
Abstract
A previous knockout of the transcription factor gene nuclear factor IX (NFIX) in mice produced impaired development of the corpus callosum and severe skeletal defects. A recent paper in BMC Developmental Biology reports an apparently similar NFIX knockout that produced marked differences in phenotype, raising intriguing general questions about the possible causes of such differences in mouse knockouts.
Similar content being viewed by others


The Nuclear Factor I (NFI) family of evolutionarily conserved transcription factors is widely expressed during development and in adulthood, in mammals but has mainly been studied in respect to brain development, where it is intimately associated with glial function [1, 2]. The family consists of four members, NFIA, NFIB, NFIC and NFIX, each having multiple splice variants [3]. NFI proteins can directly bind to the promoter and regulate the transcription activity of glial fibrillar acidic protein (GFAP), a marker of glial cells [4]. Different members of the family have been shown to have a variety of roles in neural development but taken together, loss-of-function studies of NFI members in mice reveal a common theme – a lack of development (agenesis) of the corpus callosum, the large tract of nerve fibers interconnecting the left and right hemispheres. The main feature of corpus callosum agenesis is an inability to perform tasks where a matching of visual patterns is required, for example face processing, which in turn results in social difficulties. In mild cases intelligence is mainly unaffected but low muscle tone and motor coordination are affected. In severe cases intellectual retardation, hydrocephalus, seizures and spasticity might be involved. The effect of a mutation varies from partial callosal agenesis (in the case of loss of function of NFIX) to severe agenesis (with loss of function of NFIB having a greater effect than loss of NFIA, as described later).
Less is known so far about the actions of the NFIX gene than about the other members of the family. One known property of NFIX is the regulation of expression of astrocyte-specific α1-antichymotrypsin [5]. To determine the effects of loss of function of NFIX, two groups have recently described knockouts of the NFIX gene [6, 7]. Their results turned out to be surprisingly different. The first knockout was reported by a team at the University of Freiburg (Driller et al. [6]) while the second was generated by a group from the University of New York at Buffalo and described in BMC Developmental Biology (Campbell et al. [7]). Here, we briefly review some of the possible reasons for such discrepancies. For simplicity, we will call the mutant strain generated in Freiburg 'X-Freiburg' and the one generated in New York 'X-NY'. Animals of the X-Freiburg strain suffered from hydrocephalus, partial agenesis of the corpus callosum, and spinal deformities that were due to a delay in ossification of vertebral bodies and progressive degeneration of intervertebral discs. Femoral defects were also noticed and animals usually died at around postnatal day (P) 21–28. The X-NY strain, on the other hand, did not suffer from such severe impairments. Callosal agenesis as seen in the X-Freiburg strain was not noted in X-NY NFIX-/- animals. The cingulate cortex and the entire brain are expanded along the dorsal-ventral axis, hippocampus formation is aberrant, and overabundant Pax6- and doublecortin-positive cells are found in the lateral ventricles of X-NY mice.
When the X-NY mice were fed with a soft dough chow they showed a lag in weight gain compared to non-mutant animals, but after P20 the growth rate increased and a few of the animals survived to adulthood. Skeletal deformities observed by Driller et al. and absent in the animals reported by Campbell et al. can be attributed to the severe malnutrition, which was relieved by Campbell et al. by the change in diet. Another possibility is that brain development abnormalities result in reduced appetite, leading again to skeletal defects.
Reconciling the differences
How can the discrepancies reported between the two NFIX-/- strains be reconciled? Among various possible explanations, one could be an alteration of neighboring gene expression. A case in point is the sequential generation of several prion protein (PrP) knockout strains that showed profoundly different phenotypes. Only later was this variation proved to be due to the unintentional activation of another gene in the vicinity of the PrP gene, later named Doppel [8], and which was shown to be neurotoxic.
Both reports of the NFIX knockouts [6, 7] describe the deletion of the second exon, which is uniformly present in all splice variants and carries the dimerization and DNA-binding domains (Figure 1). In both cases the targeting constructs were based on a λ phage library derived from the mouse strain 129/Sv, and transgenic animals carrying a single knockout allele were backcrossed to C57BL/6 mice for several generations. However, each research group used a slightly different embryonic stem (ES) cell line for making the mutation. In the case of the X-NY strain the targeting vector was electroporated into J1 ES cells, which are derived from the 129S4/SvJae strain and backcrossed to the C57BL/6 mouse strain for two to five generations. The X-Freiburg targeting construct was electroporated into CJ7 ES cells, which originate from the 129S1/Sv strain (129S1/Sv-p+Tyr+KitlSl-J) and transgenic animals were backcrossed to C57BL/6. Driller et al. [6] do not specify the number of backcrossings to C57BL/6, which raises the possibility that their knockout strains, although apparently congenic with those of Campbell et al., contain a substantial segment of ES-cell-derived chromosome still flanking the knockout allele – a 'congenic footprint'.

