
Abstract
Background
BRCA1 mutation carriers have an 85% risk of developing breast cancer but the risk of developing non-hereditary breast cancer is difficult to assess. Our objective is to test whether a DNA methylation (DNAme) signature derived from BRCA1 mutation carriers is able to predict non-hereditary breast cancer.
Methods
In a case/control setting (72 BRCA1 mutation carriers and 72 BRCA1/2 wild type controls) blood cell DNA samples were profiled on the Illumina 27 k methylation array. Using the Elastic Net classification algorithm, a BRCA1-mutation DNAme signature was derived and tested in two cohorts: (1) The NSHD (19 breast cancers developed within 12 years after sample donation and 77 controls) and (2) the UKCTOCS trial (119 oestrogen receptor positive breast cancers developed within 5 years after sample donation and 122 controls).
Results
We found that our blood-based BRCA1-mutation DNAme signature applied to blood cell DNA from women in the NSHD resulted in a receiver operating characteristics (ROC) area under the curve (AUC) of 0.65 (95% CI 0.51 to 0.78, P = 0.02) which did not validate in buccal cells from the same individuals. Applying the signature in blood DNA from UKCTOCS volunteers resulted in AUC of 0.57 (95% CI 0.50 to 0.64; P = 0.03) and is independent of family history or any other known risk factors. Importantly the BRCA1-mutation DNAme signature was able to predict breast cancer mortality (AUC = 0.67; 95% CI 0.51 to 0.83; P = 0.02). We also found that the 1,074 CpGs which are hypermethylated in BRCA1 mutation carriers are significantly enriched for stem cell polycomb group target genes (P <10-20).
Conclusions
A DNAme signature derived from BRCA1 carriers is able to predict breast cancer risk and death years in advance of diagnosis. Future studies may need to focus on DNAme profiles in epithelial cells in order to reach the AUC thresholds required of preventative measures or early detection strategies.
Similar content being viewed by others

Background
Breast cancer is the most common cancer in women, affecting at least 1 in 10 women in the western world. The potential to predict breast cancer and offer preventive measures is an effective intervention in women with an inherited predisposition to breast cancer due to mutations in BRCA1/2 genes [1]. However, these account for less than 10% of breast cancers [2]. While extensive genome-wide association studies have identified a number of single nucleotide polymorphisms (SNPs) associated with breast cancer risk [3], epidemiological models that include risk associated SNPs yield a receiver-operating-characteristic (ROC) area under the curve (AUC) of only 62%, a modest 4% improvement over the AUC of epidemiological models [4].
Predicting the likelihood of breast cancer development is therefore still challenging not only because the sensitivity of current strategies is low [4] but also because 11% to 52% of screen-detected breast cancers may be an over diagnosis of cancers which would have never become clinically evident [5–7]. Hence a biomarker that could predict the risk of developing breast cancer particularly in those with a poor prognosis and which is also independent of familial predisposition is urgently needed.
It is known that epigenetic variation contributes to inter-individual variation in gene expression and thus may contribute to variation in cancer susceptibility [8–10]. DNAme is the most studied mechanism of epigenetic gene regulation and represents a biologically and chemically stable signal. Aberrant DNA methylation is also a hallmark of cancer [9, 11], in particular increased promoter DNAme at stem cell differentiation genes (Polycomb-Repressive Complex 2 (PRC2) Group Target genes (PCGTs)) [12–19]. Initial evidence suggests that BRCA1 is a key negative modulator of PRC2 and that loss of BRCA1 inhibits stem cell differentiation and enhances an aggressive breast cancer phenotype by affecting PRC2 function [20]. Several proof of principle studies using a target gene approach or assessment of global DNA methylation analysing samples collected at the time of diagnosis provided the first evidence for the feasibility of breast cancer risk prediction using DNA methylation based markers [21–32]. It was also recently demonstrated that DNAme profiles in blood are able to predict cancer risk (on average 1.3 years in advance) within a group of women whose sisters had developed breast cancer [33].
Here we tested the hypothesis that women with an extremely high breast cancer risk (due to a BRCA1 mutation) carry a specific methylation signature in peripheral blood cells, which is also able to predict sporadic breast cancer incidence and death. We also tested whether this signature is tissue-specific.
Methods
Data from three different studies were used.
BRCA1 study
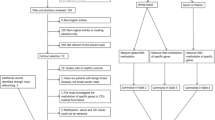
We analysed whole blood samples from two cohorts of BRCA1 mutation carriers and controls without a BRCA1 mutation (see Figure 1 and Additional file 1).

Study design and identification/validation of the BRCA1 -mutation DNAme risk signature. AUC, receiver operating characteristics area under the curve; BC, breast cancer; FDR, false discovery rate; inv., invasive; WBC, white blood cells.
