
Abstract
Human pluripotent cells such as human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) and their in vitro differentiation models hold great promise for regenerative medicine as they provide both a model for investigating mechanisms underlying human development and disease and a potential source of replacement cells in cellular transplantation approaches. The remarkable developmental plasticity of pluripotent cells is reflected in their unique chromatin marking and organization patterns, or epigenomes. Pluripotent cell epigenomes must organize genetic information in a way that is compatible with both the maintenance of self-renewal programs and the retention of multilineage differentiation potential. In this review, we give a brief overview of the recent technological advances in genomics that are allowing scientists to characterize and compare epigenomes of different cell types at an unprecedented scale and resolution. We then discuss how utilizing these technologies for studies of hESCs has demonstrated that certain chromatin features, including bivalent promoters, poised enhancers, and unique DNA modification patterns, are particularly pervasive in hESCs compared with differentiated cell types. We outline these unique characteristics and discuss the extent to which they are recapitulated in iPSCs. Finally, we envision broad applications of epigenomics in characterizing the quality and differentiation potential of individual pluripotent lines, and we discuss how epigenomic profiling of regulatory elements in hESCs, iPSCs and their derivatives can improve our understanding of complex human diseases and their underlying genetic variants.
Similar content being viewed by others


One genome, many epigenomes
Embryonic stem cells (ESCs) and the early developmental stage embryo share a unique property called pluripotency, which is the ability to give rise to the three germ layers (endoderm, ectoderm and mesoderm) and, consequently, all tissues represented in the adult organism [1, 2]. Pluripotency can also be induced in somatic cells during in vitro reprogramming, leading to the formation of so-called induced pluripotent stem cells (iPSCs; extensively reviewed in [3–7]). In order to fulfill the therapeutic potential of human ESCs (hESCs) and iPSCs, an understanding of the fundamental molecular properties underlying the nature of pluripotency and commitment is required, along with the development of methods for assessing biological equivalency among different cell populations.
Functional complexity of the human body, with over 200 specialized cell types, and intricately built tissues and organs, arises from a single set of instructions: the human genome. How, then, do distinct cellular phenotypes emerge from this genetic homogeneity? Interactions between the genome and its cellular and signaling environments are the key to understanding how cell-type-specific gene expression patterns arise during differentiation and development [8]. These interactions ultimately occur at the level of the chromatin, which comprises the DNA polymer repeatedly wrapped around histone octamers, forming a nucleosomal array that is further compacted into the higher-order structure. Regulatory variation is introduced to the chromatin via alterations within the nucleosome itself - for example, through methylation and hydroxymethylation of DNA, various post-translational modifications (PTMs) of histones, and inclusion or exclusion of specific histone variants [9–15] - as well as via changes in nucleosomal occupancy, mobility and organization [16, 17]. In turn, these alterations modulate access of sequence-dependent transcriptional regulators to the underlying DNA, the level of chromatin compaction, and communication between distant chromosomal regions [18]. The entirety of chromatin regulatory variation in a specific cellular state is often referred to as the 'epigenome' [19].
Technological advances have made the exploration of epigenomes feasible in a rapidly increasing number of cell types and tissues. Systematic efforts at such analyses had been undertaken by the human ENCyclopedia Of DNA Elements (ENCODE) and NIH Roadmap Epigenomics projects [20, 21]. These and other studies have already produced, and will generate in the near future, an overwhelming amount of genome-wide datasets that are often not readily comprehensible to many biologists and physicians. However, given the importance of epigenetic patterns in defining cell identity, understanding and utilizing epigenomic mapping will become a necessity in both basic and translational stem cell research. In this review, we strive to provide an overview of the main concepts, technologies and outputs of epigenomics in a form that is accessible to a broad audience. We summarize how epigenomes are studied, discuss what we have learned so far about unique epigenetic properties of hESCs and iPSCs, and envision direct implications of epigenomics in translational research and medicine.
Technological advances in genomics and epigenomics
Epigenomics is defined here as genomic-scale studies of chromatin regulatory variation, including patterns of histone PTMs, DNA methylation, nucleosome positioning and long-range chromosomal interactions. Over the past 20 years, many methods have been developed to probe different forms of this variation. For example, a plethora of antibodies recognizing specific histone modifications has been developed and used in chromatin immunoprecipitation (ChIP) assays for studying the local enrichment of histone PTMs at specific loci [22, 23]. Similarly, bisulfite-sequencing (BS-seq)-based, restriction enzyme-based and affinity-based approaches for analyzing DNA methylation have been established [24, 25], in addition to methods to identify genomic regions with low-nucleosomal content (for example, DNAse I hypersensitivity assay) [26] and to probe long-range chromosomal interactions (such as chromosomal conformation capture or 3C [27]).
Although these approaches were first established for low- to medium-throughput studies (for example, interrogation of a selected subset of genomic loci), recent breakthroughs in next-generation sequencing have allowed rapid adaptation and expansion of existing technologies for genome-wide analyses of chromatin features with an unprecedented resolution and coverage [28–44]. These methodologies include, among others, the ChIP-sequencing (ChIP-seq) approach to map histone modification patterns and occupancy of chromatin modifiers in a genome-wide manner, and MethylC sequencing (MethylC-seq) and BS-seq techniques for large-scale analysis of DNA methylation at single-nucleotide resolution. The main epigenomic technologies have been reviewed recently [45–47] and are listed in Table 1. The burgeoning field of epigenomics has already begun to reveal the enormous predictive power of chromatin profiling in annotating functional genomic elements in specific cell types. Indeed, chromatin signatures that characterize different classes of regulatory elements, including promoters, enhancers, insulators and long non-coding RNAs, have been uncovered (summarized in Table 2). Additional signatures that further specify and distinguish unique classes of genomic regulatory elements are likely to be discovered over the next few years. In the following section we summarize epigenomic studies of hESCs and pinpoint unique characteristics of the pluripotent cell epigenome that they reveal.
Epigenomic features of hESCs
ESCs provide a robust, genomically tractable in vitro model to investigate the molecular basis of pluripotency and embryonic development [1, 2]. In addition to sharing many fundamental properties with chromatin of somatic cells, chromatin of pluripotent cells appears to have unique features, such as the increased mobility of many structural chromatin proteins, including histones and heterochromatin protein 1 [48], and differences in nuclear organization suggestive of a less compacted chromatin structure [48–51]. Recent epigenomic profiling of hESCs has uncovered several characteristics that, although not absolutely unique to hESCs, appear particularly pervasive in these cells [52–54]. Below, we focus on these characteristics and their potential role in mediating the epigenetic plasticity of hESCs.
