
Abstract
Perhaps as much as two-thirds of the mammalian genome is composed of mobile genetic elements ('jumping genes'), a fraction of which is still active or can be reactivated. By their sheer number and mobility, retrotransposons, DNA transposons and endogenous retroviruses have shaped our genotype and phenotype both on an evolutionary scale and on an individual level. Notably, at least the non-long terminal repeat retrotransposons are still able to cause disease by insertional mutagenesis, recombination, providing enzymatic activities for other mobile DNA, and perhaps by transcriptional overactivation and epigenetic effects. Currently, there are nearly 100 examples of known retroelement insertions that cause disease. In this review, we highlight those genome-scale technologies that have expanded our knowledge of the diseases that these mobile elements can elicit, and we discuss the potential impact of these findings for medicine. It is now likely that at least some types of cancer and neurological disorders arise as a result of retrotransposon mutagenesis.
Similar content being viewed by others
The emerging importance of mobile DNA elements in disease
It has been generally held that about half of the human genome is derived from mobile genomic elements, but according to a recent estimate over two-thirds of our genome may result from the presence or ancient activity of 'jumping genes' [1]. This massive amount of DNA includes domesticated elements with evolutionarily spectacular functions, such as the RAG (recombination activating) genes that form the basis of V(D)J recombination in our immune system [2, 3], and the ERVWE1 (endogenous retrovirus group W, member 1) gene that plays a role in placental development [4]. Contrary to their evolutionary gifts, mobile elements are most notorious for being junk or for causing life-threatening human disorders by insertional mutagenesis and homologous recombination. As we usher in the era of genome-scale studies, it is clear that these elements have the potential to cause intra-individual and inter-individual variation and probably common disease through structural variation, deregulated transcriptional activity or epigenetic effects. Furthermore, large-scale studies have expanded the pool of human disorders resulting from retrotransposon-mediated insertional mutagenesis. Recent reviews have discussed the technical aspects of these new methods [5–8]. We focus here on the known, as well as inferred, potential health impact of their novel findings.
Types of mobile elements in the human genome and the main disease mechanisms
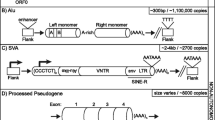
Human mobile elements can be categorized as DNA transposons or retrotransposons. DNA transposons move by a cut-and-paste mechanism, while retrotransposons mobilize by a copy-and-paste mechanism via an RNA intermediate, a process called retrotransposition. Retrotransposons can be further subdivided into long terminal repeat (LTR) and non-LTR elements. LTR retrotransposons are human endogenous retroviruses (HERVs) that have an intracellular existence as a result of a non-functional envelope gene. Non-LTR elements are classified as long interspersed elements (LINEs; the prototype of which is the RNA polymerase II transcribed LINE-1 (L1)), and short interspersed elements (SINEs), the latter consisting essentially of RNA polymerase III transcribed Alus. SVAs (SINE-R/VNTR (variable number of tandem repeat)/Alu) are also active non-LTR retrotransposable elements that are intermediate in size relative to Alus and L1s, and are likely to be transcribed by RNA polymerase II. Thus, they are best thought of as neither LINEs nor SINEs. L1s are the only known autonomously active human retrotransposons, as only they encode proteins (open reading frame 1 (ORF1) and ORF2) with which they can be mobilized. Retrotransposition occurs through a process called target primed reverse transcription (TPRT) [9]. L1s are also responsible for the mobilization of the non-autonomous Alus [10], SVAs [10–13] and processed pseudogenes, which are cellular mRNAs that are reverse transcribed and inserted into the genome [14].
DNA transposons are considered to be immobile in the human genome. Accordingly, no human disease is known to arise as a result of their activity. Also, HERVs are thought to be retrotransposition defective in humans, but some may have retained their ability to move. For example, HERV-K113 has intact ORFs and has insertional polymorphisms in the human population, implying recent evolutionary activity [15]. Although no insertional mutagenesis by HERVs has been described, oncogenic ETV1 (ets variant 1)-HERV-K fusions generated by chromosomal translocation have been observed in prostate cancer [16, 17], and HERV expression has been suggested as a potential contributor to autoimmune diseases [18]. Furthermore, syncytin, an endogenous retroviral envelope protein playing a role in placental trophoblast cell fusion, is involved in breast cancer-endothelial cell and endometrial carcinoma cell fusions [19, 20]. Also, a LTR of a MaLR human endogenous retrovirus has been shown to aberrantly activate a proto-oncogene, thereby causing lymphoma [21].
The predominant mechanism by which L1s cause disease is insertional mutagenesis into or near genes [22, 23]. L1 insertions are often accompanied by 3' transduction, the co-mobilization of DNA sequences downstream of an L1 as a consequence of transcriptional read-through resulting from a weak L1 poly(A) signal [24–27]. Alu sequences predominantly cause disease by homologous recombination between two Alu sequences, but insertion into or near exons, and aberrant Alu splicing from introns, also frequently result in pathological conditions [28–30]. Furthermore, Alu RNA toxicity, a new disease-causing phenomenon, has been recently proposed to result in macular degeneration by DICER1 deficit [31]. Regarding the role of Alu elements in eye disorders, it is interesting that an Alu insertion polymorphism in the ACE (angiotensin I converting enzyme) gene has been associated with protection from the dry/atrophic form of age-related macular degeneration [32]. SVA elements also have the ability to interrupt genes through insertional mutagenesis that can be coupled with 3' transduction, genomic deletion or aberrant splicing [11, 33–35]. The wide spectrum of disease cases caused by retrotransposons ranges from hemophilia to muscular dystrophy and cancer, and has been thoroughly reviewed [36–38]. There have been 96 known retrotransposon insertions in disease cases, of which 25 are caused by L1s, while the other 71 are also L1-mediated. Among the latter, 60 cases are attributable to Alus, 7 to SVAs, and 4 to truncated inserts with only poly(A) sequence remaining [38]. Overall, retrotransposon insertions account for about 1 in 250 (0.4%) of disease-causing mutations [29]. Processed pseudogenes have not yet been found to cause human disease by de novo insertional mutagenesis, but facioscapulohumeral dystrophy has been demonstrated to arise as a result of the contraction of macrosatellite repeats leading to aberrant expression of an array of DUX4 retrogenes residing within the repeats [39, 40]. In addition, mutations in functional processed pseudogenes can cause disease. For instance, mutations in UTP14C have been associated with male infertility [41], mutations in TACSTD2/M1S1 result in gelatinous drop-like corneal dystrophy [42, 43], and PTENP1 is selectively lost in human cancers [44, 45]. The main characteristics of mobile elements capable of causing human disease are summarized in Figure 1.