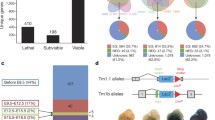
For simplicity the same structure is drawn for all four NFI genes. (a) The organization of the NFI genes. They can all use an alternative exon 1, here denoted as a single box labeled 1a/1b. The DNA-binding and dimerization domains are located in exon 2. (b) In general, two approaches are used for knockouts of these genes. The first relies on complete deletion of the second exon (including 5' and 3' splice acceptor sites of proximal introns), as shown here in the X-NY knockout. The second strategy is to insert LacZ (or a LacZ-neo hybrid or PGK-neo hybrid) in-frame into the second exon, leading to production of a fusion protein composed of a few amino acids derived from exons 1 and 2 of the NFI and LacZ genes. In all cases an alternative splice variant joining the first and the third exon of the NFI gene will be formed. The third exon is not in frame with the first, and so premature termination of translation will occur. Whether a peptide produced from the joining of exons 1 and 3 has any physiological function was never analyzed, but judging from the very different phenotypes of the different knockout strains it seems rather unlikely. The NFIB constructs are reported in [15, 16], the NFIA knockout in [17] and the NFIC knockout in [18].
In a study of congenic knockouts at another gene, Schalkwyk et al. [9] found that at least 10 genes across 40 Mb around the targeted locus show differences in expression in the different knockout strains, due to the congenic footprint effect. Genome-wide analysis of gene expression in different tissues of knockout animals by microarray profiling also indicates that a significant proportion of changes are found in the proximity of the targeted gene [10, 11]. This little excursion into the theory of induced mutation experiments does not seem so trivial in the light of several studies describing corpus callosum defects in the 129/Sv strain itself [12, 13], which vary between 129 substrains studied [14]. Callosal agenesis is one of the phenotypic features ascribed to the X-Freiburg strain, while at the same time complete callosal agenesis was not seen in X-NY strain. The locus (or loci) responsible for callosal agenesis in the 129/Sv strain is not characterized and it is not unreasonable to speculate that such a region might be present in the proximity of NFIX in the X-Freiburg strain, whereas in the X-NY strain this locus had been removed by outbreeding.
Another possible source of variation emanates from the targeting strategy used. Campbell et al. completely deleted the second exon along with proximal parts of neighboring introns, whereas Driller et al. replaced the second exon with a coding sequence of the LacZ gene fused to a coding sequence of NFIX (Figure 1b). In this regard, a comparison of all the available NFI gene knockouts is perhaps more informative (Figure 1b). An intriguing feature that emerges from this comparison is that mice in which the 3' splice acceptor of the first intron is removed somehow have a milder phenotype. Without further experimental evidence it is difficult to explain this observation, which could be purely coincidental. Formation of an alternatively spliced gene variant (which was not looked for), as with the activation of Doppel [8], is one possibility. Alternatively, the fusion of the first few amino acids of NFIX (or NFIB) to LacZ might lead to a toxic gain-of-function protein. The peptide in question is quite short but even so could endow the fusion protein with toxic properties. This hypothesis is rather easy to test by overexpressing recombinant NFIXexon1-LacZ protein in glial or neuronal cells.
A full explanation of these intriguing phenotypes will require experimental testing and a proper analysis of the ideas put forward here, as well as other possibilities. Thorough analysis of all available knockouts might reveal surprising new functions of NFI proteins and further enhance our understanding of their biological functions.
References
Shu T, Butz KG, Plachez C, Gronostajski RM, Richards LJ: Abnormal development of forebrain midline glia and commissural projections in Nfia knock-out mice. J Neurosci. 2003, 23: 203-212.
Deneen B, Ho R, Lukaszewicz A, Hochstim CJ, Gronostajski RM, Anderson DJ: The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron. 2006, 52: 953-968. 10.1016/j.neuron.2006.11.019.
Gründer A, Qian F, Ebel TT, Mincheva A, Lichter P, Kruse U, Sippel AE: Genomic organization, splice products and mouse chromosomal localization of genes for transcription factor Nuclear Factor One. Gene. 2003, 304: 171-181. 10.1016/S0378-1119(02)01204-0.