MRC National Survey of Health and Development (NSHD)
We analysed both blood cells and buccal cells from a sample of women from the NSHD, a birth cohort study of men and women born in Britain in March 1946 [34–36]. A total of 152 (75 cancer cases and 77 controls) women were selected from those who provided both a peripheral blood and a buccal cell sample at the age of 53 years in 1999, who had not previously developed any cancer and who had complete information on epidemiological variables of interest and follow-up. We analysed >480,000 CpGs (using the Illumina 450 k array) in the 46 women who developed an invasive non-skin cancer (19 breast cancer, 5 reproductive tract and 22 other cancers; diagnosed 1 to 7 years after 53 years and an average of 4.75 years) and in the women (n = 77) who did not develop any cancer during the 12-year follow-up (for descriptive analysis see Additional file 2).
United Kingdom Collaborative Trial of Ovarian Cancer Screening (UKCTOCS)
We analysed serum DNA samples (which largely represent white blood cell DNA in this cohort - see Additional files 3 and 4) from postmenopausal women who developed breast cancer (n = 119) or remained cancer-free during the follow-up period (n = 122, maximum of 12 year follow-up (2001 to 2013)).
Ethics
All studies were approved by the relevant research ethics committee or institutional review board. Informed consent was obtained by all volunteers and conforms with the Declaration of Helsinki. The BRCA1 study was approved by the ethics committee of the General University Hospital, Prague (No. 1199/07 S-IV). The NSHD epigenetics study was approved by the Central Manchester Research Ethics Committee (REC reference: 07/H1008/168). UKCTOCS was approved by the UK North West Multicentre Research Ethics Committees (North West MREC 00/8/34). Ethical approval for this nested case control study was obtained from the Joint UCL/UCLH Committees on the Ethics of Human Research (REC reference: 06/Q0505/102).
DNA methylation analysis
The DNA from whole blood and tissues was extracted at UCL [36] and at Gen-Probe [37]. Methylation analysis was performed using the validated Illumina Infinium Human Methylation27 BeadChip [16] or the Illumina Infinium Human Methylation450 BeadChip for NSHD samples. The methylation status of a specific CpG site was calculated from the intensity of the methylated (M) and unmethylated (U) alleles, as the ratio of fluorescent signals β = Max(M,0)/(Max(M,0) + Max(U,0) + 100). On this scale, 0 < β < 1, with β values close to 1 (0) indicating 100% methylation (no methylation) (see Additional file 4).
Data availability
Data from two of the studies in this manuscript have been deposited in the Gene Expression Omnibus repository under the accession numbers (GSE58119), (GSE57285), (GSE32396). The NSHD data are made available to researchers who submit data requests to mrclha.swiftinfo@ucl.ac.uk; see full policy documents at [38]. Managed access is in place for this 68-year-old study to ensure that use of the data is within the bounds of consent given previously by participants, and to safeguard any potential threat to anonymity since the participants are all born in the same week.
Statistical analyses
Differential methylation analysis
From the BRCA1 study, differentially methylated CpGs, with false discovery rate (FDR) corrected P values, between BRCA1 mutant carriers and BRCA1 wild type samples were identified via a multivariate logistic regression that was adjusted for age, batch and the presence of cancer.
Ensemble signature identification
The elastic net classification method was chosen for our study as it has been shown to be particularly effective when the number of predictors is far greater than the number of training points [39]. The elastic net method, as implemented in the glmnet R-package [40], identified a classifier comprising 1,829 CpGs with non-zero regression coefficients (see Additional file 4).
Validation
To evaluate its predictive accuracy, the identified classifier was tested on two independent datasets: (1) NSHD, and (2) UKCTOCS. For each individual, risk scores, based on their methylation profiles, were estimated and correlated to their disease status. An AUC value was then obtained via Somers’ Dxy rank correlation [41] (see Additional file 4).
Results
DNA methylation signature in white blood cells (WBC) associated with BRCA1 mutation status
We analysed DNAme of 27,578 CpGs in WBC samples from a total of 72 women with a known BRCA1 mutation and 72 women with no mutation in the BRCA1 or BRCA2 gene (Figure 1 and Additional file 1). The presence of a cancer has been shown to modulate the composition of WBCs and DNAme profiles in peripheral blood [42] and hence we used a mixture of women who did and who did not develop breast cancer in order to be able to adjust for this. Using a multivariate regression model that included age, cohort and cancer status as covariates we were able to rank CpGs according to the significance of the association between their DNAme profile and mutation status. On applying a relaxed threshold of FDR <0.3 we observed a total of 2,514 BRCA1-mutation associated CpGs, of which 1,422 (57%) were hypermethylated (hyperM) and 1,092 (43%) were hypomethylated (hypoM) in women who had a BRCA1 mutation (Figure 1, Additional file 5), representing a highly significant skew towards hypermethylated CpGs (Binomial test P < 1e-10). To arrive at a specific DNAme signature, which would allow classification of independent samples, we used the elastic net (ELNET) framework (see Additional file 4), which resulted in a signature consisting of 1,829 CpGs (Figure 2, Additional file 6).