Types of retroelements implicated in human disease. ENV, envelope; GAG, group specific antigen; HERV, human endogenous retrovirus; kb, kilobase; LTR, long terminal repeat; ORF, open reading frame; POL, polymerase; SINE, short interspersed element; SVA, SINE-R/VNTR/Alu; TPRT, target primed reverse transcription; UTR, untranslated region; VNTR, variable number of tandem repeats. [99]. Black triangles indicate target site duplications.
In the next two sections, we discuss large-scale genome, transcriptome and methylation profiling studies of mobile elements in human diseases. We also discuss some non-genome-scale studies that support or contradict the implications of these novel findings.
Genome-scale approaches to identify new retrotransposon insertions
High-throughput sequencing has increased our capacity to generate large datasets at an unprecedented resolution. It is now possible to characterize genome sequences of scarce samples or even single cells. A next-generation sequencing technique with a high coverage of germline polymorphic human-specific L1 (L1Hs) retrotransposition events has been developed by Ewing and Kazazian, comprising hemi-specific PCR coupled to Illumina sequencing [46]. Using this approach, it has been demonstrated that many L1Hs elements are population-specific [46, 47], and recapitulate genetic ancestry similar to Alu insertion polymorphisms [48]. Retrotransposons are not only excellent markers for exploring population history, but can also give rise to population-specific diseases. For example, a homozygous Alu insertion in an exon of the MAK (male germ-cell-associated kinase) gene has been identified in 21 patients of Jewish ancestry who were diagnosed with retinitis pigmentosa [49]. Oddly, the discovery of this mutation using Agilent exome capture and subsequent Illumina and ABI sequencing was paradoxical, as attempts to remove repetitive sequences from the analysis led to the identification of the insertion [49]. Another population-specific disease caused by retrotransposon mutagenesis is Fukuyama-type congenital muscular dystrophy. It is one of the most common autosomal recessive disorders in Japan and was the first human disease found to result from ancestral insertion of an SVA element [35, 50, 51]. A similar example of an apparently ethnic-specific retrotransposon allele-mediated disease is an L1-mediated orphan 3' transduction into the dystrophin gene leading to Duchenne muscular dystrophy in a Japanese boy [52, 53].
An unexpected finding of state-of-the-art, large-scale approaches to study retrotransposon insertions has been that highly active (or 'hot' [54]) L1s are much more abundant in humans than previously appreciated. The outcome of a fosmid-based paired-end DNA sequencing strategy, coupled with a cell culture assay for retrotransposition, was that over half of newly identified L1s are hot, expanding the number of known hot L1s from 6 to 43 [55]. These L1s are not only expected to be a major source of inter-individual genetic variation [55], but hot L1s account for most examples of disease-causing insertions [54].
Another unusual finding of genome-scale approaches in retrotransposon biology has been that retrotransposition occurs at a very high frequency in somatic cells. Specifically, the brain was announced to be a bona fide territory for retrotransposition. Among three somatic tissues tested, this organ supported the highest level of endogenous L1 copy number, as assessed by quantitative PCR (qPCR) [56]. In another study that awaits further validation, over 7,700 L1s, 13,600 Alus and 1,300 SVA putative somatic insertions were found in the hippocampus of three individuals using retrotransposon capture sequencing, which is based on transposon array capture followed by Illumina paired-end sequencing [57]. Surprisingly, in this study, L1 and Alu insertions were over-represented in protein-coding genes and targeted genes, such as HDAC1 (histone deacetylase 1) and RAI1 (retinoic acid induced 1), which are known to be mutated in neurological disorders [57]. These findings suggest that if a retrotransposon inserts into a gene that functions in neurological development or psychological functioning early in development, it might affect a large enough area of the brain to lead to disease. One might further speculate that retrotransposition in a single brain cell could have some physiological consequences or impact memory formation through altered extracellular signaling to neighboring neurons. If such neuronal plasticity exists, it could affect behavioral phenotypes, and could be modulated by environmental factors [58]. Conversely, knockdown of an L1 regulating cellular factor has demonstrated an effect on L1 retrotransposition in the neurodevelopmental disorder Rett syndrome [59]. MeCP2 (methyl CpG binding protein 2) has been shown to repress L1 expression and retrotransposition [60], and increased L1 retrotransposition has been observed in induced pluripotent stem cells of patients with Rett syndrome who carry MECP2 mutations [59].
Ten brain tumors were examined for somatic L1 insertions by 454 pyrosequencing, but interestingly no retrotransposon insertions were discovered [61]. However, nine somatic L1 insertions were found in 6 out of 20 lung tumors with the same technique [61]. It was not determined though whether the normal tissues also contained some number of L1 insertions relative to the tumor tissue, and thus whether insertions in the cancer represented an elevated level of retrotransposon mobilization. Furthermore, it is not known if these insertions are transcribed or affect gene expression, and whether they were drivers or merely passengers of the tumorigenic process. The genome-wide methylation status of the lung tumors and adjacent normal tissue was also examined using an Illumina platform. All 6 patient DNA samples exhibiting tumor-specific L1 insertions were clustered together as hypomethylated, compared with 13 out of the remaining 14 samples that lacked somatic insertions. These data imply that a methylation signature distinguishes L1-permissive tumors from non-permissive tumors [61].