Bivalent domains at promoters
The term 'bivalent domains' is used to describe chromatin regions that are concomitantly modified by the trimethylation of lysine 4 of histone H3 (H3K4me3), a modification generally associated with transcriptional initiation, and trimethylation of lysine 27 of histone H3 (H3K27me3), a modification associated with Polycomb-mediated gene silencing. Although first described and most extensively characterized in mouse ESCs (mESCs) [55, 56], bivalent domains are also present in hESCs [57, 58], and in both species they mark transcription start sites of key developmental genes that are poorly expressed in ESCs, but induced upon differentiation. Albeit defined by the presence of H3K27me3 and H3K4me3, bivalent promoters are also characterized by other features, such as the occupancy of the histone variant H2AZ [59]. Upon differentiation, bivalent domains at specific promoters resolve into a transcriptionally active H3K4me3-marked monovalent state, or a transcriptionally silent H3K27me3-marked monovalent state, depending on the lineage commitment [42, 56]. However, a subset of bivalent domains is retained upon differentiation [42, 60], and bivalently marked promoters have been observed in many progenitor cell populations, perhaps reflecting their remaining epigenetic plasticity [60]. Nevertheless, promoter bivalency seems considerably less abundant in differentiated cells, and appears to be further diminished in unipotent cells [42, 54, 56]. These observations led to the hypothesis that bivalent domains are important for pluripotency, allowing early developmental genes to remain silent yet able to rapidly respond to differentiation cues. A similar function of promoter bivalency can be hypothesized for multipotent or oligopotent progenitor cell types. However, it needs to be more rigorously established how many of the apparently 'bivalent' promoters observed in progenitor cells truly posses this chromatin state, and how many reflect heterogeneity of the analyzed cell populations, in which some cells display H4K4me3-only and others H3K27me3-only signatures at specific promoters.
Poised enhancers
In multicellular organisms, distal regulatory elements, such as enhancers, play a central role in cell-type and signaling-dependent gene regulation [61, 62]. Although embedded within the vast non-coding genomic regions, active enhancers can be identified by epigenomic profiling of certain histone modifications and chromatin regulators [63–65]. A recent study revealed that unique chromatin signatures distinguish two functional enhancer classes in hESCs: active and poised [66]. Both classes are bound by coactivators (such as p300 and BRG1) and marked by H3K4me1, but while the active class is enriched in acetylation of lysine 27 of histone H3 (H3K27ac), the poised enhancer class is marked by H3K27me3 instead. Active enhancers are typically associated with genes expressed in hESCs and in the epiblast, whereas poised enhancers are located in proximity to genes that are inactive in hESCs, but which play critical roles during early stages of post-implantation development (for example, gastrulation, neurulation, early somitogenesis). Importantly, upon signaling stimuli, poised enhancers switch to an active chromatin state in a lineage-specific manner and are then able to drive cell-type-specific gene expression patterns. It remains to be determined whether H3K27me3-mediated enhancer poising represents a unique feature of hESCs. Recent work by Creighton et al. [67] suggests that poised enhancers are also present in mESCs and in various differentiated mouse cells, although in this case the poised enhancer signature did not involve H3K27me3, but H3K4me1 only. Nevertheless, our unpublished data indicate that, similar to the bivalent domains at promoters, simultaneous H3K4me1/H3K27me3 marking at enhancers is much less prevalent in more restricted cell types compared with both human and mouse ESCs (A Rada-Iglesias, R Bajpai and J Wysocka, unpublished observations). Future studies should clarify whether poised enhancers are marked by the same chromatin signature in hESCs, mESCs and differentiated cell types, and evaluate the functional relevance of the Polycomb-mediated H3K27 methylation at enhancers.
Unique DNA methylation patterns
Mammalian DNA methylation occurs at position 5 of cytosine residues, generally in the context of CG dinucleotides (that is, CpG dinucleotides), and has been associated with transcriptional silencing both at repetitive DNA, including transposon elements, and at gene promoters [13, 14]. Initial DNA methylation studies of mESCs revealed that most CpG-island-rich gene promoters, which are typically associated with house-keeping and developmental genes, are DNA hypomethylated, whereas CpG-island-poor promoters, typically associated with tissue-specific genes, are hypermethylated [41, 60]. Moreover, methylation of H3K4 at both promoter-proximal and distal regulatory regions is anti-correlated with their DNA methylation level, even at CpG-island-poor promoters [60]. Nevertheless, these general correlations are not ESC-specific features as they have also been observed in a variety of other cell types [25, 60, 68]. On the other hand, recent comparisons of DNA methylation in early pre- and postimplantation mouse embryos with those of mESCs revealed that, surprisingly, mESCs accumulate promoter DNA methylation that is more characteristic of the postimplantation stage embryos rather than the blastocyst from which they are derived [69].
Although the coverage and resolution of mammalian DNA methylome maps have been steadily increasing, whole-genome analyses of human methylomes at single-nucleotide resolution require an enormous sequencing effort and have been reported only recently [70]. These analyses revealed that in hESCs, but not in differentiated cells, a significant proportion (approximately 25%) of methylated cytosines are found in a non-CG context. Non-CG methylation is a common feature of plant epigenomes [40] and, while it has been previously reported to occur in mammalian cells [71], its contribution to as much as a quarter of all cytosine methylation in hESCs had not been anticipated. It remains to be established whether non-CG methylation in hESCs is functionally relevant or, alternatively, is simply a by-product of high levels of de novo DNA methyltransferases and a hyperdynamic chromatin state that characterizes hESCs [49, 50, 72]. Regardless, its prevalence in hESC methylomes emphasizes unique properties of pluripotent cell chromatin. However, one caveat to the aforementioned study and all other BS-seq-based analyses of DNA methylation is their inability to distinguish between methylcytosine (5mC) and hydroxymethylcytosine (5hmC), as both are refractory to bisulfite conversion [15, 73], and thus it remains unclear how much of what has been mapped as DNA methylation in fact represents hydroxymethylation.
DNA hydroxymethylation
Another, previously unappreciated modification of DNA, hydroxymethylation, has become a subject of considerable attention. DNA hydroxymethylation is mediated by the TET family enzymes [15], which convert 5mC to 5hmC. Recent studies have shown that mESCs express high levels of TET proteins, and consequently their chromatin is 5hmC-rich [74, 75], a property that, to date, has only been observed in a limited number of other cell types - for example, in Purkinje neurons [76]. Although the functionality of 5hmC is still unclear, it has been suggested that it represents a first step in either active or passive removal of DNA methylation from select genomic loci. New insights into 5hmC genomic distribution in mESCs have been obtained from studies that utilized immunoprecipitation with 5hmC-specific antibodies coupled to next-generation sequencing or microarray technology, respectively [77, 78], revealing that a significant fraction of 5hmC occurs within gene bodies of transcriptionally active genes and, in contrast to 5mC, also at CpG-rich promoters [77], where it overlaps with the occupancy of the Polycomb complex PRC2 [78]. Intriguingly, a significant fraction of the intra-genic 5hmC occurs within a non-CG context [77], which prompts investigating whether a subset of the reported non-CG methylation in hESCs might actually represent 5hmC. Future studies should establish whether hESCs show a similar 5hmC distribution to mESCs. More importantly, it will be essential to re-evaluate the extent to which cytosine residues that have been mapped as methylated in hESCs are indeed hydroxymethylated, and to determine the functional relevance of this novel epigenetic mark.
