
Abstract
The mycobiome, referring primarily to the fungal biota in an environment, is an important component of the human microbiome. Despite its importance, it has remained understudied. New culture-independent approaches to determine microbial diversity, such as next-generation sequencing methods, are greatly broadening our view of fungal importance. An integrative analysis of current studies shows that different body sites harbor specific fungal populations, and that diverse mycobiome patterns are associated with various diseases. By interfacing with other biomes, as well as with the host, the mycobiome probably contributes to the progression of fungus-associated diseases and plays an important role in health and disease.
Similar content being viewed by others

Introduction
Studies of fungi have been ongoing for more than 150 years, beginning with the first mycological paper published in 1852 [1, 2]. It is only in the past 10 years, however, that the microbiome as a novel microbiological concept, in which all microbes (bacteria, archaea, viruses and fungi) in an environment are taken into account, has drawn closer attention [3]. In particular, two large government-backed projects launched in the past five years - the Human Microbiome Project (HMP) in the United States and Metagenomics of the Human Intestinal Tract (MetaHit) in Europe - have provided progress in this new field of research [4–6]. These projects have defined what constitutes the normal bacterial microbiome of various human body sites, such as the oral cavity, skin, vagina, and gut. Exciting as the results have been, the characterization of fungal diversity in these body sites is still lacking. In fact, the word 'microbiome' has implied reference to only commensal and pathogenic bacteria [7]. This connotation changed in 2010, when the term 'mycobiome' (a combination of the words 'mycology' and 'microbiome') was first used to refer to the fungal microbiome [8]. Still, in a recent search of PubMed (6 July 2013), the term 'mycobiome' appeared in only 10 publications, and relevant studies - with or without using this specific word - numbered fewer than 40. Clearly, this field is still in its infancy.
There are several reasons to include the mycobiome in assessments of the biota of specific environments. First, the incidence of fungal infection has increased greatly in the past two decades, primarily in opportunistic infections of immunosuppressed populations, such as in those who have AIDS or have undergone organ transplantation or cancer chemotherapy [9]. Second, several diseases that were formerly considered to have no association with fungi, such as hepatitis B [10], cystic fibrosis [11, 12] and inflammatory bowel disease (IBD) [13, 14], are now found to be associated with particular mycobiomes. Finally, the interaction between different biomes [15], and between the host and the mycobiome [16], are critical in disease progression. In this review, we summarize the methodology used to study the mycobiome, its composition and role in health and disease, and the outlook for this field.
Tools used to explore the fungal biome
The most basic fungal culture technique, dating back to the 1920s, involved growing fungal mats in flasks of sterile liquid media. The experimental results were evaluated by looking at the growth structures and assessing their composition within the media [17]. Fungal culture methodology has improved over the decades, but even now, organisms that occur in low abundance and those that require microbe-microbe interactions to grow cannot be cultivated optimally [18].
Like unculturable bacteria in the microbiome, unculturable fungi comprise the largest part of the human mycobiome. In a study of the bacterial and fungal microbiome of patients with cystic fibrosis, more than 60% of the species or genera were not detected by culture [19]. A study focusing on the mycobiome of the oral cavity reported that 11 of the 85 fungal genera identified could not be cultured [6]. In the gastrointestinal mycobiome, culture-independent methods identified 37 different fungal groups compared to only 5 species found by culture-dependent analyses [10].
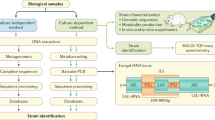
The limitations of culture-dependent methods for mycobiome studies have led to the introduction, over the past 20 years, of culture-independent approaches. Methods for classifying fungi that do not rely on microbial culture include restriction fragment length polymorphism (RFLP) analysis, oligonucleotide fingerprinting of rRNA genes (OFRG), denaturing gradient gel electrophoresis (DGGE), and in-situ hybridization (Table 1). These techniques are useful for comparing fungal diversities between different groups, but they lack the specificity necessary to identify the different fungal species in a large-scale study.
Direct sequencing of fungal genes has proven to be the most efficient method for classifying the mycobiome. Furthermore, with the development of next-generation sequencing (NGS) platforms, direct sequencing has become more cost-effective than was the case when only classical Sanger sequencing was available. Selecting target genomic regions to serve as proxy for the full-length genome is a popular approach for studies of fungal diversity, as it is in the determination of bacterial diversity in microbiome studies. The fungal ribosomal RNA gene cluster (rRNA) is the region most commonly selected as proxy, with sequencing efforts primarily targeting the 18S small subunit rDNA (SSU), 28S large subunit rDNA (LSU) or the internal transcribed spacer (ITS) [20]. Although 28S has gradually been eliminated as a target sequence because of its lack of discrimination power for many species, the debate over whether the 18S or the ITS sequences are most useful is still fierce. Compared with 18S, ITS is more diverse and enables greater genus-level phylogenetic placement. However, in our own experience, the higher conservation of the 18S facilitates the amplification of rDNA from various fungi, and also enables the detection of non-fungal eukaryotes, such as the parasitic protozoa Leishmania and Toxoplasma [21, 22].