Another genome-scale method to genotype common retrotransposon insertion polymorphisms (RIPs) to identify genotype-phenotype associations uses array-based technology. Commonly, single nucleotide polymorphisms (SNPs) and copy-number variants have been used as markers in genome-wide association studies (GWASs) to map loci involved in human disease. RIPs are a valuable resource to investigate the role of these elements in phenotypic variation and disease. Also, generally a RIP is much more likely to be the causal variant than a SNP, because a large insertion is more likely to be disruptive of gene function than a single nucleotide alteration, and retrotransposons have many features that can interfere with gene expression (reviewed by Goodier and Kazazian [62]). On the other hand, strong selection exists against retroelement insertions into coding regions, where they are under-represented compared with SNPs [63]. Currently, one array-based approach has been conducted to detect retrotransposon insertions in human disease. Using transposon insertion profiling by microarray (TIP-chip), several novel L1 insertions on the X chromosome were discovered in male probands with presumptively X-linked intellectual disability [64]. Interestingly, one of the insertions occurred in the NHS gene, which is mutated in Nance-Horan syndrome, a condition associated with intellectual disability. Another promising insertion occurred in the DACH2 (dachshund homolog 2) gene that regulates neuronal differentiation [64]. However, confirmation studies are needed to demonstrate whether these insertions are the underlying cause of intellectual disability in these patients.
Except for the Baillie et al. study [57], which analyzed L1, Alu and SVA somatic insertions, the studies mentioned above concentrated on genome-wide detection of new L1 insertions. A notable study by Witherspoon et al. [65] developed a robust technique, termed mobile element scanning, to find new insertions of young Alu elements using PCR methods coupled with high-throughput sequencing. The group found approximately 500 de novo Alu insertions [65]. Their technique is applicable to all mobile elements, and is amenable to significant multiplexing of a number of DNA samples in one sequencing run.
Genome-wide methylation studies and transcriptome analysis of retrotransposons
It is speculated that one of the main roles of DNA methylation, in addition to epigenetic reprogramming, is to silence transposable elements [66]. Most methylation studies of human transposons have investigated malignancies and showed consistent hypomethylation (for example, [67, 68]), the extent of which, however, was variable in different tissues [69]. As the malignant phenotype is inherently associated with global as well as tumor-type-specific methylation changes [70], and transposable elements comprise the majority of the human genome, it is difficult to establish the role of transposon demethylation per se in tumorigenesis, especially without accompanying functional studies. It is possible that pathogenic cellular stress responses could result in local or global transposon deregulation - for example, via demethylation or chromatin modification. Once out of control, such an epigenetic deregulation might result in single or multiple retrotransposition events.
Retrotransposons located 5' of protein coding loci frequently function as alternative promoters. They might also express non-coding RNAs, and retrotransposons in the 3' UTR (untranslated region) of genes show strong evidence of reducing the expression of the respective gene, as assessed by cap analysis gene expression and pyrosequencing [71]. Thus, an altered retrotransposon methylation state is expected to affect either the transcription of the retrotransposon itself or that of nearby genes. Accordingly, it has been shown that hypomethylation of L1s can cause altered gene expression. Specifically, an L1 is located in the MET (hepatocyte growth factor receptor) oncogene, and hypomethylation of a promoter in this L1 induced an alternative MET transcript within the urothelium of tumor-bearing bladders. At the same time, in the bladder epithelium of cancer-free donors the methylation level of this L1 promoter was high and expression of the alternative MET transcript was low [72].
There are few studies that correlate human retrotransposon methylation with their transcription level on a genome-wide scale. According to one study, expression of L1 5' and 3' UTR sequences in prostate cancer was rather decreased, despite significant hypomethylation of the L1 promoter. Different HERV-K families showed opposite trends in expression levels, and the expression of evolutionarily young Alu families was restricted to individual prostate tumors as assessed by RT-qPCR and pyrosequencing [73]. In agreement with that study, transcriptional activation of L1s was not observed in globally hypomethylated hepatocellular carcinoma compared with matched normal tissue, as assessed by RT-qPCR [74]. Of note, the quantification of some types of expressed retroelements by using classical methods may prove ambiguous. Since Alu sequences are abundant in RNA polymerase II transcripts, quantification of the relatively rare RNA polymerase III transcribed Alu transcripts by RT-qPCR is fraught with potential error [75]. Transcribed L1 sequences embedded in genes may similarly confound the results of such quantitative measurements of L1 expression.
Using a custom GeneChip microarray for transcriptome analysis of several HERV families, it was shown that numerous HERV-W loci were overexpressed in testicular cancer [76]. Interestingly, one of these was an ERVWE1 transcript whose expression is usually restricted to the placenta. Methylation was severely or completely diminished at HERV-W sequences in the tumor DNA, suggesting that DNA methylation and HERV-W expression is interrelated in this tumor context [76]. With a genome-wide technique termed selective differential display of RNAs containing interspersed repeats and with its modified version, termed L1 chimera display, it has been also demonstrated that the levels of many HERV-K LTR transcripts differ between normal and testicular germ cell tumor tissues [77], and that the L1 antisense promoter gives rise to novel chimeric transcripts that are unique in tumor samples [78]. Furthermore, the cancer-specific chimeric L1 transcripts could be induced in non-malignant cells by using the demethylating drug 5-azacytidine [78].