CpGs (n = 1829), which are differentially methylated in WBCs between BRCA1 mutation carriers and wild type controls and which comprise the ‘ BRCA1 -mutation DNA methylation signature’. Heatmap of normalised methylation values (blue = relative high methylation, yellow = relative low methylation) of CpGs comprising the BRCA1-mutation DNAme signature. The first colour bar at the top denotes the two main clusters where ‘red’ reflects the samples with a BRCA1 mutation whereas ‘green’ reflects samples without a mutation in BRCA1 or BRCA2 gene. The distribution of cancer cases is given in the second colour bar indicating women who had developed a breast cancer in purple. Right panel shows the enrichment of the top components of the gene set enrichment analysis in the hyper- and hypomethylated subset of CpGs; PCGT; Polycomb repressor complex 2 Group Target. Dashed line separates hypermethylated from hypomethylated CpGs.
Given that PCGT methylation is a hallmark of almost all cancers and that a BRCA1 defect in normal non-neoplastic cells is likely to silence PCGTs and compromise cell differentiation [20], we posited that our BRCA1 DNAme signature may be able to predict sporadic breast cancer. Interestingly, Gene Set Enrichment Analysis (GSEA) [43, 44] on the 1,074 hypermethylated (Additional file 7) and 755 hypomethylated (Additional file 8) CpGs of the BRCA1- mutation signature demonstrated the association of BRCA1 mutation with promoter hypermethylation of PCGTs. Indeed, the top categories of genes, associated with the hypermethylated CpGs in BRCA1 mutation carriers, were significantly (P <10-10) enriched for stem cell PCGTs irrespective of the definition used (Figure 2, Additional file 7). In contrast, none of the gene categories associated with those CpGs which are hypomethylated in BRCA1 mutation carriers reached significance based on adjusted P values (Additional file 8). Even the GSEA on the 105 CpGs with a more stringent FDR (<=0.05) associated with BRCA1 mutation in white blood cells demonstrated the enrichment of PCGTs (P < =0.02) (Additional file 9).
BRCA1-mutation DNAme signature and breast cancer risk in peripheral blood cells in the NSHD
In order to test whether the BRCA1-mutation DNAme signature is able to identify women who will develop breast cancer we analysed one of the best available characterised longitudinal cohorts (Additional file 2). Applying the BRCA1-mutation DNAme signature (out of the 1,829 BRCA1 CpGs, 1,722 were present on the 450 k Illumina methylation array), yielded a breast cancer risk AUC = 0.65 (0.51 to 0.78, P = 0.02) (Figure 3A). Interestingly, the BRCA1 signature also significantly predicted the future development of invasive non-breast cancers (AUC = 0.62; 0.50 to 0.74; P = 0.04) (Additional file 10A).

Validation of the BRCA1 -mutation DNAme signature in two independent prospective cohorts. ROC curves and AUC statistics to predict future breast cancer (BC) incidence applying the BRCA1-mutation DNAme signature in white blood cells (WBCs) (A) and in buccal (BUCC) cells (B) of the NSHD cohort and in serum DNA of the UKCTOCS cohort (C). Overlap of the top CpGs differently methylated in WBC between BRCA1 mutant and wild type (BRCA1 study) and the top CpGs differently methylated in serum DNA between women who have developed oestrogen receptor positive BCs and women who remained cancer-free (D). ROC curve and AUC statistics to predict deadly BCs applying the BRCA1-mutation DNAme signature in serum DNA in the UKCTOCS cohort (E) and Kaplan Meier curve (and hazard ratio (HR)) of future breast cancer patients with a high and low BRCA1-mutation DNAme score in serum DNA (F).
Consistent with the view that DNAme is tissue-specific, our DNAme signature - derived from peripheral blood cells from women with known BRCA1 status - was not able to predict invasive breast cancer (Figure 3B) or invasive non-breast cancer (Additional file 10B) in the buccal cell DNAme profiles obtained at the same time from the same women who provided blood DNA.