Reduced genomic blocks marked by repressive histone modifications
A comprehensive study of epigenomic profiles in hESCs and human fibroblasts showed that, in differentiated cells, regions enriched in histone modifications associated with heterochromatin formation and gene repression, such as H3K9me2/3 and H3K27me3, are significantly expanded [79]. These two histone methylation marks cover only 4% of the hESC genome, but well over 10% of the human fibroblast genome. Parallel observations have been made independently in mice, where large H3K9me2-marked regions are more frequent in adult tissues in comparison with mESCs [80]. Interestingly, H3K9me2-marked regions largely overlap with the recently described nuclear lamina-associated domains [81], suggesting that the appearance or expansion of the repressive histone methylation marks might reflect a profound three-dimensional reorganization of chromatin during differentiation [82]. Indeed, heterochromatic foci increase in size and number upon ESC differentiation, and it has been proposed that an 'open', hyperdynamic chromatin structure is a crucial component of pluripotency maintenance [48–50].
Are hESCs and iPSCs epigenetically equivalent?
Since Yamanaka's seminal discovery in 2006 showing that introduction of the four transcription factors Oct4, Sox2, Klf4 and c-Myc is sufficient to reprogram fibroblasts to a pluripotent state, progress in the iPSC field has been breathtaking [4, 83, 84]. iPSCs have now been generated from a variety of adult and fetal somatic cell types using a myriad of alternative protocols [3, 6, 7]. Remarkably, the resulting iPSCs seem to share phenotypic and molecular properties of ESCs; these properties include pluripotency, self-renewal and similar gene expression profiles. However, an outstanding question remains: to what extent are hESCs and iPSCs functionally equivalent? The most stringent pluripotency assay, tetraploid embryo complementation, demonstrated that mouse iPSCs can give rise to all tissues of the embryo proper [85, 86]. On the other hand, many iPSC lines do not support tetraploid complementation, and those that do remain quite inefficient in comparison with mESCs [85, 87]. Initial genome-wide comparisons between ESCs and iPSCs focused on gene expression profiles, which reflect the transcriptional state of a given cell type, but not its developmental history or differentiation potential [4, 84, 88]. These additional layers of information can be uncovered, at least partially, by examining epigenetic landscapes. In this section, we summarize studies comparing DNA methylation and histone modification patterns in ESCs and iPSCs.
Sources of variation in iPSC and hESC epigenetic landscapes
Bird's eye view comparisons show that all major features of the hESC epigenome are re-established in iPSCs [89, 90]. On the other hand, when more subtle distinctions are considered, recent studies have reported differences between iPSC and hESC DNA methylation and gene expression patterns [90–94]. Potential sources of these differences can be largely divided into three groups: (i) experimental variability in cell line derivation and culture; (ii) genetic variation among cell lines; and (iii) systematic differences representing hotspots of aberrant epigenomic reprogramming.
Although differences arising as a result of experimental variability do not constitute biologically meaningful distinctions between the two stem cell types, they can be informative when assessing the quality and differentiation potential of individual lines [91, 95]. The second source of variability is a natural consequence of the genetic variation among human cells or embryos from which iPSCs and hESCs are respectively derived. Genetic variation likely underlies many of the line-to-line differences in DNA and histone modification patterns, underscoring the need for using cohorts of cell lines and stringent statistical analyses to draw systematic comparisons between hESCs, healthy donor-derived iPSCs, and disease-specific iPSCs. In support of the significant impact of human genetic variation on epigenetic landscapes, recent studies of specific chromatin features in lymphoblastoid cells [96, 97] isolated from related and unrelated subjects showed that individual, as well as allele-specific, heritable differences in chromatin signatures can be largely explained by the underlying genetic variants. Although genetic differences make comparisons between hESC and iPSC lines less straightforward, we will discuss later how these can be harnessed to uncover the role of specific regulatory sequence variants in human disease. Finally, systematic differences between hESC and iPSC epigenomes may arise through the incomplete erasure of marks characteristic of the somatic cell type of origin (somatic memory) during iPSC reprogramming, or defects in the re-establishment of hESC-like patterns in iPSCs, or as a result of selective pressure during reprogramming and the appearance of iPSC-specific signatures [90, 98]. Regardless of the underlying sources of variation, understanding epigenetic differences between hESC and iPSC lines will be essential for harnessing the potential of these cells in regenerative medicine.
Remnants of the somatic cell epigenome in iPSCs: lessons from DNA methylomes
Studies of stringently defined models of mouse reprogramming have shown that cell-type-of-origin-specific differences in gene expression and differentiation potential exist in early passage iPSCs, leading to the hypothesis that an epigenetic memory of previous fate persists in these cells [98, 99]. This epigenetic memory has been attributed to the presence of residual somatic DNA methylation in iPSCs, most of which is retained within regions located outside of, but in proximity to, CpG islands, at so-called 'shores' [98, 100]. The incomplete erasure of somatic methylation appears to predispose iPSCs to differentiation into fates related to the cell type of origin, while restricting differentiation towards other lineages. Importantly, this residual memory of past fate appears to be transient, and diminishes upon continuous passaging, serial reprogramming or treatment with small molecule inhibitors of histone deacetylase or DNA methyltransferase activity [98, 99]. These results suggest that remnants of somatic DNA methylation are not actively maintained in iPSCs during replication and thus can be erased through cell division.
More recently, whole-genome, single-base-resolution DNA methylome maps have been generated for five distinct human iPSC lines and compared with those of hESCs and somatic cells [90]. That study demonstrated that although the hESC and iPSC DNA methylation landscapes are remarkably similar overall, hundreds of differentially methylated regions (DMRs) exist. Nevertheless, only a small fraction of DMRs represents failure in erasure of somatic DNA methylation, whereas the vast majority corresponds to either hypomethylation (defects in the methylation of genomic regions that are marked in hESCs) or the appearance of iPSC-specific methylation patterns, not present in hESCs or the somatic cell type of origin. Moreover, these DMRs are likely to be resistant to passaging, as the methylome analyses were performed using relatively late passage iPSCs [80]. Due to a limited number of iPSC and hESC lines used in the study, genetic and experimental variation among individual lines may be a big contributor to the reported DMRs. However, a significant subset of DMRs is shared among iPSC lines of different genetic background and cell type of origin, and is transmitted through differentiation, suggesting that at least some DMRs may represent non-stochastic epigenomic hotspots that are refractive to reprogramming.
Reprogramming resistance of subtelomeric and subcentromeric regions?
