
Abstract
Aberrant DNA methylation, in particular promoter hypermethylation and transcriptional silencing of tumor suppressor genes, has an important role in the development of many human cancers, including renal cell carcinoma (RCC). Indeed, apart from mutations in the well studied von Hippel-Lindau gene (VHL), the mutation frequency rates of known tumor suppressor genes in RCC are generally low, but the number of genes found to show frequent inactivation by promoter methylation in RCC continues to grow. Here, we review the genes identified as epigenetically silenced in RCC and their relationship to pathways of tumor development. Increased understanding of RCC epigenetics provides new insights into the molecular pathogenesis of RCC and opportunities for developing novel strategies for the diagnosis, prognosis and management of RCC.
Similar content being viewed by others


Background
Epidemiology and pathogenesis of renal cell carcinoma
Kidney cancers account for about 2% of all cancers, and more than 200,000 new cases of kidney cancer are diagnosed worldwide each year [1]. The most common form of kidney cancer in adults is renal cell carcinoma (RCC). Most RCC cases (approximately 75%) are classified as clear cell (conventional) RCC (ccRCC), and the next most frequent subtype is papillary RCC (pRCC; approximately 15% of all cases) [2]. The most common genetic event in the evolution of sporadic ccRCC is inactivation of the von Hippel-Lindau (VHL) tumor suppressor gene (TSG) [3–6]. VHL inactivation leads to stabilization of the hypoxia-inducible transcription factors HIF-1 and HIF-2 and activation of a wide repertoire of hypoxia response genes [7]. The frequency of VHL mutations in sporadic ccRCC has been reported to be as high as 75% (although VHL mutations are rare in non-clear-cell forms of RCC). In addition to VHL mutations, VHL allele loss of 3p25, resulting in biallelic VHL inactivation, is the most frequent copy number abnormality in ccRCC (as predicted by a classical 'two hit' model of tumorigenesis, where loss of the second allele of a key tumour suppressor is required for tumour formation to occur) [8, 9].
Although the VHL mutations in primary RCC were detected about 16 years ago, attempts to identify other frequently mutated RCC genes have been unsuccessful, with none of the thousands of genes tested so far mutated in over 15% of tumors [10]. TSG inactivation may result from genetic or epigenetic events, and it is well recognized that epigenetic silencing of TSGs has a significant role in the pathogenesis of many, if not all, human cancers. Indeed, promoter methylation and epigenetic silencing of VHL in RCC [5] was one of the first examples of this phenomenon and so far approximately 60 genes have been suggested to be epigenetically dysregulated in RCC (Table 1).
Epigenetics and cancer
There are two major, interrelated modes of epigenetic regulation in the mammalian genome: cytosine methylation and histone modification. Only cytosine bases located 5' to a guanosine can be methylated, and CpG dinucleotides are generally underrepresented in the genome. However, short regions found frequently in proximal promoter regions are CpG rich [11]. These regions (CpG islands, 0.4 to 4 kb long and found in over 50% of all genes) are generally unmethylated in normal cells but may be hypermethylated in tumors, where CpG island methylation is also associated with histone modification and chromatin remodeling resulting in transcriptional silencing [12–16]. Epigenetic states are, like gene mutations, inherited in cell division but, unlike mutations, DNA methylation and other epigenetic changes are potentially reversible [17, 18].
In a non-disease setting, gene silencing by promoter methylation occurs to regulate the expression of germline and tissue-specific genes and to regulate the monoallelic expression of imprinted genes [19–22]. However, in the past decade it has become accepted that aberrant promoter methylation and the resultant gene silencing can provide a selective advantage to neoplastic cells in the same manner that mutations do [22–26]. Thus, epigenetic silencing of 'gatekeeper' or 'caretaker' TSGs can occur frequently at the earliest stages of cancer initiation, resulting in the clonal evolution of a population of cells at risk of obtaining further genetic or epigenetic lesions [27, 28]. In inherited cancer syndromes such as von Hippel-Lindau disease (associated with susceptibility to RCC) de novo VHL promoter hypermethylation can provide the 'second hit' that initiates tumor development [29]. In such cases methylation is specific to the wild-type allele, suggesting clonal selection for the epigenetic loss of expression.