BRCA1- mutation DNA methylation signature and breast cancer risk in serum DNA in the UKCTOCS cohort
Less than 10% of invasive breast cancers are due to a BRCA1 mutation [45] and therefore it is unlikely that the predictive capacity of the BRCA1-mutation DNAme signature in the NSHD cohort was due to the correct identification of BRCA1 mutation carriers. Nevertheless in order to further substantiate that the BRCA1-mutation DNAme signature identifies sporadic cancers, we performed a nested case–control study within the UKCTOCS cohort (a 202,638 postmenopausal women cohort, who based on their family history were not at an increased risk of ovarian or breast cancer - see Additional files 3 and 4). As BRCA1-associated cancers are far more likely (75%) to be oestrogen receptor (ER) negative [46], we solely focused our analysis on women who provided a blood sample between 0.42 and 4.18 years (average 2 years) before they developed an ER positive invasive breast cancer (n = 119) and matched (on age at blood donation and recruitment centre) them to 122 women who did not develop a breast cancer during the follow-up period (5.61 to 12 years, average follow-up 11.92 years). As there was no whole blood DNA samples available from the women in UKCTOCS, we used serum-free DNA as a source of material for this analysis. Since >95% of blood samples were only spun down 24 to 48 h after the blood draw, it was important for us to identify the likely source of DNA in the serum samples. Although we were not able to definitely identify the source, the evidence clearly pointed towards an enriched for WBC DNA (see Additional file 11). The BRCA1-mutation DNAme signature predicted the development of an ER positive breast cancer with an AUC = 0.57 (0.50 to 0.64; P = 0.03) (Figure 3C) independent of whether the sample was taken less or more than 2 years prior to diagnosis (see Additional file 12). Importantly, the BRCA1- mutation DNAme signature also substantially overlapped with an ER + breast cancer specific risk signature (Additional file 13), which we derived de novo in the UKCTOCS cohort (P <2 x 10-33, Figure 3D). Of note, in the breast cancer specific risk signature, we also observed enrichment of biological terms, all crucially involved in stem cell differentiation and biology (Additional file 14). Again, these stem cell gene categories were only enriched among CpGs hypermethylated in cases, but not among CpGs hypomethylated in cases (Additional file 15). This observation is particularly pertinent given that NIPP1, PRC2, MSX1 and NANOG all suppress differentiation through occupation and suppression of specific gene sets.
BRCA1-mutation DNAme signature identifies women years in advance of fatal breast cancer diagnosis
In order to test whether the BRCA1-mutation DNAme signature is able to predict not only incidence but also breast cancer mortality we performed ROC statistics in the UKCTOCS set comparing women who died from breast cancer (n = 10) during the follow-up period with women who did not develop breast cancer (Figure 3E) and found an AUC = 0.67 (0.51 to 0.83; P = 0.02). In line with these findings women with a higher than average BRCA1-mutation DNAme signature score were 8.46 (95% CI 1.06 to 67.69) -fold more likely to die from breast cancer (P = 0.04) than those with lower than average scores (Figure 3F). Interestingly, apart from the number of nodes, none of the other clinico-pathological features or treatment modalities was associated with the BRCA1-mutation DNAme signature in these ER positive breast cancers (Additional file 16).
BRCA1-mutation DNAme signature and association with epidemiological and hormonal risk markers
Next, we were interested whether our DNAme signature could be explained by any of the breast cancer risk factors we had available for the UKCTOCS cohort. Interestingly, neither any of the epidemiological breast cancer risk factors nor any of the hormones (Tables 1, 2 and 3) we have analysed in the same serum samples was associated with our BRCA1-mutation DNAme signature. Interestingly, when we analysed women with and without a family history [47] separately, both BC incidence and death was predicted by our BRCA1-DNAme signature only in the group without family history (Additional file 17), but not in the (obviously very small) group of women with any family history (Additional file 18).
Discussion
Here we have provided several novel lines of evidence indicating that DNAme profiles obtained in cells from women with a BRCA1 mutation have the potential to indicate future breast cancer development (and death) many years in advance of diagnosis. Our findings also show that genes encoding developmental transcription factors integral for stem cell differentiation and biology are hypermethylated in women predisposed to breast cancer.
Our data suggest that the BRCA1-associated DNAme signature is a risk predicting signature rather than an early detection signature, because: (1) the DNAme signature was derived from WBCs in women with a known BRCA1 status and was adjusted for cancer status (analysis included BRCA1 carriers without cancer at the time of sample draw); (2) the time from sample draw to diagnosis had no dramatic impact on the strength of association between DNAme and potential for breast cancer development; (3) the signature was validated in two independent cohorts; (4) we observed a very strong overlap of CpGs associated with BRCA1 mutation (BRCA1 study) and CpGs indicating future breast cancer risk (UKCTOCS); and finally (5) the signature was also associated with invasive non-breast cancers.
The observation that the top ranked hypermethylated BRCA1-mutation associated CpGs are highly enriched for PCGTs which we and others have previously shown to be an epigenetic hallmark of cancer tissue [12–18] and which are among the earliest, if not the earliest, molecular changes in human carcinogenesis [18] was an exciting finding because it fully supports recent data demonstrating that a BRCA1 defect leads to retargeting of the PRC2 and reduces cell differentiation.
Two key issues remain unclear. First, which factors lead to a BRCA1-mutation DNAme pattern in the absence of a BRCA1 mutation? It is likely that a combination of risk factors or factors which we have not captured (for example, early life events, transgenerational inheritance, and so on) contribute to epigenetic modifications which are in common to those associated with BRCA1 mutation. Second, is the BRCA1-mutation DNAme signature in WBCs functionally relevant or just simply an indicator of breast cancer risk? The fact that the signature is indicative of breast cancer mortality would support the view that subtle epigenetic mis-programming of immune cells may lead to general immune defects which in turn supports the development and proliferation of cancers. However, all these suggestions are highly speculative and need validation in further independent cohorts using well-defined subsets of blood cells or epithelial cells.