Which region within the ITS is selected as a target also remains a matter of choice. Several studies amplify the ITS1 or ITS2 regions, whereas others favor amplifying the entire length of ITS1-5.8S-ITS2 (Table 2; Figure 1). This selection should be made carefully because some primers, such as the ITS1 and ITS1-F primers, are biased toward amplification of Basidiomycetes, whereas others, such as the ITS2 and ITS4 primers, are biased toward Ascomycetes [23]. The quantitative evaluation and rational design of improved ITS primers are still badly needed, and experience gained in the evaluation of 16S primer sets for bacterial microbiome studies could provide a good model to follow in this endeavor [24]. To judge the accuracy of different primer pairs in taxonomic classification, it would be worth sequencing the full-length ITS, trimming it to simulate the different amplicons that would be obtained by various primer pairs, and comparing them back with the full-length ITS. Similarly, and as has been done for bacterial species [25], it may be interesting to construct a 'mock' community (MC) with a known composition of fungal species, amplify the rDNA with various primer pairs, and compare the abundance of each species detected with the actual original proportions.

Schematic representation of the fungal ribosomal gene cluster, with binding locations of PCR primers. Within the fungal rDNA, 18S, 5.8S and 28S are separated by ITS1 and ITS2. Several primer sets that target specific regions of the fungal rDNA have been designed and used in previous studies.
Thus, at present, no common view exists as to the single best fungal rDNA region to select for deep-sequencing analysis. If the goal of the study is to measure the abundance of specific fungi, then using the same set of primers for each mycobiome included in the study is important; but if the intent is to characterize fungal diversity, then a combination of different regions would probably cover more fungal species and thus produce more comprehensive results. Our experience leads us to believe that the efficiency of amplification and the size of the amplicon generated also dictate which portion is the most productive, with shorter amplicons being more consistently generated.
Several NGS platforms that vary in their characteristics are available for mycobiome sequencing. Despite its relatively high cost, pyrosequencing on the Roche/454 GS-FLX is the method most commonly used in mycobiome studies because it achieves the longest sequence reads (500 bp). Other, newer NGS platforms, including Illumina's HiSeq and MiSeq and Life Technologies' Ion Torrent, have also demonstrated their potential recently. Based on results obtained in bacterial microbiome studies, the HiSeq platform provides the highest data output at the lowest cost (50 times less expensive than 454 pyrosequencing), whereas MiSeq is more appropriate when longer read length and quick turn-around time are the priority [26, 27]. The Ion Torrent (Ion PGM™ Sequencer and Ion Proton™ Sequencer), with its new protocols leading to 400 bp sequence reads, has also become competitive, providing a low-cost, scalable and high-throughput solution [28].
Analysis of sequence data also presents a number of issues relating to methodology. First, the pipeline must be selected. Two of the most commonly used pipelines in the analysis of microbiome sequencing data are QIIME (http://qiime.org) and mothur (http://www.mothur.org). Built upon a series of bioinformatic tools, both pipelines allow: the trimming, screening, and alignment of sequences; the assignment of operational taxonomic units (OTUs); phylogenetic analyses; and determination of fungal diversity within and across groups (referred to as α and β diversities) [29, 30]. In addition, pipelines that are specific for mycobiome studies, such as CloVR-ITS and BROCC, have been developed recently [21, 31]. A database against which the amplified sequences can be compared must also be selected, but a database as rich as that for bacterial 16S rDNA is still lacking for fungi, as is the capability to categorize fungal rRNA sequences at the level of subspecies. Research groups currently use the fungal rDNA databases of UNITE (http://unite.ut.ee/), which includes 6,816 ITS sequences from 1,977 species within 418 genera of fungi [32, 33], and SILVA (http://www.arb-silva.de/), which includes 6,571 18S and 1,753 28S sequences from fungi in its release 111. Useful as these databases are, they do have some limitations that affect taxonomic assignments. For example, many synonyms and misclassifications are found in the fungal nomenclature; and sexual and asexual forms of a fungal species can be classified as different taxa [21]. Recently, Findley and colleagues optimized the current ITS database by fixing many of the inconsistencies described in taxonomic entries [22]. They also implemented a species-level resolution to skin-associated Malassezia within the software pplacer [34], which provides phylogenetic placement of the sequences. Despite these advancements, we still need to improve the reliability of fungal analyses by pursuing a more systematic evaluation of current databases to determine whether the mycobiomes analyzed to date are indeed well characterized.