It will be interesting to learn if tumor-specific retrotransposon profiles reveal enhanced retroelement mobility. For example, L1 retrotransposition is associated with genetic instability [79], a hallmark of cancer [80]. Thus, local or global overactivation of L1s could have the potential to contribute to tumorigenesis. In particular, germ cell tumors are good candidates to examine cancer-specific retroelement activity, because the genome of germ cells goes through epigenetic reprogramming through methylation at CpG sites. Thus, deregulation of this process might easily lead to the derepression of transposable elements, and potentially to germ cell tumors. In support of this hypothesis, the L1 ORF1 protein was overexpressed in all 62 cases of investigated childhood malignant germ cell tumors relative to adjacent normal tissue and was associated with poor differentiation [81]. Testicular germ cell tumors should also be examined for L1-conferred hereditary disease, as no high penetrance susceptibility genes have been identified in this condition. With pyrosequencing of bisulfite-treated DNA using L1-specific primers, transgenerational L1 methylation inheritance was implicated to be associated with testicular cancer risk [82]. Thus, L1s are attractive candidates for both somatic drivers and hereditary predisposition factors in germ cell tumors and possibly in other cancer types. However, currently their functional impact in malignancy is poorly understood.
Concluding remarks and future directions
Genome-scale technologies now provide us with the opportunity to investigate retrotransposon biology in unprecedented detail. Ultimately, it will be important to test the functional consequences of these results, such as the effect of RIPs on gene function, and their role in cancer and neurological disorders. This outcome might be accomplished by classical functional studies, or by combining the results of several genome-scale experiments. For instance, if comprehensive RIP profiles were coupled with next-generation RNA sequencing data, it would allow testing of hypotheses pertaining to retrotransposons and their effects upon gene expression. Such platforms would also be useful to explore whether there is a role for common RIPs in common disease and if these RIPs convey the disease phenotype through expression. In a similar manner, one could incorporate chromatin/methyl-seq/RNA/ChIP-seq profiles for DNA-binding or RNA-binding proteins with the respective RIP profiles. It would also be advantageous to carry out studies to explore whether any overlap between a GWAS hit and a known RIP exists, as the RIP might indeed be the causal variant.
As an alternative genome-scale approach to understand the impact of human transposons on disease, functional genetic screening strategies could be developed in cell culture. For instance, haploid cell lines [83, 84] and BLM (Bloom syndrome, RecQ helicase-like)-deficient cells that can be converted to generate a genome-wide library of homozygous mutant cells [85–87] are available to be mutagenized and screened for any desirable phenotype, such as altered retrotransposition activity, using suitable read-out systems. One such system could be a retrotransposition assay, where an L1 reporter construct has been designed so that translation of the reporter (drug-resistance gene or enhanced green fluorescent protein) occurs only after L1 reverse transcription and insertion of its cDNA copy into the genome [88, 89]. Also, genome-wide mutagenesis might be accomplished with mobile elements themselves, such as retroviruses or DNA transposons [85–87]. Similarly, large-scale small interfering RNA (siRNA) and cDNA functional genetics screening strategies could be designed to identify host cell factors modulating L1 activity. One should also investigate whether some host factors elicit a disease phenotype through deregulated retrotransposon activity. For example, the remarkable finding of the role of Alu RNA toxicity due to DICER1 deficiency in macular degeneration [31] needs to be replicated by alternative methods. Those methods should exclude the possibility that what is really being detected is amplification of the closely related 7SL RNA or Alu sequences contained in RNA polymerase II transcripts, which vastly outnumber the RNA polymerase III-transcribed Alu elements [75]. Also, functional genetic follow-up studies should circumvent - if at all feasible - non-specific toxicity arising as a result of ectopic Alu overexpression or antisense oligo-mediated downregulation of essential RNA polymerase II transcripts with embedded Alu sequences. DICER1 may also have a role in tumorigenesis through retrotransposon overexpression, as germline mutations in this gene have been found in familial pleuropulmonary blastoma [90] and in familial multinodular goiter with ovarian Sertoli-Leydig cell tumors [91]. Sense and antisense transcripts derived from L1 promoters could be processed to siRNAs that might suppress retrotransposition by RNA interference [92], and DICER1 has been implicated in this process [93]. These data raise the possibility that genomic instability in some malignancies could arise - at least partly - from retrotransposon overdose as a consequence of a mutated small non-coding RNA pathway. This could lead eventually to retrotransposon RNA toxicity [31], genotoxic stress through DNA nicking by ORF2 [94], or elevated insertional mutagenesis [61].
For the future of personalized medicine it will be vital not to exclude the transposon profile of patients, as exemplified by the case of an Alu insertion in a retinitis pigmentosa proband [49]. Another aspect of personalized medicine is gene therapy. In one form of gene therapy, antisense oligonucleotides that block aberrant splicing into an intronic SVA that causes Fukuyama muscular dystrophy has been suggested [35]. Another aspect of gene therapy is the use of DNA transposons that hold the promise of lower immunogenicity, enhanced safety profile and reduced manufacturing costs compared with viral vectors [95]. Two DNA transposons from non-mammalian species have emerged as gene therapy tools based on their efficient transposition in humans: the reconstructed Tc1/mariner element Sleeping Beauty from salmonid fish and piggyBac from the baculovirus genome [95, 96]. The first ex vivo gene therapy clinical trial using Sleeping Beauty has been approved [97], and induced pluripotent stem cells are now being generated after targeted gene correction using piggyBac technology [98]. Once the potential side-effects of these therapies - such as secondary mutagenesis resulting from transposon hopping or activation of nearby genes - are overcome, the roles of mobile elements can be redefined from being just 'junk' or 'enemy' to 'life-guards' of our genomes.
Abbreviations
- GWAS:
-
genome-wide association study
- HERV:
-
human endogenous retrovirus
- L1Hs:
-
human-specific L1
- LINE-1/L1:
-
long interspersed element
- LTR:
-
long terminal repeat
- ORF:
-
open reading frame
- qPCR:
-
quantitative PCR
- RIP:
-
retrotransposon insertion polymorphism
- SINE:
-
short interspersed element
- siRNA:
-
small interfering RNA
- SNP:
-
single nucleotide polymorphism
- SVA:
-
SINE-R/VNTR/Alu
- TPRT:
-
target primed reverse transcription
- UTR:
-
untranslated region
- VNTR:
-
variable number of tandem repeat.