There are limitations to this study. First, we analysed whole blood DNA or serum DNA representing whole blood DNA and not a specific subset of peripheral blood cells. Second, although we found some good preliminary evidence that DNAme profiles in buccal cells are better at predicting future breast cancer risk (data not shown), we did not analyse buccal cells from BRCA1 mutation carriers, nor did we have access to independent prospective buccal cell data. Third, we used the 27 k array, instead of the 450 k array, to generate the BRCA1-mutation DNAme signature.
In summary, our data highlight DNAme analysis as a promising tool to predict future breast cancer development. Future epigenome-wide studies should focus on using epithelial cells like buccal - or epithelial cells from the uterine cervix which are hormone sensitive and more likely to capture an ‘epigenetic record’ of breast cancer risk factors. Such studies are more likely to provide the level of specificity and sensitivity which is required for a clinically useful risk prediction tool.
Conclusions
In summary, our DNAme signature derived from blood cells from BRCA1 carriers is able to predict breast cancer risk and death years in advance of diagnosis albeit with a modest AUC. Our data further support the notion that DNAme modification at stem-cell differentiation genes, even in unrelated tissues, is an early event associated with carcinogenesis.
Abbreviations
- AUC:
-
Area under the curve
- DNAme:
-
DNA methylation
- FDR:
-
False discovery rate
- GSEA:
-
Gene Set Enrichment Analysis
- hyperM:
-
Hypermethylated
- hypoM:
-
Hypomethylated
- ROC:
-
Receiver operating characteristics.
References
Domchek SM, Friebel TM, Singer CF, Evans DG, Lynch HT, Isaacs C, Garber JE, Neuhausen SL, Matloff E, Eeles R, Pichert G, Van t’veer L, Tung N, Weitzel JN, Couch FJ, Rubinstein WS, Ganz PA, Daly MB, Olopade OI, Tomlinson G, Schildkraut J, Blum JL, Rebbeck TR: Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA. 2010, 304: 967-975.
Fackenthal JD, Olopade OI: Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat Rev Cancer. 2007, 7: 937-948.
Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R, Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Le Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Peto J, , et al: Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007, 447: 1087-1093.
Wacholder S, Hartge P, Prentice R, Garcia-Closas M, Feigelson HS, Diver WR, Thun MJ, Cox DG, Hankinson SE, Kraft P, Rosner B, Berg CD, Brinton LA, Lissowska J, Sherman ME, Chlebowski R, Kooperberg C, Jackson RD, Buckman DW, Hui P, Pfeiffer R, Jacobs KB, Thomas GD, Hoover RN, Gail MH, Chanock SJ, Hunter DJ: Performance of common genetic variants in breast-cancer risk models. N Engl J Med. 2010, 362: 986-993.
Independent UK Panel on Breast Cancer Screening: The benefits and harms of breast cancer screening: an independent review. Lancet. 2012, 380: 1778-1786.
Jorgensen KJ, Gotzsche PC: Overdiagnosis in publicly organised mammography screening programmes: systematic review of incidence trends. BMJ. 2009, 339: b2587-
Zahl PH, Gotzsche PC, Maehlen J: Natural history of breast cancers detected in the Swedish mammography screening programme: a cohort study. Lancet Oncol. 2011, 12: 1118-1124.
Cui H, Cruz-Correa M, Giardiello FM, Hutcheon DF, Kafonek DR, Brandenburg S, Wu Y, He X, Powe NR, Feinberg AP: Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003, 299: 1753-1755.
Jones PA, Baylin SB: The epigenomics of cancer. Cell. 2007, 128: 683-692.
Teschendorff AE, Jones A, Fiegl H, Sargent A, Zhuang JJ, Kitchener HC, Widschwendter M: Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med. 2012, 4: 24-
Widschwendter M, Jones PA: DNA methylation and breast carcinogenesis. Oncogene. 2002, 21: 5462-5482.
Baylin SB, Jones PA: A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011, 11: 726-734.
Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, Issa JP: Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010, 20: 332-340.
O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, Clements EG, Cai Y, Van Neste L, Easwaran H, Casero RA, Sears CL, Baylin SB: Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011, 20: 606-619.
Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, Bergman Y, Simon I, Cedar H: Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007, 39: 232-236.
Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, Savage DA, Mueller-Holzner E, Marth C, Kocjan G, Gayther SA, Jones A, Beck S, Wagner W, Laird PW, Jacobs IJ, Widschwendter M: Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010, 20: 440-446.
Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW: Epigenetic stem cell signature in cancer. Nat Genet. 2007, 39: 157-158.