Composition and role of the mycobiome in health and disease
The ultimate aim of human mycobiome studies is to uncover the role that fungal populations play in affecting health. Unfortunately, owing to the limitation of culture-dependent methods in mycology, most early studies were restricted to the analysis of a few species: Candida albicans, Candida glabrata, Cryptococcus neoformans, Aspergillus fumigatus and the dimorphic fungi (Coccidioides, Histoplasma and Blastomyces) [9]. The development of culture-independent methods has expanded our knowledge of the mycobiomes found in different body sites and their association with disease (Table 3). For example, specific organs that were previously thought to be sterile, such as the lung [19], are now known to harbor a variety of fungi. In other body sites previously known to be colonized with fungus, the fungal variety detected by classical methods is much less than that discovered by newly developed methods. Figure 2 shows an integrated analysis of different mycobiomes reported in the literature. Fungal distributions show significant differences among distant body sites, whereas similar patterns of distribution were found in mycobiomes from nearby sites. For example, Cladosporium species, Aspergillus species and Penicillium species all dominate other fungal genera in both in both oral and nasal cavities [8, 35].

Distribution of fungal genera in different body sites. Fungal distributions differ significantly among distant body sites, whereas similar patterns are found in nearby sites. For example, Cladosporium spp., Aspergillus spp., and Penecillium spp. all dominate other fungal genera in both oral and nasal cavities.
Although the association of many mycobiomes with various diseases has been reported, more associations will undoubtedly be characterized in the future. For example, immune-suppressed (for example, HIV-positive) individuals are more likely to contract opportunistic fungal (and protozoa) infections than are healthy persons; thus, mycobiome studies on immune-suppressed individuals are needed to uncover more relevant fungal species, and possibly to identify the mechanistic link between fungal pathogenesis and immune suppression. In lung disease, several studies have attempted to characterize the mycobiome in cystic fibrosis (CF) [11, 12, 19]. These studies suggest that the fungal diversity is lower in CF patients than in healthy people. The lung mycobiome has not yet been determined in those with chronic obstructive pulmonary disease (COPD) or asthma, although it may have an effect on the progression of these conditions [18].
The mycobiomes present in different body sites potentially interact with each other. At present, the gastrointestinal-respiratory interaction is understood most clearly: a disturbance of the gut mycobiome by C. albicans impacts allergic pulmonary disease induced by A. fumigatus in the lung mycobiome [36]. Translocation into the bloodstream and subsequent circulation of molecules from fungi, such as RNA, DNA or peptidoglycans, may initiate systemic immune responses and lead to disease remote from the initial site of fungal infection [37, 38].
Interactions between the mycobiome and the bacterial microbiome may also play a role in health and disease (Table 4). In some cases, the occurrence of bacteria correlates positively with the presence of fungi; for example, Mycobacterium superinfection sometimes occurs along with aspergillosis [39]. In other cases, bacteria compete with fungi; the growth of Candida species and possibly other fungi is suppressed when Pseudomonas aeruginosa dominates in CF [40]. Various hypotheses have been proposed to explain the dual nature of this interaction. In the case of Mycobacterium and aspergillosis, the commensalism of bacteria and fungi may synergistically strengthen their resistance to environmental pressure, such as antimicrobial agents [41]. In the case of Candida and Pseudomonas, the inhibition of fungal growth by the bacteria may better meet the nutritional requirements of the bacteria, allowing them to secure more attachment sites on the host cell [12]. Regardless of whether the interaction between a mycobiome and a bacterial microbiome is synergistic or competitive, it has the potential to alter both the intrinsic host immune response to pathogens and the susceptibility of the mycobiome and the bacterial microbiome to medical therapy.
Interactions between the host and the mycobiome are likely to be carefully balanced, leading to clearance, asymptomatic infection, latency, or disease [9]. Several factors in the host will have an effect on mycobiome composition and variations, including host genotype, physiology, immune system, and lifestyle (Figure 3) [42, 43]. As some of these factors might change over time, the mycobiome will fluctuate accordingly. The skin mycobiome, for example, is thought to change drastically early in life. Using the genotyping of Malassezia species as an indicator, one study demonstrated that it takes 30 days for the skin microbiota to change from its initial state to an adult type [44].

Host and environmental factors that contribute to diversity observed in the human mycobiome.
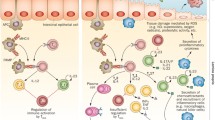
The host immune response influences the composition of the mycobiome (Figure 4). Several pattern-recognition receptors (PRRs) on phagocytes, including TLR-2, TLR-4, dectin-1, dectin-2, and galectin-3, specifically recognize pathogen-associated molecular patterns (PAMPs) of fungi, such as α-mannans, β-mannans, and β-glucans [45–48]. Following this pattern recognition, macrophages and dendritic cells mature and activate T cells through an antigen-presenting process. Depending on which cytokines are stimulated, activated T cells differentiate into either Th-1, which promotes the phagocytosis of fungi, or Th-2, which activates B cells to release fungi-specific antibodies [16]. Two studies also reported the differentiation of Th-17 following C. albicans infection, indicating a potential role for Th-17 in host defense against fungi [47, 49]. Despite our depth of knowledge of fungal immunology, it remains to be determined whether these interactions are ubiquitous or tissue specific, and whether some of the interactions mentioned above are due entirely to a pathogenic process or are mainly involved in retaining the homeostasis required for host immune development [7].