References
de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD: Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7: e1002384-10.1371/journal.pgen.1002384.
Zhou L, Mitra R, Atkinson PW, Hickman AB, Dyda F, Craig NL: Transposition of hAT elements links transposable elements and V(D)J recombination. Nature. 2004, 432: 995-1001. 10.1038/nature03157.
Kapitonov VV, Jurka J: RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol. 2005, 3: e181-10.1371/journal.pbio.0030181.
Blond JL, Lavillette D, Cheynet V, Bouton O, Oriol G, Chapel-Fernandes S, Mandrand B, Mallet F, Cosset FL: An envelope glycoprotein of the human endogenous retrovirus HERV-W is expressed in the human placenta and fuses cells expressing the type D mammalian retrovirus receptor. J Virol. 2000, 74: 3321-3329. 10.1128/JVI.74.7.3321-3329.2000.
O'Donnell KA, Burns KH: Mobilizing diversity: transposable element insertions in genetic variation and disease. Mob DNA. 2010, 1: 21-10.1186/1759-8753-1-21.
Beck CR, Garcia-Perez JL, Badge RM, Moran JV: LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 2011, 12: 187-215. 10.1146/annurev-genom-082509-141802.
Ray DA, Batzer MA: Reading TE leaves: new approaches to the identification of transposable element insertions. Genome Res. 2011, 21: 813-820. 10.1101/gr.110528.110.
Faulkner GJ: Retrotransposons: mobile and mutagenic from conception to death. FEBS Lett. 2011, 585: 1589-1594. 10.1016/j.febslet.2011.03.061.
Cost GJ, Feng Q, Jacquier A, Boeke JD: Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002, 21: 5899-5910. 10.1093/emboj/cdf592.
Dewannieux M, Esnault C, Heidmann T: LINE-mediated retrotransposition of marked Alu sequences. Nat Genet. 2003, 35: 41-48. 10.1038/ng1223.
Ostertag EM, Goodier JL, Zhang Y, Kazazian HH: SVA elements are nonautonomous retrotransposons that cause disease in humans. Am J Hum Genet. 2003, 73: 1444-1451. 10.1086/380207.
Hancks DC, Goodier JL, Mandal PK, Cheung LE, Kazazian HH: Retrotransposition of marked SVA elements by human L1s in cultured cells. Hum Mol Genet. 2011, 20: 3386-3400. 10.1093/hmg/ddr245.
Raiz J, Damert A, Chira S, Held U, Klawitter S, Hamdorf M, Lower J, Stratling WH, Lower R, Schumann GG: The non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res. 2012, 40: 1666-1683. 10.1093/nar/gkr863.
Esnault C, Maestre J, Heidmann T: Human LINE retrotransposons generate processed pseudogenes. Nat Genet. 2000, 24: 363-367. 10.1038/74184.
Turner G, Barbulescu M, Su M, Jensen-Seaman MI, Kidd KK, Lenz J: Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr Biol. 2001, 11: 1531-1535. 10.1016/S0960-9822(01)00455-9.
Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, Yu J, Wang L, Montie JE, Rubin MA, Pienta KJ, Roulston D, Shah RB, Varambally S, Mehra R, Chinnaiyan AM: Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007, 448: 595-599. 10.1038/nature06024.
Hermans KG, van der Korput HA, van Marion R, van de Wijngaart DJ, Ziel-van der Made A, Dits NF, Boormans JL, van der Kwast TH, van Dekken H, Bangma CH, Korsten H, Kraaij R, Jenster G, Trapman J: Truncated ETV1, fused to novel tissue-specific genes, and full-length ETV1 in prostate cancer. Cancer Res. 2008, 68: 7541-7549. 10.1158/0008-5472.CAN-07-5930.
Balada E, Vilardell-Tarres M, Ordi-Ros J: Implication of human endogenous retroviruses in the development of autoimmune diseases. Int Rev Immunol. 2010, 29: 351-370. 10.3109/08830185.2010.485333.
Bjerregaard B, Holck S, Christensen IJ, Larsson LI: Syncytin is involved in breast cancer-endothelial cell fusions. Cell Mol Life Sci. 2006, 63: 1906-1911. 10.1007/s00018-006-6201-9.
Strick R, Ackermann S, Langbein M, Swiatek J, Schubert SW, Hashemolhosseini S, Koscheck T, Fasching PA, Schild RL, Beckmann MW, Strissel PL: Proliferation and cell-cell fusion of endometrial carcinoma are induced by the human endogenous retroviral Syncytin-1 and regulated by TGF-beta. J Mol Med (Berl). 2007, 85: 23-38.
Lamprecht B, Walter K, Kreher S, Kumar R, Hummel M, Lenze D, Kochert K, Bouhlel MA, Richter J, Soler E, Stadhouders R, Johrens K, Wurster KD, Callen DF, Harte MF, Giefing M, Barlow R, Stein H, Anagnostopoulos I, Janz M, Cockerill PN, Siebert R, Dorken B, Bonifer C, Mathas S: Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat Med. 2010, 16: 571-579. 10.1038/nm.2129. 1p following 579
Kazazian HH, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE: Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature. 1988, 332: 164-166. 10.1038/332164a0.
Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, Vogelstein B, Nakamura Y: Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992, 52: 643-645.
Holmes SE, Dombroski BA, Krebs CM, Boehm CD, Kazazian HH: A new retrotransposable human L1 element from the LRE2 locus on chromosome 1q produces a chimaeric insertion. Nat Genet. 1994, 7: 143-148. 10.1038/ng0694-143.
Moran JV, DeBerardinis RJ, Kazazian HH: Exon shuffling by L1 retrotransposition. Science. 1999, 283: 1530-1534. 10.1126/science.283.5407.1530.
Goodier JL, Ostertag EM, Kazazian HH: Transduction of 3'-flanking sequences is common in L1 retrotransposition. Hum Mol Genet. 2000, 9: 653-657. 10.1093/hmg/9.4.653.