Zhuang J, Jones A, Lee SH, Ng E, Fiegl H, Zikan M, Cibula D, Sargent A, Salvesen HB, Jacobs IJ, Kitchener HC, Teschendorff AE, Widschwendter M: The dynamics and prognostic potential of DNA methylation changes at stem cell gene loci in women's cancer. PLoS Genet. 2012, 8: e1002517-
Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, Koseki H, Fuchikami T, Abe K, Murray HL, Zucker JP, Yuan B, Bell GW, Herbolsheimer E, Hannett NM, Sun K, Odom DT, Otte AP, Volkert TL, Bartel DP, Melton DA, Gifford DK, Jaenisch R, Young RA: Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006, 125: 301-313.
Wang L, Zeng X, Chen S, Ding L, Zhong J, Zhao JC, Wang L, Sarver A, Koller A, Zhi J, Ma Y, Yu J, Chen J, Huang H: BRCA1 is a negative modulator of the PRC2 complex. EMBO J. 2013, 32: 1584-1597.
Bosviel R, Garcia S, Lavediaux G, Michard E, Dravers M, Kwiatkowski F, Bignon YJ, Bernard-Gallon DJ: BRCA1 promoter methylation in peripheral blood DNA was identified in sporadic breast cancer and controls. Cancer Epidemiol. 2012, 36: e177-e182.
Brennan K, Garcia-Closas M, Orr N, Fletcher O, Jones M, Ashworth A, Swerdlow A, Thorne H, Investigators KC, Riboli E, Vineis P, Dorronsoro M, Clavel-Chapelon F, Panico S, Onland-Moret NC, Trichopoulos D, Kaaks R, Khaw KT, Brown R, Flanagan JM: Intragenic ATM methylation in peripheral blood DNA as a biomarker of breast cancer risk. Cancer Res. 2012, 72: 2304-2313.
Choi JY, James SR, Link PA, McCann SE, Hong CC, Davis W, Nesline MK, Ambrosone CB, Karpf AR: Association between global DNA hypomethylation in leukocytes and risk of breast cancer. Carcinogenesis. 2009, 30: 1889-1897.
Delgado-Cruzata L, Wu HC, Perrin M, Liao Y, Kappil MA, Ferris JS, Flom JD, Yazici H, Santella RM, Terry MB: Global DNA methylation levels in white blood cell DNA from sisters discordant for breast cancer from the New York site of the Breast Cancer Family Registry. Epigenetics. 2012, 7: 868-874.
Delgado-Cruzata L, Wu HC, Liao Y, Santella RM, Terry MB: Differences in DNA methylation by extent of breast cancer family history in unaffected women. Epigenetics. 2014, 9: 243-248.
Flanagan JM, Munoz-Alegre M, Henderson S, Tang T, Sun P, Johnson N, Fletcher O, Dos Santos SI, Peto J, Boshoff C, Narod S, Petronis A: Gene-body hypermethylation of ATM in peripheral blood DNA of bilateral breast cancer patients. Hum Mol Genet. 2009, 18: 1332-1342.
Ito Y, Koessler T, Ibrahim AE, Rai S, Vowler SL, Abu-Amero S, Silva AL, Maia AT, Huddleston JE, Uribe-Lewis S, Woodfine K, Jagodic M, Nativio R, Dunning A, Moore G, Klenova E, Bingham S, Pharoah PD, Brenton JD, Beck S, Sandhu MS, Murrell A: Somatically acquired hypomethylation of IGF2 in breast and colorectal cancer. Hum Mol Genet. 2008, 17: 2633-2643.
Iwamoto T, Yamamoto N, Taguchi T, Tamaki Y, Noguchi S: BRCA1 promoter methylation in peripheral blood cells is associated with increased risk of breast cancer with BRCA1 promoter methylation. Breast Cancer Res Treat. 2011, 129: 69-77.
Widschwendter M, Apostolidou S, Raum E, Rothenbacher D, Fiegl H, Menon U, Stegmaier C, Jacobs IJ, Brenner H: Epigenotyping in peripheral blood cell DNA and breast cancer risk: a proof of principle study. PLoS One. 2008, 3: e2656-
Woo HD, Kim J: Global DNA hypomethylation in peripheral blood leukocytes as a biomarker for cancer risk: a meta-analysis. PLoS One. 2012, 7: e34615-
Wu HC, John EM, Ferris JS, Keegan TH, Chung WK, Andrulis I, Delgado-Cruzata L, Kappil M, Gonzalez K, Santella RM, Terry MB: Global DNA methylation levels in girls with and without a family history of breast cancer. Epigenetics. 2011, 6: 29-33.
Xu X, Gammon MD, Hernandez-Vargas H, Herceg Z, Wetmur JG, Teitelbaum SL, Bradshaw PT, Neugut AI, Santella RM, Chen J: DNA methylation in peripheral blood measured by LUMA is associated with breast cancer in a population-based study. FASEB J. 2012, 26: 2657-2666.