Interaction between the mycobiome and the immune system. Several pattern recognition receptors (PRRs) on phagocytes specifically recognize the pathogen-associated molecular patterns (PAMPs) of fungi. Following this pattern recognition, macrophage and dendritic cells mature and activate T cells through an antigen-presenting process. Activated T cells differentiate into either Th-1, which promotes the phagocytosis of fungi, or Th-2, which activates B cells to release fungi-specific antibodies, depending by which cytokines they are stimulated. IFN, interferon; IL, interleukin; TNF, tumor necrosis factor. (Figure design inspired by the information reported in [16] and [53].)
Role of the mycobiome: beneficial, commensal or pathogenic?
The pathogenesis of many single fungal species has been well studied, whereas the correlation between the diversity of the whole mycobiome and disease progression is less clearly defined. It is often assumed that fungal diversity should be greater in more severe cases of a disease. For some conditions, such as in inflammatory bowel disease (IBD) [14], atopic dermatitis [50], and hepatitis B cirrhosis or chronic hepatitis B, the diversity of fungi present is proportional to the progression of disease [10]. But in other disease environments, such as the respiratory mycobiome in cystic fibrosis [12, 19] and the nasal mycobiome in allergic patients [35], the fungal diversity is inversely proportional to disease progression [35]. To further complicate the issue, the diversity of fungi identified in some diseases has no correlation with disease status, and the diversity of fungi in samples cluster more according to individuals rather than to health status [51]. Drawing a generalized conclusion about the correlation between diseases and fungal diversity is, thus, difficult. Fungal diversity is more likely to correlate positively with disease status in those with infectious diseases because fungi can exacerbate the infection; in chronic diseases, where fungal infection plays a secondary role in disease pathogenesis, however, an inverse correlation between fungal diversity and disease progression is more likely to occur because the microenvironment (such as that resulting from mucus dysfunction in CF) becomes less suitable for fungal growth.
Like the mycobiome as a whole, individual members of the mycobiome may also play a beneficial or commensal role in the host. Beneficial fungi have been found to be preventive and therapeutic agents, an example being the use of Saccharomyces boulardii for the treatment of diarrhoeal diseases [52]. Commensal fungi, such as Malassezia spp. and C. albicans, usually co-evolve with the adaptive immune system, although in certain cases they may switch from a 'friendly' relationship with the host to a pathological one [53].
Alterations in the mycobiome are frequently reported to be associated with disease progression, but it remains to be elucidated whether this variation is cause or effect. One concern is whether such an alteration in the mycobiome is primary or secondary to an imbalanced bacterial microbiome, as Ott et al. [14] proposed for the increase in mycobiome diversity of IBD. A causal relationship could be established if an antimicrobial treatment targeting certain fungal groups were to lead to either exacerbation of disease or cure; on the other hand, if the treatment of the disease were to lead concurrently to modulation of the mycobiome, then it would seem more likely that the mycobiome is being affected by the disease status [54]. Specific mycobiome patterns may be useful as diagnostic or prognostic markers of diseases.
Outlook
Targeting vital fungal species that are associated with disease progression may impact disease severity. Evidence exists to indicate that altering the mycobiome with antifungal drugs can improve certain conditions, such as gastrointestinal graft-versus-host disease (GI-GVHD) [49]. Furthermore, specifically controlling the growth of less desirable fungal species, or controlling biofilm-associated infections, may also be useful in manipulating the complex microflora in diseased body sites [19]. Another viable alternative to direct antimicrobial treatment is to introduce prebiotic and probiotic therapy to restore bacterial commensals. A clinical trial of VSL#3 in pouchitis patients showed that the bacterial diversity in the gut was increased while the fungal diversity was reduced with the use of a probiotic therapy. This effect could result from the restoration of the integrity of a 'protective' intestinal mucosa-related microbiota [13].
With a better understanding of the mechanisms of recognition and modulation in the immune response to fungi, it might become practical to administer immune therapy to treat mycobiome-associated diseases. Effective monoclonal antibodies, which promote opsonization in phagocytosis, activate the complement system or act directly on fungal cells [55], have already been developed for several fungal species, such as Cryptococcus neoformans [56], C. albicans [57], Histoplasma capsulatum [58] and A. fumigatus [59]. Fungal-targeted vaccination is another alternative, in both prophylaxis and therapeutics. Fungi that induce a long-term immunity are considered better candidates for vaccination [55], and a few vaccines have already been put forward for clinical trials, such as the killed spherule vaccine against coccidioidomycosis [53].
The limitation of today's immune therapies is that most target only a single fungal species, ignoring the overall mycobiome composition. To date, our knowledge of the mycobiome suggests that interactions among fungi within an environment and between mycobiomes found in different body sites may play an important role in pathogenesis, and that the development of a broad-spectrum monoclonal antibody or a universal vaccine targeting multiple pathogenic fungi would be more promising than therapies targeting a single fungal species. Given that many fungi share a series of PAMPs, it should be feasible to develop a universal immune therapeutic tool that targets such a widely used fungal signature.