Pickeral OK, Makalowski W, Boguski MS, Boeke JD: Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res. 2000, 10: 411-415. 10.1101/gr.10.4.411.
Halling KC, Lazzaro CR, Honchel R, Bufill JA, Powell SM, Arndt CA, Lindor NM: Hereditary desmoid disease in a family with a germline Alu I repeat mutation of the APC gene. Hum Hered. 1999, 49: 97-102. 10.1159/000022852.
Wimmer K, Callens T, Wernstedt A, Messiaen L: The NF1 gene contains hotspots for L1 endonuclease-dependent de novo insertion. PLoS Genet. 2011, 7: e1002371-10.1371/journal.pgen.1002371.
Nystrom-Lahti M, Kristo P, Nicolaides NC, Chang SY, Aaltonen LA, Moisio AL, Jarvinen HJ, Mecklin JP, Kinzler KW, Vogelstein B: Founding mutations and Alu-mediated recombination in hereditary colon cancer. Nat Med. 1995, 1: 1203-1206. 10.1038/nm1195-1203.
Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, Kleinman ME, Ponicsan SL, Hauswirth WW, Chiodo VA, Kariko K, Yoo JW, Lee DK, Hadziahmetovic M, Song Y, Misra S, Chaudhuri G, Buaas FW, Braun RE, Hinton DR, Zhang Q, Grossniklaus HE, Provis JM, Madigan MC, Milam AH, Justice NL, Albuquerque RJ, Blandford AD, Bogdanovich S, Hirano Y, et al: DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011, 471: 325-330. 10.1038/nature09830.
Hamdi HK, Reznik J, Castellon R, Atilano SR, Ong JM, Udar N, Tavis JH, Aoki AM, Nesburn AB, Boyer DS, Small KW, Brown DJ, Kenney MC: Alu DNA polymorphism in ACE gene is protective for age-related macular degeneration. Biochem Biophys Res Commun. 2002, 295: 668-672. 10.1016/S0006-291X(02)00728-3.
Conley ME, Partain JD, Norland SM, Shurtleff SA, Kazazian HH: Two independent retrotransposon insertions at the same site within the coding region of BTK. Hum Mutat. 2005, 25: 324-325.
Takasu M, Hayashi R, Maruya E, Ota M, Imura K, Kougo K, Kobayashi C, Saji H, Ishikawa Y, Asai T, Tokunaga K: Deletion of entire HLA-A gene accompanied by an insertion of a retrotransposon. Tissue Antigens. 2007, 70: 144-150. 10.1111/j.1399-0039.2007.00870.x.
Taniguchi-Ikeda M, Kobayashi K, Kanagawa M, Yu CC, Mori K, Oda T, Kuga A, Kurahashi H, Akman HO, DiMauro S, Kaji R, Yokota T, Takeda S, Toda T: Pathogenic exon-trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature. 2011, 478: 127-131. 10.1038/nature10456.
Chen JM, Stenson PD, Cooper DN, Ferec C: A systematic analysis of LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease. Hum Genet. 2005, 117: 411-427. 10.1007/s00439-005-1321-0.
Belancio VP, Hedges DJ, Deininger P: Mammalian non-LTR retrotransposons: for better or worse, in sickness and in health. Genome Res. 2008, 18: 343-358. 10.1101/gr.5558208.
Hancks DC, Kazazian HH: Active human retrotransposons: variation and disease. Curr Opin Genet Dev.
Clapp J, Mitchell LM, Bolland DJ, Fantes J, Corcoran AE, Scotting PJ, Armour JA, Hewitt JE: Evolutionary conservation of a coding function for D4Z4, the tandem DNA repeat mutated in facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2007, 81: 264-279. 10.1086/519311.
Snider L, Geng LN, Lemmers RJ, Kyba M, Ware CB, Nelson AM, Tawil R, Filippova GN, van der Maarel SM, Tapscott SJ, Miller DG: Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6: e1001181-10.1371/journal.pgen.1001181.
Rohozinski J, Lamb DJ, Bishop CE: UTP14c is a recently acquired retrogene associated with spermatogenesis and fertility in man. Biol Reprod. 2006, 74: 644-651. 10.1095/biolreprod.105.046698.
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, Nakamura Y: Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nat Genet. 1999, 21: 420-423. 10.1038/7759.
Vinckenbosch N, Dupanloup I, Kaessmann H: Evolutionary fate of retroposed gene copies in the human genome. Proc Natl Acad Sci USA. 2006, 103: 3220-3225. 10.1073/pnas.0511307103.
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP: A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010, 465: 1033-1038. 10.1038/nature09144.
Poliseno L, Haimovic A, Christos PJ, Vega Y, Saenz de Miera EC, Shapiro R, Pavlick A, Berman RS, Darvishian F, Osman I: Deletion of PTENP1 pseudogene in human melanoma. J Invest Dermatol. 2011, 131: 2497-2500. 10.1038/jid.2011.232.
Ewing AD, Kazazian HH: High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res. 2010, 20: 1262-1270. 10.1101/gr.106419.110.
Ewing AD, Kazazian HH: Whole-genome resequencing allows detection of many rare LINE-1 insertion alleles in humans. Genome Res. 2011, 21: 985-990. 10.1101/gr.114777.110.
Hormozdiari F, Alkan C, Ventura M, Hajirasouliha I, Malig M, Hach F, Yorukoglu D, Dao P, Bakhshi M, Sahinalp SC, Eichler EE: Alu repeat discovery and characterization within human genomes. Genome Res. 2011, 21: 840-849. 10.1101/gr.115956.110.
Tucker BA, Scheetz TE, Mullins RF, DeLuca AP, Hoffmann JM, Johnston RM, Jacobson SG, Sheffield VC, Stone EM: Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc Natl Acad Sci USA. 2011, 108: E569-576. 10.1073/pnas.1108918108.
Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K, Osawa M, Hamano K, Sakakihara Y, Nonaka I, Nakagome Y, Kanazawa I, Nakamura Y, Tokunaga K, Toda T: An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998, 394: 388-392. 10.1038/28653.
Watanabe M, Kobayashi K, Jin F, Park KS, Yamada T, Tokunaga K, Toda T: Founder SVA retrotransposal insertion in Fukuyama-type congenital muscular dystrophy and its origin in Japanese and Northeast Asian populations. Am J Med Genet A. 2005, 138: 344-348.
Solyom S, Ewing AD, Hancks DC, Takeshima Y, Awano H, Matsuo M, Kazazian HH: Pathogenic orphan transduction created by a non-reference LINE-1 retrotransposon. Hum Mutat. 2012, 33: 369-371. 10.1002/humu.21663.
Awano H, Malueka RG, Yagi M, Okizuka Y, Takeshima Y, Matsuo M: Contemporary retrotransposition of a novel non-coding gene induces exon-skipping in dystrophin mRNA. J Hum Genet. 2010, 55: 785-790. 10.1038/jhg.2010.111.
Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH: Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci USA. 2003, 100: 5280-5285. 10.1073/pnas.0831042100.
Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV: LINE-1 retrotransposition activity in human genomes. Cell. 2010, 141: 1159-1170. 10.1016/j.cell.2010.05.021.
Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O'Shea KS, Moran JV, Gage FH: L1 retrotransposition in human neural progenitor cells. Nature. 2009, 460: 1127-1131. 10.1038/nature08248.
Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Brennan PM, Rizzu P, Smith S, Fell M, Talbot RT, Gustincich S, Freeman TC, Mattick JS, Hume DA, Heutink P, Carninci P, Jeddeloh JA, Faulkner GJ: Somatic retrotransposition alters the genetic landscape of the human brain. Nature. 2011, 479: 534-537. 10.1038/nature10531.
Singer T, McConnell MJ, Marchetto MC, Coufal NG, Gage FH: LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes?. Trends Neurosci. 2010, 33: 345-354. 10.1016/j.tins.2010.04.001.
Muotri AR, Marchetto MC, Coufal NG, Oefner R, Yeo G, Nakashima K, Gage FH: L1 retrotransposition in neurons is modulated by MeCP2. Nature. 2010, 468: 443-446. 10.1038/nature09544.
Yu F, Zingler N, Schumann G, Stratling WH: Methyl-CpG-binding protein 2 represses LINE-1 expression and retrotransposition but not Alu transcription. Nucleic Acids Res. 2001, 29: 4493-4501. 10.1093/nar/29.21.4493.
Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, Van Meir EG, Vertino PM, Devine SE: Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010, 141: 1253-1261. 10.1016/j.cell.2010.05.020.
Goodier JL, Kazazian HH: Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008, 135: 23-35. 10.1016/j.cell.2008.09.022.
Stewart C, Kural D, Stromberg MP, Walker JA, Konkel MK, Stutz AM, Urban AE, Grubert F, Lam HY, Lee WP, Busby M, Indap AR, Garrison E, Huff C, Xing J, Snyder MP, Jorde LB, Batzer MA, Korbel JO, Marth GT, 1000 Genomes Project: A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet. 2011, 7: e1002236-10.1371/journal.pgen.1002236.
Huang CR, Schneider AM, Lu Y, Niranjan T, Shen P, Robinson MA, Steranka JP, Valle D, Civin CI, Wang T, Wheelan SJ, Ji H, Boeke JD, Burns KH: Mobile interspersed repeats are major structural variants in the human genome. Cell. 2010, 141: 1171-1182. 10.1016/j.cell.2010.05.026.
Witherspoon DJ, Xing J, Zhang Y, Watkins WS, Batzer MA, Jorde LB: Mobile element scanning (ME-Scan) by targeted high-throughput sequencing. BMC Genomics. 2010, 11: 410-10.1186/1471-2164-11-410.
Yoder JA, Walsh CP, Bestor TH: Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997, 13: 335-340. 10.1016/S0168-9525(97)01181-5.
Chalitchagorn K, Shuangshoti S, Hourpai N, Kongruttanachok N, Tangkijvanich P, Thong-ngam D, Voravud N, Sriuranpong V, Mutirangura A: Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene. 2004, 23: 8841-8846. 10.1038/sj.onc.1208137.
Rodriguez J, Vives L, Jorda M, Morales C, Munoz M, Vendrell E, Peinado MA: Genome-wide tracking of unmethylated DNA Alu repeats in normal and cancer cells. Nucleic Acids Res. 2008, 36: 770-784.
Schulz WA: L1 retrotransposons in human cancers. J Biomed Biotechnol. 2006, 2006: 83672-
Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, Lang JC, Schuller DE, Yu L, Bloomfield CD, Caligiuri MA, Yates A, Nishikawa R, Su Huang H, Petrelli NJ, Zhang X, O'Dorisio MS, Held WA, Cavenee WK, Plass C: Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000, 24: 132-138. 10.1038/72785.
Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T, Waki K, Hornig N, Arakawa T, Takahashi H, Kawai J, Forrest AR, Suzuki H, Hayashizaki Y, Hume DA, Orlando V, Grimmond SM, Carninci P: The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009, 41: 563-571. 10.1038/ng.368.
Wolff EM, Byun HM, Han HF, Sharma S, Nichols PW, Siegmund KD, Yang AS, Jones PA, Liang G: Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010, 6: e1000917-10.1371/journal.pgen.1000917.
Goering W, Ribarska T, Schulz WA: Selective changes of retroelement expression in human prostate cancer. Carcinogenesis. 2011, 32: 1484-1492. 10.1093/carcin/bgr181.
Lin CH, Hsieh SY, Sheen IS, Lee WC, Chen TC, Shyu WC, Liaw YF: Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res. 2001, 61: 4238-4243.
Deininger P: Alu elements: know the SINEs. Genome Biol. 2011, 12: 236-10.1186/gb-2011-12-12-236.
