle E, Hyde D, Tepper J: Induction of murine asthma by intratracheal IL9 and IL13. Am J Respir Crit Care Med. 2001.
Webb DC, McKenzie AN, Koskinen AM, Yang M, Mattes J, Foster PS: Integrated signals between IL-13, IL-4, IL-5 regulate airways hyperreactivity. J Immunol. 2000, 165: 108-113.
Townsend MJ, Fallon PG, Matthews DJ, Smith P, Jolin HE, McKenzie AN: IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development. Immunity. 2000, 13: 573-583.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhou, Y., McLane, M. & Levitt, R.C. Th2 cytokines and asthma — Interleukin-9 as a therapeutic target for asthma. Respir Res 2, 80 (2001). https://doi.org/10.1186/rr42
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/rr42