Along with the heterogeneity of the mycobiome, genetic polymorphism across human populations also raises different risks for mycobiome-associated diseases (summarized by Romani [53]). For instance, the dectin-1 single nucleotide polymorphism (SNP) Y238X mutation could increase the likelihood of Candida colonization and indirectly associate with GVHD [49]. With these recent findings, the significance of sequencing the genomes of hosts with or without disease has been highlighted, and personalized treatment is now often touted as the way forward. Potentially, by classifying people by known genetic markers, we will be able to stratify patients with a high susceptibility to fungal infection, and those who would benefit from antifungal agents, thereby optimizing the therapeutic effect and reducing the risk of antifungal resistance.
Conclusions
Defining the mycobiome has broadened the scope of human microbiome studies. Several mycobiomes in different body sites have been characterized, and diverse mycobiome patterns associated with various diseases. After summarizing the methods used in mycobiome studies and analyzing the role of the mycobiome in health and disease, we propose that combining fungal characterization with a generalized assessment of the microbiome will expand our understanding of the microbial environment in disease progression. The mycobiome contributes to disease through the interaction between different biomes as well as through the interaction between the mycobiome and the host. Future studies characterizing the mycobiome will be instrumental in understanding disease pathogenesis and in developing novel therapies.
Abbreviations
- CF:
-
cystic fibrosis
- DGGE:
-
denaturing gradient gel electrophoresis
- GVHD:
-
graft-versus-host disease
- ITS:
-
internal transcribed spacer
- LSU:
-
28S large subunit rDNA
- NGS:
-
next-generation sequencing
- PAMP:
-
pathogen associated molecular pattern
- RFLP:
-
restriction fragment length polymorphism
- rRNA:
-
ribosomal RNA gene cluster
- SSU:
-
18S small subunit rDNA.
References
Hassall A: On the development of Torulæ in the urine, and on the relation of these fungi to albuminous and saccharine urine. Med Chir Trans 1853, 36: 23-78.
Chiene J, Ewart JC: Do bacteria or their germs exist in the organs of healthy living animals? J Anat Physiol 1878, 12: 448-453.
Relman DA: New technologies, human-microbe interactions, and the search for previously unrecognized pathogens. J Infect Dis 2002, 186: S254-S258. 10.1086/344935
Mitreva M: Structure, function and diversity of the healthy human microbiome. Nature 2012, 486: 207-214. 10.1038/nature11234
Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C: The NIH human microbiome project. Genome Res 2009, 19: 2317-2323.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T: A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464: 59-65. 10.1038/nature08821
Moyes DL, Naglik JR: The mycobiome: influencing IBD severity. Cell Host Microbe 2012, 11: 551-552. 10.1016/j.chom.2012.05.009
Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, Naqvi A, Gillevet PM: Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog 2010, 6: e1000713. 10.1371/journal.ppat.1000713
Perfect JR, Casadevall A: Fungal molecular pathogenesis: what can it do and why do we need it? In Molecular Principles of Fungal Pathogenesis. Edited by: Heitman J, Filler SG, Edwards JE, Mitchell AP. Washington DC: ASM Press; 2006:3-12.
Chen Y, Chen Z, Guo R, Chen N, Lu H, Huang S, Wang J, Li L: Correlation between gastrointestinal fungi and varying degrees of chronic hepatitis B virus infection. Diagn Micr Infec Dis 2011, 70: 492-498. 10.1016/j.diagmicrobio.2010.04.005
Nelson A, De Soyza A, Bourke S, Perry J, Cummings S: Assessment of sample handling practices on microbial activity in sputum samples from patients with cystic fibrosis. Lett Appl Microbiol 2010, 51: 272-277. 10.1111/j.1472-765X.2010.02891.x
Harrison MJ, Twomey KB, McCarthy Y, O'Connell OJ, Alston M, Febrer M, Murphy DM, Ryan RP, Plant BJ: Fungal microbiota in the adult cystic fibrosis (CF) airway: characterization by second-generation sequencing and correlation with standard culture-based methods and clinical phenotype. Ir J Med Sci 2012, 181: S369-S437.
Kuehbacher T, Ott SJ, Helwig U, Mimura T, Rizzello F, Kleessen B, Gionchetti P, Blaut M, Campieri M, Fölsch UR, Kamm MA, Schreiber S: Bacterial and fungal microbiota in relation to probiotic therapy (VSL# 3) in pouchitis. Gut 2006, 55: 833-841. 10.1136/gut.2005.078303
Ott SJ, Kühbacher T, Musfeldt M, Rosenstiel P, Hellmig S, Rehman A, Drews O, Weichert W, Timmis KN, Schreiber S: Fungi and inflammatory bowel diseases: alterations of composition and diversity. Scand J Gastroenterol 2008, 43: 831-841. 10.1080/00365520801935434
Deborah AH, Roberto K: Fungal-bacterial interactions. In Molecular Principles of Fungal Pathogenesis. Edited by: Heitman J, Filler SG, Edwards JE, Mitchell AP. Washington DC: ASM Press; 2006:261-269.