Gimenez J, Montgiraud C, Pichon JP, Bonnaud B, Arsac M, Ruel K, Bouton O, Mallet F: Custom human endogenous retroviruses dedicated microarray identifies self-induced HERV-W family elements reactivated in testicular cancer upon methylation control. Nucleic Acids Res. 2010, 38: 2229-2246. 10.1093/nar/gkp1214.
Vinogradova T, Leppik L, Kalinina E, Zhulidov P, Grzeschik KH, Sverdlov E: Selective differential display of RNAs containing interspersed repeats: analysis of changes in the transcription of HERV-K LTRs in germ cell tumors. Mol Genet Genomics. 2002, 266: 796-805. 10.1007/s00438-001-0596-7.
Cruickshanks HA, Tufarelli C: Isolation of cancer-specific chimeric transcripts induced by hypomethylation of the LINE-1 antisense promoter. Genomics. 2009, 94: 397-406. 10.1016/j.ygeno.2009.08.013.
Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD: Human l1 retrotransposition is associated with genetic instability in vivo. Cell. 2002, 110: 327-338. 10.1016/S0092-8674(02)00839-5.
Negrini S, Gorgoulis VG, Halazonetis TD: Genomic instability - an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010, 11: 220-228. 10.1038/nrm2858.
Su Y, Davies S, Davis M, Lu H, Giller R, Krailo M, Cai Q, Robison L, Shu XO, Children's Oncology Group: Expression of LINE-1 p40 protein in pediatric malignant germ cell tumors and its association with clinicopathological parameters: a report from the Children's Oncology Group. Cancer Lett. 2007, 247: 204-212. 10.1016/j.canlet.2006.04.010.
Mirabello L, Savage SA, Korde L, Gadalla SM, Greene MH: LINE-1 methylation is inherited in familial testicular cancer kindreds. BMC Med Genet. 2010, 11: 77-
Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, Brummelkamp TR: Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009, 326: 1231-1235. 10.1126/science.1178955.
Leeb M, Wutz A: Derivation of haploid embryonic stem cells from mouse embryos. Nature. 2011, 479: 131-134. 10.1038/nature10448.
Guo G, Wang W, Bradley A: Mismatch repair genes identified using genetic screens in Blm-deficient embryonic stem cells. Nature. 2004, 429: 891-895. 10.1038/nature02653.
Wang W, Bradley A, Huang Y: A piggyBac transposon-based genome-wide library of insertionally mutated Blm-deficient murine ES cells. Genome Res. 2009, 19: 667-673. 10.1101/gr.085621.108.
Trombly MI, Su H, Wang X: A genetic screen for components of the mammalian RNA interference pathway in Bloom-deficient mouse embryonic stem cells. Nucleic Acids Res. 2009, 37: e34-
Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH: High frequency retrotransposition in cultured mammalian cells. Cell. 1996, 87: 917-927. 10.1016/S0092-8674(00)81998-4.
Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, Kazazian HH: Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Res. 2000, 28: 1418-1423. 10.1093/nar/28.6.1418.
Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, Jarzembowski JA, Wikenheiser-Brokamp KA, Suarez BK, Whelan AJ, Williams G, Bracamontes D, Messinger Y, Goodfellow PJ: DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009, 325: 965-10.1126/science.1174334.
Rio Frio T, Bahubeshi A, Kanellopoulou C, Hamel N, Niedziela M, Sabbaghian N, Pouchet C, Gilbert L, O'Brien PK, Serfas K, Broderick P, Houlston RS, Lesueur F, Bonora E, Muljo S, Schimke RN, Bouron-Dal Soglio D, Arseneau J, Schultz KA, Priest JR, Nguyen VH, Harach HR, Livingston DM, Foulkes WD, Tischkowitz M: DICER1 mutations in familial multinodular goiter with and without ovarian Sertoli-Leydig cell tumors. JAMA. 2011, 305: 68-77. 10.1001/jama.2010.1910.
Yang N, Kazazian HH: L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat Struct Mol Biol. 2006, 13: 763-771. 10.1038/nsmb1141.
Soifer HS, Zaragoza A, Peyvan M, Behlke MA, Rossi JJ: A potential role for RNA interference in controlling the activity of the human LINE-1 retrotransposon. Nucleic Acids Res. 2005, 33: 846-856. 10.1093/nar/gki223.
Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, Zhang J, Rose DW, Fu XD, Glass CK, Rosenfeld MG: Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009, 139: 1069-1083. 10.1016/j.cell.2009.11.030.
Ivics Z, Izsvak Z: Nonviral gene delivery with the sleeping beauty transposon system. Hum Gene Ther. 2011, 22: 1043-1051. 10.1089/hum.2011.143.
VandenDriessche T, Ivics Z, Izsvak Z, Chuah MK: Emerging potential of transposons for gene therapy and generation of induced pluripotent stem cells. Blood. 2009, 114: 1461-1468. 10.1182/blood-2009-04-210427.
Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H, Hackett PB, Kohn DB, Shpall EJ, Champlin RE, Cooper LJ: Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008, 68: 2961-2971. 10.1158/0008-5472.CAN-07-5600.
Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, Paschon DE, Miranda E, Ordonez A, Hannan NR, Rouhani FJ, Darche S, Alexander G, Marciniak SJ, Fusaki N, Hasegawa M, Holmes MC, Di Santo JP, Lomas DA, Bradley A, Vallier L: Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011, 478: 391-394. 10.1038/nature10424.
Economou-Pachnis A, Tsichlis PN: Insertion of an Alu SINE in the human homologue of the Mlvi-2 locus. Nucleic Acids Res. 1985, 13: 8379-8387. 10.1093/nar/13.23.8379.
Acknowledgements
We thank Dustin C Hancks, John L Goodier, Adam D Ewing and Tara Doucet for their comments on the manuscript. Research in the Kazazian laboratory is funded by grants from the National Institutes of Health awarded to HHK.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.