Xu Z, Bolick SC, DeRoo LA, Weinberg CR, Sandler DP, Taylor JA: Epigenome-wide association study of breast cancer using prospectively collected sister study samples. J Natl Cancer Inst. 2013, 105: 694-700.
Wadsworth M, Kuh D, Richards M, Hardy R: Cohort Profile: The 1946 National Birth Cohort (MRC National Survey of Health and Development). Int J Epidemiol. 2006, 35: 49-54.
Kuh D, Pierce M, Adams J, Deanfield J, Ekelund U, Friberg P, Ghosh AK, Harwood N, Hughes A, Macfarlane PW, Mishra G, Pellerin D, Wong A, Stephen AM, Richards M, Hardy R, : Cohort profile: updating the cohort profile for the MRC National Survey of Health and Development: a new clinic-based data collection for ageing research. Int J Epidemiol. 2011, 40: e1-e9.
Rousseau K, Vinall LE, Butterworth SL, Hardy RJ, Holloway J, Wadsworth ME, Swallow DM: MUC7 haplotype analysis: results from a longitudinal birth cohort support protective effect of the MUC7*5 allele on respiratory function. Ann Hum Genet. 2006, 70: 417-427.
Hologic Gen-Probe – DNA extraction. [http://www.gen-probe.com], []
The National Survey of Health and development (NSHD) data archive. [http://www.nshd.mrc.ac.uk/data.aspx], []
Zou H, Hastie T: Regularization and variable selection via the elastic net. J R Stat Soc. 2005, B.67: 301-320.
Friedman JH, Hastie T, Tibshirani R: Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010, 33: 1-22.
Newson R: Confidence intervals for rank statistics: Percentile slopes, differences, and ratios. Stata J. 2006, 6: 497-520.
Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Gayther SA, Apostolidou S, Jones A, Lechner M, Beck S, Jacobs IJ, Widschwendter M: An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS One. 2009, 4: e8274-
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC: PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003, 34: 267-273.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP: Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005, 102: 15545-15550.
Huzarski T, Byrski T, Gronwald J, Gorski B, Domagala P, Cybulski C, Oszurek O, Szwiec M, Gugala K, Stawicka M, Morawiec Z, Mierzwa T, Janiszewska H, Kilar E, Marczyk E, Kozak-Klonowska B, Siołek M, Surdyka D, Wiśniowski R, Posmyk M, Sun P, Lubiński J, Narod SA: Ten-year survival in patients with BRCA1-negative and BRCA1-positive breast cancer. J Clin Oncol. 2013, 31: 3191-3196.
Narod SA, Foulkes WD: BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer. 2004, 4: 665-676.
Menon U, Gentry-Maharaj A, Ryan A, Sharma A, Burnell M, Hallett R, Lewis S, Lopez A, Godfrey K, Oram D, Herod J, Williamson K, Seif M, Scott I, Mould T, Woolas R, Murdoch J, Dobbs S, Amso N, Leeson S, Cruickshank D, McGuire A, Campbell S, Fallowfield L, Skates S, Parmar M, Jacobs I: Recruitment to multicentre trials–lessons from UKCTOCS: descriptive study. BMJ. 2008, 337: a2079-
Acknowledgments
This work was supported by the European Union's Seventh Framework Programme (FP7/2007-2013) under grant agreement number 305428 (Project EpiFemCare), the Eve Appeal and a grant from the UCLH/UCL Comprehensive Biomedical Research Centre project No 152 and by the Charles University in Prague, projects PRVOUK P27 and UNCE 204024 and undertaken at UCLH/UCL, which received a proportion of its funding from the Department of Health NIHR Biomedical Research Centres funding scheme. UKCTOCS was core funded by the Medical Research Council, Cancer Research UK and the Department of Health (G9901012 and G0801228) with additional support from the Eve Appeal, Special Trustees of UCLH and Special Trustees of Bart’s and the London. DK, RH and AW are supported by the Medical Research Council (MC_UU_12019/1). AET was supported by a Heller Research Fellowship, the Chinese Academy of Sciences and the Max-Planck Gesellschaft No funding bodies had any role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
IJ had a consultancy arrangement with Becton Dickinson in the field of tumour markers and ovarian cancer. Both IJ and UM have a financial interest through UCL Business and Abcodia Ltd. in the third party exploitation of clinical trials biobanks which have been developed through the research at UCL. The remaining authors declare that they have no competing interests.
Authors’ contributions
MW and SA had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: MW. Acquisition of data: EOF, MZ, AW, AGM, RH, DC, DK, UM, IJ. Analysis and interpretation of data: SA, AJ, AET, MW. Drafting of the manuscript: MW. Critical revision of the manuscript for important intellectual content: AET, SA, EOF, MZ, AW, AGM, AJ, RH, DC, DK, UM, IJ. Statistical analysis: SA. Obtained funding: MW, IJ, UM, DK. Administrative, technical or material support: SA. Study supervision: MW, AET, DK, IJ, UM. All authors read and approved the final manuscript.