Cutler JE, Deepe GS Jr, Klein BS: Advances in combating fungal diseases: vaccines on the threshold. Nat Rev Microbiol 2007, 5: 13-28. 10.1038/nrmicro1537
Marloth RH: An apparatus for the study of matforming fungi in culture solutions. Science 1929, 69: 524-525. 10.1126/science.69.1794.524
Beck JM, Young VB, Huffnagle GB: The microbiome of the lung. Transl Res 2012, 160: 258-266. 10.1016/j.trsl.2012.02.005
Delhaes L, Monchy S, Fréalle E, Hubans C, Salleron J, Leroy S, Prevotat A, Wallet F, Wallaert B, Dei-Cas E: The airway microbiota in cystic fibrosis: a complex fungal and bacterial community-implications for therapeutic management. PLoS One 2012, 7: e36313. 10.1371/journal.pone.0036313
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Bolchacova E, Voigt K, Crous PW: Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc Natl Acad Sci USA 2012, 109: 6241-6246. 10.1073/pnas.1117018109
Dollive S, Peterfreund GL, Sherrill-Mix S, Bittinger K, Sinha R, Hoffmann C, Nabel C, Hill DA, Artis D, Bachman MA: A tool kit for quantifying eukaryotic rRNA gene sequences from human microbiome samples. Genome Biol 2012, 13: R60. 10.1186/gb-2012-13-7-r60
Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, Schoenfeld D, Nomicos E, Park M, NIH Intramural Sequencing Center Comparative Sequencing Program, Kong HH, Segre JA: Topographic diversity of fungal and bacterial communities in human skin. Nature 2013, 498: 367-370. 10.1038/nature12171
Bellemain E, Carlsen T, Brochmann C, Coissac E, Taberlet P, Kauserud H: ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol 2010, 10: 189-197. 10.1186/1471-2180-10-189
Nossa CW, Oberdorf WE, Yang L, Aas JA, Paster BJ, DeSantis TZ, Brodie EL, Malamud D, Poles MA, Pei Z: Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World J Gastroenterol 2010, 16: 4135-4144. 10.3748/wjg.v16.i33.4135
Jumpstart Consortium Human Microbiome Project Data Generation Working Group: Evaluation of 16S rDNA-based community profiling for human microbiome research. PLoS One 2012, 7: e39315. 10.1371/journal.pone.0039315
Bartram AK, Lynch MD, Stearns JC, Moreno-Hagelsieb G, Neufeld JD: Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumina reads. Appl Environ Microbiol 2011, 77: 3846-3852. 10.1128/AEM.02772-10
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R: Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 2012, 6: 1621-1624. 10.1038/ismej.2012.8
Whiteley AS, Jenkins S, Waite I, Kresoje N, Payne H, Mullan B, Allcock R, O'Donnell A: Microbial 16S rRNA ion tag and community metagenome sequencing using the Ion Torrent (PGM) Platform. J Microbiol Methods 2012, 91: 80-88. 10.1016/j.mimet.2012.07.008
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R: QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010, 7: 335-336. 10.1038/nmeth.f.303
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF: Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009, 75: 7537-7541. 10.1128/AEM.01541-09
White JR, Maddox C, White O, Angiuoli SV, Fricke WF: CloVR-ITS: automated internal transcribed spacer amplicon sequence analysis pipeline for the characterization of fungal microbiota. Microbiome 2013, 1: 1-11. 10.1186/2049-2618-1-1
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO: The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 2013, 41: D590-D596. 10.1093/nar/gks1219
Abarenkov K, Henrik Nilsson R, Larsson KH, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T: The UNITE database for molecular identification of fungi-recent updates and future perspectives. New Phytol 2010, 186: 281-285. 10.1111/j.1469-8137.2009.03160.x
Matsen F, Kodner R, Armbrust EV: pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 2010, 11: 538. 10.1186/1471-2105-11-538
Sellart-Altisent M, Torres-Rodríguez JM, Gómez de Ana S, Alvarado-Ramírez E: Nasal fungal microbiota in allergic and healthy subjects. Rev Iberoam Micol 2007, 24: 125-130. 10.1016/S1130-1406(07)70027-X
Noverr MC, Falkowski NR, McDonald RA, McKenzie AN, Huffnagle GB: Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: role of host genetics, antigen, and interleukin-13. Infect Immun 2005, 73: 30-38. 10.1128/IAI.73.1.30-38.2005
Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC: Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 2006, 12: 1365-1371.
Morris A, Hillenbrand M, Finkelman M, George MP, Singh V, Kessinger C, Lucht L, Busch M, McMahon D, Weinman R: Serum (1→3)-β-D-Glucan levels in HIV-infected individuals are associated with immunosuppression, inflammation, and cardiopulmonary function. J Acquir Immune Defic Syndr 2012, 61: 462-468. 10.1097/QAI.0b013e318271799b
Darling WM: Co-cultivation of mycobacteria and fungus. Lancet 1976, 2: 740.