In addition to erasing somatic epigenetic marks, an essential component of reprogramming is the faithful re-establishment of hESC-like epigenomic features. Although, as discussed above, most of the DNA methylation is correctly re-established during reprogramming, large megabase-scale regions of reduced methylation can be detected in iPSCs, often within the vicinity of centromeres and telomeres [90]. Biased depletion of DNA methylation from subcentromeric and subtelomeric regions correlates with blocks of H3K9me3 that mark these loci in iPSCs and somatic cells, but not in hESCs [79, 90]. Aberrant DNA methylation in proximity to centromeres and telomeres suggests that these chromosomal territories may have features that render them more resistant to epigenetic changes. Intriguingly, histone variant H3.3, which is generally implicated in transcription-associated and replication-independent histone deposition, was recently found to also occupy subtelomeric and subcentromeric regions in mESCs and mouse embryo [36, 101, 102]. It has been previously suggested that H3.3 plays a critical role in the maintenance of transcriptional memory during reprogramming of somatic nuclei by the egg environment (that is, reprogramming by somatic cell nuclear transfer) [103], and it is tempting to speculate that a similar mechanism may contribute to the resistance of the subtelomeric and subcentromeric regions to reprogramming in iPSCs.
Anticipating future fates: reprogramming at regulatory elements
Pluripotent cells are in a state of permanent anticipation of many alternative developmental fates, and this is reflected in the prevalence of the poised promoters and enhancers in their epigenomes [42, 66]. Although multiple studies have demonstrated that bivalent domains at promoters are re-established in iPSCs with high fidelity [89], the extent to which chromatin signatures associated with poised developmental enhancers in hESCs are recapitulated in iPSCs remains unclear. However, the existence of a large class of poised developmental enhancers linked to genes that are inactive in hESCs, but involved in postimplantation steps of human embryogenesis [66], suggest that proper enhancer rewiring to a hESC-like state may be central to the differentiation potential of iPSCs. Defective epigenetic marking of developmental enhancers to a poised state may result in impaired or delayed ability of iPSCs to respond to differentiation cues, without manifesting itself at the transcriptional or promoter modification level in the undifferentiated state. Therefore, we would argue that epigenomic profiling of enhancer repertoires should be a critical component in evaluating iPSC quality and differentiation potential (Figure 1) and could be incorporated into already existing pipelines [91, 95].

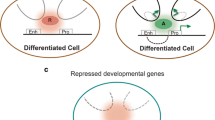
Epigenomics as a tool to assess iPSC identity. Chromatin signatures obtained by epigenomic profiling of a cohort of human embryonic stem cell (hESC) lines can be used to generate hESC reference epigenomes (left panels). The extent of reprogramming and differentiation potential of individual induced pluripotent stem cell (iPSC) lines can be assessed by comparing iPSC epigenomes (right panels) to the reference hESC epigenomes. (a-c) Such comparisons should evaluate epigenetic states at regulatory elements of self-renewal genes that are active in hESCs (a), developmental genes that are poised in hESCs (b), and tissue-specific genes that are inactive in hESCs, but are expressed in the cell type of origin used to derive iPSC (c). H3K4me1, methylation of lysine 4 of histone H3; H3K4me3, trimethylation of lysine 4 of histone H3; H3K27ac, acetylation of lysine 27 of histone H3; H3K27me3, trimethylation of lysine 27 of histone H3; meC, methylcytosine.
Relevance of epigenomics for human disease and regenerative medicine
In this section, we envision how recent advances in epigenomics can be used to gain insight into human development and disease, and to facilitate the transition of stem cell technologies towards clinical applications.
Using epigenomics to predict developmental robustness of iPSC lines for translational applications
As discussed earlier, epigenomic profiling can be used to annotate functional genomic elements in a genome-wide and cell-type specific manner. Distinct chromatin signatures can distinguish active and poised enhancers and promoters, identify insulator elements and uncover non-coding RNAs transcribed in a given cell type [42, 56, 63, 64, 66, 104, 105] (Table 2). Given that developmental potential is likely to be reflected in the epigenetic marking of promoters and enhancers linked to poised states, epigenomic maps should be more predictive of iPSC differentiation capacity than transcriptome profiling alone (Figure 1). However, before epigenomics can be used as a standard tool in assessing iPSC and hESC quality in translational applications, the appropriate resources need to be developed. For example, although ChIP-seq analysis of chromatin signatures is extremely informative, its reliance on antibody quality requires the development of renewable, standardized reagents. Also, importantly, to assess the significance of epigenomic pattern variation, sufficient numbers of reference epigenomes need to be obtained from hESC and iPSC lines that are representative of genetic variation and have been rigorously tested in a variety of differentiation assays. The first forays towards the development of such tools and resources have already been made [89, 91, 106, 107].
Annotating regulatory elements that orchestrate human differentiation and development
As a result of ethical and practical limitations, we know very little about the regulatory mechanisms that govern early human embryogenesis. hESC-based differentiation models offer a unique opportunity to isolate and study cells that correspond to transient progenitor states arising during human development. Subsequent epigenomic profiling of hESCs that have been differentiated in vitro along specific lineages can be used to define the functional genomic regulatory space, or 'regulatome', of a given cell lineage (Figure 2a). This approach is particularly relevant for genome-wide identification of tissue-specific enhancers and silencers, which are highly variable among different, even closely related, cell types. Characterizing cell-type-specific regulatomes will be useful for comparative analyses of gene expression circuitries. In addition, through bioinformatic analysis of the underlying DNA sequence, they can be used to predict novel master regulators of specific cell fate decisions, and these can then serve as candidates in direct transdifferentiation approaches. Moreover, mapping enhancer repertoires provides an enormous resource for the development of reporters for isolation and characterization of rare human cell populations, such as the progenitor cells that arise only transiently in the developmental process [66]. Ultimately, this knowledge will allow refinement of the current differentiation protocols and derivation of well-defined, and thus safer and more appropriate, cells for replacement therapies [3, 108–110]. Furthermore, as discussed below, characterizing cell-type specific regulatomes will be essential for understanding non-coding variation in human disease.

The combination of stem cell models and epigenomics in studies of the role of non-coding mutations in human disease. Epigenomic analyses of cells derived through in vitro stem cell differentiation models can be used to define the functional regulatory space, or 'regulatome', of a given cell type and to study the significance of the non-coding genetic variation in human disease. (a) The vast non-coding fraction of the human genome can be significantly reduced by defining the regulatome of a given cell type via epigenomic profiling of chromatin signatures that define different types of regulatory elements, such as enhancers, promoters and insulators. Regulatome maps obtained in the disease-relevant cell types define genomic space that can be subsequently searched for the recurrent disease-associated genetic variants. (b) Most genetic variants associated with complex human diseases appear to reside in non-coding regions of the human genome. To assess functional consequences of such variants, disease-relevant cell types can be derived from healthy and disease-affected donor induced pluripotent stem cells (iPSCs) and epigenomic profiling can be used to evaluate how these genetic variants affect chromatin signatures, and transcription factor and coactivator occupancy at regulatory elements. CTCF, CCCTC-binding factor, insulator associated protein; ESC, embryonic stem cell; H3K4me1, methylation of lysine 4 of histone H3; H3K4me3, trimethylation of lysine 4 of histone H3; H3K27ac, acetylation of lysine 27 of histone H3; H3K27me3, trimethylation of lysine 27 of histone H3; meC, methylcytosine.