Shahzia Anjum, Evangelia-Ourania Fourkala, Michal Zikan, Andrew Wong contributed equally to this work.
Electronic supplementary material
13073_2014_545_MOESM1_ESM.xls
Additional file 1:Description of the BRCA1 Study (White Blood Cell DNA samples from the Charles University in Prague).(XLS 52 KB)
13073_2014_545_MOESM2_ESM.xls
Additional file 2:Characteristics of the sample from the MRC National Survey of Health and Development study (NSHD).(XLS 29 KB)
13073_2014_545_MOESM3_ESM.xls
Additional file 3:Characteristics of the samples used from the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS). Statistical significance was tested using a two-sided, Fisher’s exact test. The missing values were not included in the analysis. (XLS 30 KB)
13073_2014_545_MOESM5_ESM.xls
Additional file 5:Top ranked 2514 CpGs in white blood cells associated with BRCA1 mutation. These differentially methylated CpGs were identified based on their β-value methylation profiles. This was done via a multivariate logistic regression that was adjusted for age, batch and the presence of cancer. To correct for multiple hypothesis testing, the false discovery rates (FDR) were estimated using the q-value analytical procedure available through the R qvalue Bioconductor package. (XLS 452 KB)
13073_2014_545_MOESM6_ESM.xls
Additional file 6:BRCA1 -mutation associated DNA methylation signature consisting 1829 CpGs. The signature was derived by combining, via stacked generalisation, 100 cross-validated classifiers using ElasticNet with an alpha = 0.1. (XLS 336 KB)
13073_2014_545_MOESM7_ESM.xls
Additional file 7:Gene Set Enrichment Analysis on the top 1074 hypermethylated CpGs of the BRCA1 DNA methylation signature.(XLS 170 KB)
13073_2014_545_MOESM8_ESM.xls
Additional file 8:Gene Set Enrichment Analysis on the top 755 hypomethylated CpGs of the BRCA1 DNA methylation signature.(XLS 135 KB)
13073_2014_545_MOESM9_ESM.xls
Additional file 9:Gene Set Enrichment Analysis on the 105 differentially methylated CpGs (FDR < =0.05) in white blood cells associated with BRCA1 mutation.(XLS 106 KB)
13073_2014_545_MOESM10_ESM.pdf
Additional file 10:ROC curve for the identified signature in invasive non-breast cancer samples of the NSHD dataset.(PDF 145 KB)
13073_2014_545_MOESM11_ESM.xls
Additional file 11:Average number of differentially methylated CpGs between tissue types. UKOPS WB = whole blood from postmenopausal healthy women; T1D WB = whole blood from pre and postmenopausal women with type-1 diabetes. (XLS 26 KB)
13073_2014_545_MOESM12_ESM.pdf
Additional file 12:ROC curve of the identified signature on the UKCTOCS dataset, separated around the diagnosis time of less than/greater than 2 years.(PDF 147 KB)
13073_2014_545_MOESM13_ESM.xls
Additional file 13:Top ranked 5482 CpGs associated with future breast cancer risk from serum DNA in the UKCTOCS set.(XLS 962 KB)
13073_2014_545_MOESM14_ESM.xls
Additional file 14:Gene Set Enrichment Analysis on the top 3395 hypermethylated CpGs in the UKCTOCS cohort using a multivariate linear regression framework.(XLS 99 KB)
13073_2014_545_MOESM15_ESM.xls
Additional file 15:Gene Set Enrichment Analysis on the top 2087 hypomethylated CpGs in the UKCTOCS cohort using a multivariate linear regression framework.(XLS 64 KB)
13073_2014_545_MOESM16_ESM.xls
Additional file 16:Characteristics of the samples used from the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS). The samples were categorised according to their individual risk scores. These risk scores are the product of the methylation profile with the regression coefficients of the signature. The statistical significance was assessed by a two-sided, Fisher’s exact test. The missing values were not included in the analysis. (XLS 29 KB)
13073_2014_545_MOESM17_ESM.pdf
Additional file 17:ROC curve of the identified signature on the UKCTOCS dataset samples, without a family history of breast cancer, separated into breast cancer incidence and breast cancer mortality.(PDF 137 KB)
13073_2014_545_MOESM18_ESM.pdf
Additional file 18:ROC curve of the identified signature on the UKCTOCS dataset samples, with a family history of breast cancer, separated into breast cancer incidence and breast cancer mortality.(PDF 138 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Anjum, S., Fourkala, EO., Zikan, M. et al. A BRCA1-mutation associated DNA methylation signature in blood cells predicts sporadic breast cancer incidence and survival. Genome Med 6, 47 (2014). https://doi.org/10.1186/gm567
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/gm567