Kerr J: Inhibition of fungal growth by Pseudomonas aeruginosa and Pseudomonas cepacia isolated from patients with cystic fibrosis. J Infect 1994, 28: 305-310.
Adam B, Baillie GS, Douglas LJ: Mixed species biofilms of Candida albicans and Staphylococcus epidermidis . J Med Microbiol 2002, 51: 344-349.
Grice EA, Segre JA: The skin microbiome. Nat Rev Microbiol 2011, 9: 244-253. 10.1038/nrmicro2537
Jacobs PH: An overview: antifungal treatment. In Fungal Disease: Biology, Immunology, and Diagnosis. Edited by: Jacobs PH, Nall L. New York: CRC Press; 1997:1-5.
Nagata R, Nagano H, Ogishima D, Nakamura Y, Hiruma M, Sugita T: Transmission of the major skin microbiota, Malassezia , from mother to neonate. Pediatr Int 2012, 54: 350-355. 10.1111/j.1442-200X.2012.03563.x
Jawhara S, Mogensen E, Maggiotto F, Fradin C, Sarazin A, Dubuquoy L, Maes E, Guérardel Y, Janbon G, Poulain D: A murine model of dextran sulfate sodium-induced colitis reveals Candida glabrata virulence and contribution of β-mannosyltransferases. J Biol Chem 2012, 287: 11313-11324. 10.1074/jbc.M111.329300
Netea MG, Gow NA, Munro CA, Bates S, Collins C, Ferwerda G, Hobson RP, Bertram G, Hughes HB, Jansen T, Jacobs L, Buurman ET, Gijzen K, Williams DL, Torensma R, McKinnon A, MacCallum DM, Odds FC, Van der Meer JW, Brown AJ, Kullberg BJ: Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J Clin Invest 2006, 116: 1642-1650. 10.1172/JCI27114
Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, Fujikado N, Kusaka T, Kubo S, Chung S-h, Komatsu R, Miura N, Adachi Y, Ohno N, Shibuya K, Yamamoto N, Kawakami K, Yamasaki S, Saito T, Akira S, Iwakura Y: Dectin-2 recognition of α-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans . Immunity 2010, 32: 681-691. 10.1016/j.immuni.2010.05.001
Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, Brown J, Becker CA, Fleshner PR, Dubinsky M: Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 2012, 336: 1314-1317. 10.1126/science.1221789
van der Velden WJ, Netea MG, de Haan AF, Huls GA, Donnelly JP, Blijlevens N: The role of the mycobiome in human acute graft-versus-host disease. Biol Blood Marrow Transplant 2013, 19: 329-332. 10.1016/j.bbmt.2012.11.008
Zhang E, Tanaka T, Tajima M, Tsuboi R, Nishikawa A, Sugita T: Characterization of the skin fungal microbiota in patients with atopic dermatitis and in healthy subjects. Microbiol Immunol 2011, 55: 625-632. 10.1111/j.1348-0421.2011.00364.x
Paulino LC, Tseng CH, Blaser MJ: Analysis of Malassezia microbiota in healthy superficial human skin and in psoriatic lesions by multiplex real-time PCR. FEMS Yeast Res 2008, 8: 460-471. 10.1111/j.1567-1364.2008.00359.x
McFarland L, Bernasconi P: Saccharomyces boulardii : a review of an innovative biotherapeutic agent. Microb Ecol Health Dis 1993, 6: 157-171. 10.3109/08910609309141323
Romani L: Immunity to fungal infections. Nat Rev Immunol 2011, 11: 275-288. 10.1038/nri2939
Cho I, Blaser MJ: The human microbiome: at the interface of health and disease. Nat Rev Genet 2012, 13: 260-270.
Ostrosky-Zeichner L, Casadevall A, Galgiani JN, Odds FC, Rex JH: An insight into the antifungal pipeline: selected new molecules and beyond. Nat Rev Drug Discov 2010, 9: 719-727. 10.1038/nrd3074
Fleuridor R, Zhong Z, Pirofski L: A human IgM monoclonal antibody prolongs survival of mice with lethal cryptococcosis. J Infect Dis 1998, 178: 1213-1216. 10.1086/515688
Moragues MD, Omaetxebarria MJ, Elguezabal N, Sevilla MJ, Conti S, Polonelli L, Pontón J: A monoclonal antibody directed against a Candida albicans cell wall mannoprotein exerts three anti- C. albicans activities. Infect Immun 2003, 71: 5273-5279. 10.1128/IAI.71.9.5273-5279.2003
Nosanchuk JD, Steenbergen JN, Shi L, Deepe GS, Casadevall A: Antibodies to a cell surface histone-like protein protect against Histoplasma capsulatum . J Clin Invest 2003, 112: 1164-1175.