Cell-type-specific regulatomes as a tool for understanding the role of non-coding mutations in human disease
During the past few years, genome-wide association studies have dramatically expanded the catalog of genetic variants associated with some of the most common human disorders, such as various cancer types, type 2 diabetes, obesity, cardiovascular disease, Crohn's disease and cleft lip/palate [111–118]. One recurrent observation is that most disease-associated variants occur in non-coding parts of the human genome, suggesting a large non-coding component in human phenotypic variation and disease. Indeed, several studies document a critical role for genetic aberrations occurring within individual distal enhancer elements in human pathogenesis [119–121]. To date, the role of regulatory sequence mutation in human disease has not been systematically examined. However, given the rapidly decreasing cost of high-throughput sequencing and the multiple disease-oriented whole genome sequencing projects that are under way, the next years will bring the opportunity and challenge to ascribe functional significance to disease-associated non-coding mutations [122]. Doing so will require both an ability to identify and obtain cell types relevant to disease, and the ability to characterize their specific regulatomes.
We envision that combining pluripotent cell differentiation models with epigenomic profiling will provide an important tool for uncovering the role of non-coding mutations in human disease. For example, if the disease of interest affects a particular cell type that can be derived in vitro from hESCs, characterizing the reference regulatome of this cell type, as described above, will shrink the vast genomic regions that might be implicated in disease into a much smaller regulatory space that can be more effectively examined for recurrent variants that are associated with disease (Figure 2a). The function of these regulatory variants can be further studied using in vitro and in vivo models, of which iPSC-based 'disease in a dish' models appear particularly promising [123]. For example, disease-relevant cell types obtained from patient-derived and healthy-donor-derived iPSCs can be used to study the effects of the disease genotype on cell-type-specific regulatomes (Figure 2b). Moreover, given that many, if not most, regulatory variants are likely to be heterozygous in patients, loss or gain of chromatin features associated with those variants (such as p300 binding, histone modifications and nucleosome occupancy) can be assayed independently for each allele within the same iPSC line. Indeed, allele-specific sequencing assays are already being developed [42, 96, 97, 124] (Table 1). Moreover, these results can be compared with allele-specific RNA-seq transcriptome analyses from the same cells [125], yielding insights into the effects of disease-associated regulatory alleles on the transcription of genes located in relative chromosomal proximity [96, 125].
Conclusions and future perspective
Analyses of hESC and iPSC chromatin landscapes have already provided important insights into the molecular basis of pluripotency, reprogramming and early human development. Our current view of the pluripotent cell epigenome has been largely acquired due to recent advances in next-generation sequencing technologies, such as ChIP-seq or MethylC-seq. Several chromatin features, including bivalent promoters, poised enhancers and pervasive non-CG methylation seem to be more abundant in hESCs compared with differentiated cells. It will be important in future studies to dissect the molecular function of these epigenomic attributes and their relevance for hESC biology. Epigenomic tools are also being widely used in the evaluation of iPSC identity. In general, the epigenomes of iPSC lines seem highly similar to those of hESC lines, albeit recent reports suggest that differences in DNA methylation patterns exist between the two pluripotent cell types. It will be important to understand the origins of these differences (that is, somatic memory, experimental variability, genetic variation), as well as their impact on iPSC differentiation potential or clinical applications. Moreover, additional epigenetic features other than DNA methylation should be thoroughly compared, including proper re-establishment of poised enhancer patterns. As a more complete picture of the epigenomes of ESCs, iPSCs and other cell types emerges, important lessons regarding early developmental decisions in humans will be learnt, facilitating not only our understanding of human development, but also the establishment of robust in vitro differentiation protocols. These advancements will in turn allow for generation of replacement cells for cellular transplantation approaches and for development of the appropriate 'disease in a dish' models. Within such models, epigenomic profiling could be especially helpful in understanding the genetic basis of complex human disorders, where most of the causative variants are predicted to occur within the vast non-coding fraction of the human genome.
Abbreviations
- BS-seq:
-
bisulfite sequencing
- ChIP:
-
chromatin immunoprecipitation
- ChIP-seq:
-
ChIP sequencing
- DMR:
-
differentially methylated region
- ESC:
-
embryonic stem cell
- hESC:
-
human embryonic stem cell
- H3K4me3:
-
trimethylation of lysine 4 of histone H3
- H3K27ac:
-
acetylation of lysine 27 of histone H3
- H3K27me3:
-
trimethylation of lysine 27 of histone H3
- iPSC:
-
induced pluripotent stem cell
- MethylC-seq:
-
MethylC sequencing
- 5mC:
-
methylcytosine
- 5hmC:
-
hydroxymethylcytosine
- PTM:
-
post-translational modification.
References
Hanna JH, Saha K, Jaenisch R: Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 2010, 143: 508-525. 10.1016/j.cell.2010.10.008.
Jaenisch R, Young R: Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008, 132: 567-582. 10.1016/j.cell.2008.01.015.
Gonzalez F, Boue S, Belmonte JC: Methods for making induced pluripotent stem cells: reprogramming a la carte. Nat Rev Genet. 2011, 12: 231-242.
Takahashi K, Yamanaka S: Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006, 126: 663-676. 10.1016/j.cell.2006.07.024.
Yamanaka S: Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell. 2007, 1: 39-49. 10.1016/j.stem.2007.05.012.
Yamanaka S, Blau HM: Nuclear reprogramming to a pluripotent state by three approaches. Nature. 2010, 465: 704-712. 10.1038/nature09229.
Hochedlinger K, Plath K: Epigenetic reprogramming and induced pluripotency. Development. 2009, 136: 509-523. 10.1242/dev.020867.
Busser BW, Bulyk ML, Michelson AM: Toward a systems-level understanding of developmental regulatory networks. Curr Opin Genet Dev. 2008, 18: 521-529. 10.1016/j.gde.2008.09.003.
Banaszynski LA, Allis CD, Lewis PW: Histone variants in metazoan development. Dev Cell. 2010, 19: 662-674. 10.1016/j.devcel.2010.10.014.
Goldberg AD, Allis CD, Bernstein E: Epigenetics: a landscape takes shape. Cell. 2007, 128: 635-638. 10.1016/j.cell.2007.02.006.
Jenuwein T, Allis CD: Translating the histone code. Science. 2001, 293: 1074-1080. 10.1126/science.1063127.
Kouzarides T: Chromatin modifications and their function. Cell. 2007, 128: 693-705. 10.1016/j.cell.2007.02.005.
Ooi SK, O'Donnell AH, Bestor TH: Mammalian cytosine methylation at a glance. J Cell Sci. 2009, 122: 2787-2791. 10.1242/jcs.015123.
Suzuki MM, Bird A: DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008, 9: 465-476.
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A: Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009, 324: 930-935. 10.1126/science.1170116.
Jiang C, Pugh BF: Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009, 10: 161-172.
Segal E, Widom J: What controls nucleosome positions?. Trends Genet. 2009, 25: 335-343. 10.1016/j.tig.2009.06.002.