Cebolla B, Vallejo M: Nuclear factor-I regulates glial fibrillary acidic protein gene expression in astrocytes differentiated from cortical precursor cells. J Neurochem. 2006, 97: 1057-1070. 10.1111/j.1471-4159.2006.03804.x.
Gopalan SM, Wilczynska KM, Konik BS, Bryan L, Kordula T: Nuclear factor-1-X regulates astrocyte-specific expression of the alpha1-antichymotrypsin and glial fibrillary acidic protein genes. J Biol Chem. 2006, 281: 13126-13133. 10.1074/jbc.M601194200.
Driller K, Pagenstecher A, Uhl M, Omran H, Berlis A, Grunder A, Sippel AE: Nuclear factor I X deficiency causes brain malformation and severe skeletal defects. Mol Cell Biol. 2007, 27: 3855-3867. 10.1128/MCB.02293-06.
Campbell CE, Piper M, Plachez C, Yeh YT, Baizer JS, Osinski JM, Litwack ED, Richards LJ, Gronostajski RM: The transcription factor Nfix is essential for normal brain development. BMC Dev Biol. 2008, 8: 52-10.1186/1471-213X-8-52.
Weissmann C, Flechsig E: PrP knock-out and PrP transgenic mice in prion research. Br Med Bull. 2003, 66: 43-60. 10.1093/bmb/66.1.43.
Schalkwyk LC, Fernandes C, Nash MW, Kurrikoff K, Vasar E, Kõks S: Interpretation of knockout experiments: the congenic footprint. Genes Brain Behav. 2007, 6: 299-303. 10.1111/j.1601-183X.2007.00304.x.
Valor LM, Grant SG: Clustered gene expression changes flank targeted gene loci in knockout mice. PLoS ONE. 2007, 2: e1303-10.1371/journal.pone.0001303.
Crusio WE: Flanking gene and genetic background problems in genetically manipulated mice. Biol Psychiatry. 2004, 56: 381-385. 10.1016/j.biopsych.2003.12.026.
Livy DJ, Wahlsten D: Tests of genetic allelism between four inbred mouse strains with absent corpus callosum. J Hered. 1991, 82: 459-464.
Wahlsten D, Schalomon PM: A new hybrid mouse model for agenesis of the corpus callosum. Behav Brain Res. 1994, 64: 111-117. 10.1016/0166-4328(94)90123-6.
Wahlsten D: Deficiency of corpus callosum varies with strain and supplier of the mice. Brain Res. 1982, 239: 329-347. 10.1016/0006-8993(82)90513-3.
Steele-Perkins G, Plachez C, Butz KG, Yang G, Bachurski CJ, Kinsman SL, Litwack ED, Richards LJ, Gronostajski RM: The transcription factor gene Nfib is essential for both lung maturation and brain development. Mol Cell Biol. 2005, 25: 685-698. 10.1128/MCB.25.2.685-698.2005.
Gründer A, Ebel TT, Mallo M, Schwarzkopf G, Shimizu T, Sippel AE, Schrewe H: Nuclear factor I-B (Nfib) deficient mice have severe lung hypoplasia. Mech Dev. 2002, 112: 69-77. 10.1016/S0925-4773(01)00640-2.
das Neves L, Duchala CS, Tolentino-Silva F, Haxhiu MA, Colmenares C, Macklin WB, Campbell CE, Butz KG, Gronostajski RM: Disruption of the murine nuclear factor I-A gene (Nfia) results in perinatal lethality, hydrocephalus, and agenesis of the corpus callosum. Proc Natl Acad Sci USA. 1999, 96: 11946-11951. 10.1073/pnas.96.21.11946.
Steele-Perkins G, Butz KG, Lyons GE, Zeichner-David M, Kim HJ, Cho MI, Gronostajski RM: Essential role for NFI-C/CTF transcription-replication factor in tooth root development. Mol Cell Biol. 2003, 23: 1075-1084. 10.1128/MCB.23.3.1075-1084.2003.
Acknowledgements
Work in the laboratory of Juan Carlos Izpisua Belmonte was supported by funds from the G Harold and Leila Y Mathers Charitable Foundation, Fundacion Cellex and the Marato.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Pekarik, V., Belmonte, J.C.I. NFIX - one gene, two knockouts, multiple effects. J Biol 7, 29 (2008). https://doi.org/10.1186/jbiol94
Published:
DOI: https://doi.org/10.1186/jbiol94