Chaturvedi AK, Kavishwar A, Keshava GS, Shukla P: Monoclonal immunoglobulin G1 directed against Aspergillus fumigatus cell wall glycoprotein protects against experimental murine aspergillosis. Clin Diagn Lab Immunol 2005, 12: 1063-1068.
Korabečná M, Liška V, Fajfrlik K: PrimersITS1, ITS2 andITS4 detect the intraspecies variability in the internal transcribed spacers and 5.8 S rRNA gene region in clinical isolates of fungi. Folia Microbiol 2003, 48: 233-238. 10.1007/BF02930961
Esteve-Zarzoso B, Belloch C, Uruburu F, Querol A: Identification of yeasts by RFLP analysis of the 5.8 S rRNA gene and the two ribosomal internal transcribed spacers. Int J Syst Bacteriol 1999, 49: 329-337. 10.1099/00207713-49-1-329
Scupham AJ, Presley LL, Wei B, Bent E, Griffith N, McPherson M, Zhu F, Oluwadara O, Rao N, Braun J: Abundant and diverse fungal microbiota in the murine intestine. Appl Environ Microbiol 2006, 72: 793-801. 10.1128/AEM.72.1.793-801.2006
Drell T, Lillsaar T, Tummeleht L, Simm J, Aaspõllu A, Väin E, Saarma I, Salumets A, Donders GG, Metsis M: Characterization of the vaginal micro- and mycobiome in asymptomatic reproductive-age Estonian women. PLoS One 2013, 8: e54379. 10.1371/journal.pone.0054379
Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M: Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012, 2012: 1-11.
Amend AS, Seifert KA, Samson R, Bruns TD: Indoor fungal composition is geographically patterned and more diverse in temperate zones than in the tropics. Proc Natl Acad Sci USA 2010, 107: 13748-13753. 10.1073/pnas.1000454107
Park HK, Ha M-H, Park S-G, Kim MN, Kim BJ, Kim W: Characterization of the fungal microbiota (mycobiome) in healthy and dandruff-afflicted human scalps. PLoS One 2012, 7: e32847. 10.1371/journal.pone.0032847
Bosco VL, Birman EG, Cury AE, Paula CR: Yeasts from the oral cavity of children with AIDS: exoenzyme production and antifungal resistance. Pediatr Dent 2003, 17: 217-222.
Paulino LC, Tseng C-H, Strober BE, Blaser MJ: Molecular analysis of fungal microbiota in samples from healthy human skin and psoriatic lesions. J Clin Microbiol 2006, 44: 2933-2941. 10.1128/JCM.00785-06
Purim K, Bordignon G, Queiroz-Telles F: Fungal infection of the feet in soccer players and non-athlete individuals. Rev Iberoam Micol 2005, 22: 34-38. 10.1016/S1130-1406(05)70004-8
Santos PM, Melo CM, Martins SA, Chaves Ade A, Sá DSP, Santos RC: Study of ocular fungal microbiota in patients with Hansen's disease and in individuals who deal with them. Arq Bras Oftalmol 2006, 69: 915-918. 10.1590/S0004-27492006000600022
Thomas JG, Ramage G, Lopez-Ribot JL: Biofilms and implant infections. In Microbial Biofilms. Edited by: Ghannoum MA, O'Toole GA. Washington DC: ASM Press; 2004:269-293.
Grimaudo N, Nesbitt W: Coaggregation of Candida albicans with oral Fusobacterium species. Oral Microbiol Immunol 1997, 12: 168-173. 10.1111/j.1399-302X.1997.tb00374.x
Holmes AR, McNAB R, Jenkinson HF: Candida albicans binding to the oral bacterium Streptococcus gordonii involves multiple adhesin-receptor interactions. Infect Immun 1996, 64: 4680-4685.
Branting C, Sund M-L, Linder L: The influence of Streptococcus mutans on adhesion of Candida albicans to acrylic surfaces in vitro . Arch Oral Biol 1989, 34: 347-353. 10.1016/0003-9969(89)90108-8
Wagner RD, Pierson C, Warner T, Dohnalek M, Hilty M, Balish E: Probiotic effects of feeding heat-killed Lactobacillus acidophilus and Lactobacillus casei to Candida albicans -colonized immunodeficient mice. J Food Prot 2000, 63: 638-644.
Boris S, Barbés C: Role played by Lactobacilli in controlling the population of vaginal pathogens. Microbes Infect 2000, 2: 543-546. 10.1016/S1286-4579(00)00313-0
Acknowledgements
This work was supported in part by the National Heart, Lung, and Blood Institute of the National Institutes of Health (NHLBI/NIH) grant number U01HL098962-04 (EG, AM), and R21HL114231-02 (AM). LC is a medical student at Tsinghua University and a visiting scholar at the University of Pittsburgh School of Medicine.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing Interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Cui, L., Morris, A. & Ghedin, E. The human mycobiome in health and disease. Genome Med 5, 63 (2013). https://doi.org/10.1186/gm467
Published:
DOI: https://doi.org/10.1186/gm467