Gondor A, Ohlsson R: Chromosome crosstalk in three dimensions. Nature. 2009, 461: 212-217. 10.1038/nature08453.
Murrell A, Rakyan VK, Beck S: From genome to epigenome. Hum Mol Genet. 2005, 14 Spec No 1: R3-R10.
Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA: The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010, 28: 1045-1048. 10.1038/nbt1010-1045.
ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H: Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007, 447: 799-816. 10.1038/nature05874.
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K: High-resolution profiling of histone methylations in the human genome. Cell. 2007, 129: 823-837. 10.1016/j.cell.2007.05.009.
Solomon MJ, Larsen PL, Varshavsky A: Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell. 1988, 53: 937-947. 10.1016/S0092-8674(88)90469-2.
Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL: A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992, 89: 1827-1831. 10.1073/pnas.89.5.1827.
Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D: Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005, 37: 853-862. 10.1038/ng1598.
Stalder J, Larsen A, Engel JD, Dolan M, Groudine M, Weintraub H: Tissue-specific DNA cleavages in the globin chromatin domain introduced by DNAase I. Cell. 1980, 20: 451-460. 10.1016/0092-8674(80)90631-5.
Dekker J, Rippe K, Dekker M, Kleckner N: Capturing chromosome conformation. Science. 2002, 295: 1306-1311. 10.1126/science.1067799.
Auerbach RK, Euskirchen G, Rozowsky J, Lamarre-Vincent N, Moqtaderi Z, Lefrancois P, Struhl K, Gerstein M, Snyder M: Mapping accessible chromatin regions using Sono-Seq. Proc Natl Acad Sci USA. 2009, 106: 14926-14931. 10.1073/pnas.0905443106.
Boyle AP, Song L, Lee BK, London D, Keefe D, Birney E, Iyer VR, Crawford GE, Furey TS: High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome Res. 2011, 21: 456-464. 10.1101/gr.112656.110.
Brinkman AB, Simmer F, Ma K, Kaan A, Zhu J, Stunnenberg HG: Whole-genome DNA methylation profiling using MethylCap-seq. Methods. 2010, 52: 232-236. 10.1016/j.ymeth.2010.06.012.
Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE: Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008, 452: 215-219. 10.1038/nature06745.
Deal RB, Henikoff JG, Henikoff S: Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 2010, 328: 1161-1164. 10.1126/science.1186777.
Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Gräf S, Johnson N, Herrero J, Tomazou EM, Thorne NP, Bäckdahl L, Herberth M, Howe KL, Jackson DK, Miretti MM, Marioni JC, Birney E, Hubbard TJ, Durbin R, Tavaré S, Beck S: A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol. 2008, 26: 779-785. 10.1038/nbt1414.
Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, Chew EG, Huang PY, Welboren WJ, Han Y, Ooi HS, Ariyaratne PN, Vega VB, Luo Y, Tan PY, Choy PY, Wansa KD, Zhao B, Lim KS, Leow SC, Yow JS, Joseph R, Li H, Desai KV, Thomsen JS, Lee YK, et al: An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009, 462: 58-64. 10.1038/nature08497.
Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD: FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17: 877-885. 10.1101/gr.5533506.
Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, Wen D, Chapgier A, DeKelver RC, Miller JC, Lee YL, Boydston EA, Holmes MC, Gregory PD, Greally JM, Rafii S, Yang C, Scambler PJ, Garrick D, Gibbons RJ, Higgs DR, Cristea IM, Urnov FD, Zheng D, Allis CD: Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010, 140: 678-691. 10.1016/j.cell.2010.01.003.
Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, Olshen A, Ballinger T, Zhou X, Forsberg KJ, Gu J, Echipare L, O'Geen H, Lister R, Pelizzola M, Xi Y, Epstein CB, Bernstein BE, Hawkins RD, Ren B, Chung WY, Gu H, Bock C, Gnirke A, Zhang MQ, Haussler D, et al: Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 2010, 28: 1097-1105. 10.1038/nbt.1682.
Ho L, Jothi R, Ronan JL, Cui K, Zhao K, Crabtree GR: An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc Natl Acad Sci USA. 2009, 106: 5187-5191. 10.1073/pnas.0812888106.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J: Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009, 326: 289-293. 10.1126/science.1181369.
Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR: Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008, 133: 523-536. 10.1016/j.cell.2008.03.029.
Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, Gnirke A, Jaenisch R, Lander ES: Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008, 454: 766-770.
Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O'Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE: Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007, 448: 553-560. 10.1038/nature06008.
Schnetz MP, Handoko L, Akhtar-Zaidi B, Bartels CF, Pereira CF, Fisher AG, Adams DJ, Flicek P, Crawford GE, Laframboise T, Tesar P, Wei CL, Scacheri PC: CHD7 targets active gene enhancer elements to modulate ES cell-specific gene expression. PLoS Genet. 2010, 6: e1001023-10.1371/journal.pgen.1001023.
Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K: Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008, 132: 887-898. 10.1016/j.cell.2008.02.022.
Bock C, Tomazou EM, Brinkman AB, Muller F, Simmer F, Gu H, Jager N, Gnirke A, Stunnenberg HG, Meissner A: Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 2010, 28: 1106-1114. 10.1038/nbt.1681.
Hawkins RD, Hon GC, Ren B: Next-generation genomics: an integrative approach. Nat Rev Genet. 2010, 11: 476-486.
Zhou VW, Goren A, Bernstein BE: Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011, 12: 7-18.
Meshorer E, Yellajoshula D, George E, Scambler PJ, Brown DT, Misteli T: Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell. 2006, 10: 105-116. 10.1016/j.devcel.2005.10.017.
Gaspar-Maia A, Alajem A, Meshorer E, Ramalho-Santos M: Open chromatin in pluripotency and reprogramming. Nat Rev Mol Cell Biol. 2011, 12: 36-47. 10.1038/nrm3036.
Meshorer E, Misteli T: Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol. 2006, 7: 540-546. 10.1038/nrm1938.
Park SH, Kook MC, Kim EY, Park S, Lim JH: Ultrastructure of human embryonic stem cells and spontaneous and retinoic acid-induced differentiating cells. Ultrastruct Pathol. 2004, 28: 229-238. 10.1080/01913120490515595.
Fisher CL, Fisher AG: Chromatin states in pluripotent, differentiated, and reprogrammed cells. Curr Opin Genet Dev. 2011, 21: 140-146. 10.1016/j.gde.2011.01.015.
Meissner A: Epigenetic modifications in pluripotent and differentiated cells. Nat Biotechnol. 2010, 28: 1079-1088. 10.1038/nbt.1684.
Sha K, Boyer LA: The chromatin signature of pluripotent cells. StemBook. Edited by: Girard L. 2008, Massachusetts: Harvard Stem Cell Institute
Azuara V, Perry P, Sauer S, Spivakov M, Jorgensen HF, John RM, Gouti M, Casanova M, Warnes G, Merkenschlager M, Fisher AG: Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006, 8: 532-538. 10.1038/ncb1403.
Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES: A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006, 125: 315-326. 10.1016/j.cell.2006.02.041.
Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA: Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell. 2007, 1: 299-312. 10.1016/j.stem.2007.08.003.
Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, Orlov YL, Sung WK, Shahab A, Kuznetsov VA, Bourque G, Oh S, Ruan Y, Ng HH, Wei CL: Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell. 2007, 1: 286-298. 10.1016/j.stem.2007.08.004.
Creyghton MP, Markoulaki S, Levine SS, Hanna J, Lodato MA, Sha K, Young RA, Jaenisch R, Boyer LA: H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell. 2008, 135: 649-661. 10.1016/j.cell.2008.09.056.
Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schubeler D: Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell. 2008, 30: 755-766. 10.1016/j.molcel.2008.05.007.
Bulger M, Groudine M: Enhancers: the abundance and function of regulatory sequences beyond promoters. Dev Biol. 2010, 339: 250-257. 10.1016/j.ydbio.2009.11.035.
Levine M: Transcriptional enhancers in animal development and evolution. Curr Biol. 2010, 20: R754-763. 10.1016/j.cub.2010.06.070.
Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B: Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009, 459: 108-112. 10.1038/nature07829.
Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B: Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007, 39: 311-318. 10.1038/ng1966.
Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, Afzal V, Ren B, Rubin EM, Pennacchio LA: ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009, 457: 854-858. 10.1038/nature07730.
Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J: A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011, 470: 279-283. 10.1038/nature09692.
Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R: Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010, 107: 21931-21936. 10.1073/pnas.1016071107.
Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D: Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007, 39: 457-466. 10.1038/ng1990.
Borgel J, Guibert S, Li Y, Chiba H, Schubeler D, Sasaki H, Forne T, Weber M: Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet. 2010, 42: 1093-1100. 10.1038/ng.708.
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR: Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009, 462: 315-322. 10.1038/nature08514.
Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R: Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci USA. 2000, 97: 5237-5242. 10.1073/pnas.97.10.5237.
Altun G, Loring JF, Laurent LC: DNA methylation in embryonic stem cells. J Cell Biochem. 2010, 109: 1-6.
Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A: The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 2010, 5: e8888-10.1371/journal.pone.0008888.
Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y: Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010, 466: 1129-1133. 10.1038/nature09303.
Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, Lahesmaa R, Orkin SH, Rodig SJ, Daley GQ, Rao A: Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011, 8: 200-213. 10.1016/j.stem.2011.01.008.
Kriaucionis S, Heintz N: The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009, 324: 929-930. 10.1126/science.1169786.
Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W: Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011, in press
Wu H, D'Alessio AC, Ito S, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y: Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011, 25: 679-684. 10.1101/gad.2036011.
Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, Antosiewicz-Bourget J, Ye Z, Espinoza C, Agarwahl S, Shen L, Ruotti V, Wang W, Stewart R, Thomson JA, Ecker JR, Ren B: Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010, 6: 479-491. 10.1016/j.stem.2010.03.018.
Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP: Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet. 2009, 41: 246-250. 10.1038/ng.297.
Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B: Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008, 453: 948-951. 10.1038/nature06947.
Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Gräf S, Flicek P, Kerkhoven RM, van Lohuizen M, Reinders M, Wessels L, van Steensel B: Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell. 2010, 38: 603-613. 10.1016/j.molcel.2010.03.016.
Takahashi K, Okita K, Nakagawa M, Yamanaka S: Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc. 2007, 2: 3081-3089. 10.1038/nprot.2007.418.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S: Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007, 131: 861-872. 10.1016/j.cell.2007.11.019.
Boland MJ, Hazen JL, Nazor KL, Rodriguez AR, Gifford W, Martin G, Kupriyanov S, Baldwin KK: Adult mice generated from induced pluripotent stem cells. Nature. 2009, 461: 91-94. 10.1038/nature08310.
Zhao XY, Li W, Lv Z, Liu L, Tong M, Hai T, Hao J, Guo CL, Ma QW, Wang L, Zeng F, Zhou Q: iPS cells produce viable mice through tetraploid complementation. Nature. 2009, 461: 86-90. 10.1038/nature08267.
Kang L, Wang J, Zhang Y, Kou Z, Gao S: iPS cells can support full-term development of tetraploid blastocyst-complemented embryos. Cell Stem Cell. 2009, 5: 135-138. 10.1016/j.stem.2009.07.001.
Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R: In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007, 448: 318-324. 10.1038/nature05944.
Guenther MG, Frampton GM, Soldner F, Hockemeyer D, Mitalipova M, Jaenisch R, Young RA: Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell. 2010, 7: 249-257. 10.1016/j.stem.2010.06.015.
Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O'Malley R, Castanon R, Klugman S, Downes M, Yu R, Stewart R, Ren B, Thomson JA, Evans RM, Ecker JR: Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011, 471: 68-73. 10.1038/nature09798.
Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, Gnirke A, Eggan K, Meissner A: Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011, 144: 439-452. 10.1016/j.cell.2010.12.032.
Chin MH, Mason MJ, Xie W, Volinia S, Singer M, Peterson C, Ambartsumyan G, Aimiuwu O, Richter L, Zhang J, Khvorostov I, Ott V, Grunstein M, Lavon N, Benvenisty N, Croce CM, Clark AT, Baxter T, Pyle AD, Teitell MA, Pelegrini M, Plath K, Lowry WE: Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009, 5: 111-123. 10.1016/j.stem.2009.06.008.
Chin MH, Pellegrini M, Plath K, Lowry WE: Molecular analyses of human induced pluripotent stem cells and embryonic stem cells. Cell Stem Cell. 2010, 7: 263-269. 10.1016/j.stem.2010.06.019.
Deng J, Shoemaker R, Xie B, Gore A, LeProust EM, Antosiewicz-Bourget J, Egli D, Maherali N, Park IH, Yu J, Daley GQ, Eggan K, Hochedlinger K, Thomson J, Wang W, Gao Y, Zhang K: Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009, 27: 353-360. 10.1038/nbt.1530.
Müller FJ, Schuldt BM, Williams R, Mason D, Altun G, Papapetrou EP, Danner S, Goldmann JE, Herbst A, Schmidt NO, Aldenhoff JB, Laurent LC, Loring JF: A bioinformatic assay for pluripotency in human cells. Nat Methods. 2011, 8: 315-317. 10.1038/nmeth.1580.
Birney E, Lieb JD, Furey TS, Crawford GE, Iyer VR: Allele-specific and heritable chromatin signatures in humans. Hum Mol Genet. 2010, 19: R204-209. 10.1093/hmg/ddq404.
McDaniell R, Lee BK, Song L, Liu Z, Boyle AP, Erdos MR, Scott LJ, Morken MA, Kucera KS, Battenhouse A, Keefe D, Collins FS, Willard HF, Lieb JD, Furey TS, Crawford GE, Iyer VR, Birney E: Heritable individual-specific and allele-specific chromatin signatures in humans. Science. 2010, 328: 235-239. 10.1126/science.1184655.
Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, Yabuuchi A, Takeuchi A, Cunniff KC, Hongguang H, McKinney-Freeman S, Naveiras O, Yoon TJ, Irizarry RA, Jung N, Seita J, Hanna J, Murakami P, Jaenisch R, Weissleder R, Orkin SH, Weissman IL, Feinberg AP, Daley GQ: Epigenetic memory in induced pluripotent stem cells. Nature. 2010, 467: 285-290. 10.1038/nature09342.
Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, Apostolou E, Stadtfeld M, Li Y, Shioda T, Natesan S, Wagers AJ, Melnick A, Evans T, Hochedlinger K: Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol. 2010, 28: 848-855. 10.1038/nbt.1667.
Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, Miller J, Schlaeger T, Daley GQ, Feinberg AP: Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009, 41: 1350-1353. 10.1038/ng.471.
Santenard A, Ziegler-Birling C, Koch M, Tora L, Bannister AJ, Torres-Padilla ME: Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nat Cell Biol. 2010, 12: 853-862. 10.1038/ncb2089.
Wong LH, Ren H, Williams E, McGhie J, Ahn S, Sim M, Tam A, Earle E, Anderson MA, Mann J, Choo KH: Histone H3.3 incorporation provides a unique and functionally essential telomeric chromatin in embryonic stem cells. Genome Res. 2009, 19: 404-414.
Ng RK, Gurdon JB: Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat Cell Biol. 2008, 10: 102-109. 10.1038/ncb1674.
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES: Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009, 458: 223-227. 10.1038/nature07672.
Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B: Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007, 128: 1231-1245. 10.1016/j.cell.2006.12.048.
Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, Cheung MS, Day DS, Gadel S, Gorchakov AA, Gu T, Kharchenko PV, Kuan S, Latorre I, Linder-Basso D, Luu Y, Ngo Q, Perry M, Rechtsteiner A, Riddle NC, Schwartz YB, Shanower GA, Vielle A, Ahringer J, Elgin SC, Kuroda MI, Pirrotta V, Ren B, Strome S, Park PJ, et al: An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 2011, 18: 91-93. 10.1038/nsmb.1972.
Fuchs SM, Krajewski K, Baker RW, Miller VL, Strahl BD: Influence of combinatorial histone modifications on antibody and effector protein recognition. Curr Biol. 2011, 21: 53-58. 10.1016/j.cub.2010.11.058.
Lee G, Studer L: Induced pluripotent stem cell technology for the study of human disease. Nat Methods. 2010, 7: 25-27. 10.1038/nmeth.f.283.
Sun N, Longaker MT, Wu JC: Human iPS cell-based therapy: considerations before clinical applications. Cell Cycle. 2010, 9: 880-885. 10.4161/cc.9.5.10827.
Zhang F, Citra F, Wang DA: Prospects of induced pluripotent stem cell technology in regenerative medicine. Tissue Eng Part B Rev. 2011, 17: 115-124.
The Wellcome Trust Case Control Consortium: Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007, 447: 661-678. 10.1038/nature05911.
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, NIDDK IBD Genetics Consortium, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I: Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008, 40: 955-962. 10.1038/ng.175.
Mangold E, Ludwig KU, Birnbaum S, Baluardo C, Ferrian M, Herms S, Reutter H, de Assis NA, Chawa TA, Mattheisen M, Steffens M, Barth S, Kluck N, Paul A, Becker J, Lauster C, Schmidt G, Braumann B, Scheer M, Reich RH, Hemprich A, Pötzsch S, Blaumeiser B, Moebus S, Krawczak M, Schreiber S, Meitinger T, Wichmann HE, Steegers-Theunissen RP, Kramer FJ: Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nat Genet. 2010, 42: 24-26. 10.1038/ng.506.
Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research, Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson Boström K, Isomaa B, Lettre G, Lindblad U, Lyon HN, Melander O, Newton-Cheh C, Nilsson P, Orho-Melander M, Råstam L, Speliotes EK, Taskinen MR: Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007, 316: 1331-1336.
Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J: A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007, 316: 1341-1345. 10.1126/science.1142382.
Scuteri A, Sanna S, Chen WM, Uda M, Albai G, Strait J, Najjar S, Nagaraja R, Orrú M, Usala G, Dei M, Lai S, Maschio A, Busonero F, Mulas A, Ehret GB, Fink AA, Weder AB, Cooper RS, Galan P, Chakravarti A, Schlessinger D, Cao A, Lakatta E, Abecasis GR: Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007, 3: e115-10.1371/journal.pgen.0030115.
Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, Crenshaw A, Cancel-Tassin G, Staats BJ, Wang Z, Gonzalez-Bosquet J, Fang J, Deng X, Berndt SI, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cussenot O, Valeri A, et al: Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008, 40: 310-315. 10.1038/ng.91.
Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, Crenshaw A, Cancel-Tassin G, Staats BJ, Wang Z, Gonzalez-Bosquet J, Fang J, Deng X, Berndt SI, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cussenot O, Valeri A: Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet. 2007, 39: 989-994. 10.1038/ng2089.
Epstein DJ: Cis-regulatory mutations in human disease. Brief Funct Genomic Proteomic. 2009, 8: 310-316. 10.1093/bfgp/elp021.
Gaulton KJ, Nammo T, Pasquali L, Simon JM, Giresi PG, Fogarty MP, Panhuis TM, Mieczkowski P, Secchi A, Bosco D, Berney T, Montanya E, Mohlke KL, Lieb JD, Ferrer J: A map of open chromatin in human pancreatic islets. Nat Genet. 2010, 42: 255-259. 10.1038/ng.530.
Wasserman NF, Aneas I, Nobrega MA: An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res. 2010, 20: 1191-1197. 10.1101/gr.105361.110.
Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, Kellis M, Bernstein BE: Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011, 473: 43-49. 10.1038/nature09906.
Zhu H, Lensch MW, Cahan P, Daley GQ: Investigating monogenic and complex diseases with pluripotent stem cells. Nat Rev Genet. 2011, 12: 266-275. 10.1038/nrg2951.
Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, Habegger L, Rozowsky J, Shi M, Urban AE, Hong MY, Karczewski KJ, Huber W, Weissman SM, Gerstein MB, Korbel JO, Snyder M: Variation in transcription factor binding among humans. Science. 2010, 328: 232-235. 10.1126/science.1183621.
Pastinen T: Genome-wide allele-specific analysis: insights into regulatory variation. Nat Rev Genet. 2010, 11: 533-538.
Acknowledgements
We thank members of the Wysocka laboratory for ideas and manuscript comments. We apologize to all those authors whose work was not cited because of space limitations. JW acknowledges grant CIRM RN1 00579-1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AR-I and JW conceived and wrote the manuscript together.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Rada-Iglesias, A., Wysocka, J. Epigenomics of human embryonic stem cells and induced pluripotent stem cells: insights into pluripotency and implications for disease. Genome Med 3, 36 (2011). https://doi.org/10.1186/gm252
Published:
DOI: https://doi.org/10.1186/gm252