A survey of methylated genes in RCC
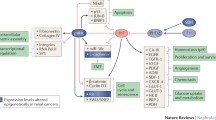
In order to catalog candidate TSGs reported to show tumor-specific region hypermethylation in RCC, we searched PubMed and other online databases (such as PubMeth) [30]. Of the 58 genes that were identified as being methylated in RCC (Table 1, Figure 1; see Table 1 for full gene names), 43 had a mean combined methylation/mutation rate of over 20% and the characteristics of these genes were analyzed in further detail (although 31 genes had been reported only by a single study).

Genes methylated in RCC are distributed across the genome. However, there is a concentration of silenced genes at 3p (see text for details). Methylated genes are also concentrated at chromosome 17 and both loss and gain of chromosome 17 have been reported in RCC.
Chromosome 3p tumor suppressors
Deletions of 3p are frequent in many adult cancers [31] and occur in 45 to 90% of sporadic RCCs [4, 32, 33]. Inactivation of the 3p25 TSG VHL is of critical importance to the pathogenesis of ccRCC and occurs in up to 86% of tumors [34]. Although VHL mutations are rare in non-clear-cell RCC, VHL methylation has been reported in pRCC and ccRCC [9, 35, 36]. VHL methylation does not associate with tumor stage, consistent with the interpretation that it is an early event in tumor formation [9, 37]. In addition to VHL, several other 3p candidate TSGs have been reported to be methylated in RCC (Figure 1). The RASSF1 gene maps to 3p21, a region of frequent allele loss in RCC and other cancers (including lung, bladder, breast and hepatocellular). Somatic RASSF1A mutations are infrequent in cancer [38], but RASSF1 is frequently methylated in sporadic RCC (and various other common cancers), either biallelically or as a second hit following 3p deletion [39, 40]. After VHL, RASSF1 methylation has been examined more than any other gene in sporadic RCC, the mean methylation frequency is 51% [34, 35, 38, 41–47]. In a study by Costa et al. [44], frequent RASSF1A methylation was detected in kidney tissue surrounding the excised tumor. Aberrant methylation in morphologically normal renal tissue adjacent to the tumor (but not in more distant normal tissue) has been interpreted as evidence that the TSG methylation is part of a 'field effect' at an early stage of tumorigenesis that produces a large number of cells with an initial epigenetic lesion that is then followed by additional genetic and/or epigenetic events that lead to tumor development. The candidate tumor suppressor gene TU3A (located at 3p21.1) is frequently downregulated in cancers, most notably prostate cancer [48] and astrocytoma [49]. In one study of 61 tumors, TU3A was methylated in 42% of ccRCC and 25% of pRCC [50].
The FHIT gene encodes a diadenosine 5',5'''-P1,P3-triphosphate hydrolase involved in purine metabolism. The gene encompasses the common fragile site FRA3B at 3p14. Loss of FHIT is common to many tumor types [51, 52]. In vivo, re-expression of FHIT has tumor suppressing activity [53]. FHIT promoter methylation is common (52 to 53%) in both ccRCC and pRCC [44, 54].
RARB regulates cell proliferation and differentiation and, in common with other 3p TSGs (RARB maps to 3p24), is frequently downregulated or lost in multiple tumor types. However, several small studies have found RARB to be methylated in less than 20% of RCC cases [35, 42, 44, 55].
WNT pathway regulators
Dysregulation of the WNT/β-catenin pathway is common in a variety of cancers, and oncogenic activation of this pathway drives the expression of genes that contribute to proliferation, survival and invasion [56, 57]. Inhibitors of WNT signaling can be divided into two functional classes: the SFRP proteins, which bind directly to WNT, preventing its binding to the FZ receptor [58], and the Dickkopf (DKK) proteins, which bind to the Low-density lipoprotein receptor-related protein 5 (LRP5)-LRP6 component of the Wnt receptor complex [59]. The SFRP1, SFRP2, SFRP4, SFRP5 and related WIF1 genes are all frequently methylated in RCC (47 to 73%) [60–64], as are the Dickkopf genes DKK1, DKK2 and DKK3 (44 to 58%) [63–66]. Recently, SFRP1 was shown to be overexpressed in metastatic RCC compared with non-metastatic tumors, in which expression was often attenuated by promoter methylation [67].
Epigenetics and familial RCC genes
As described above, germline VHL mutations cause inherited RCC and VHL inactivation is also critical to the development of most ccRCC. Similarly, a constitutional translocation associated with RCC susceptibility disrupted the NORE1A (RASSF5) and LSAMP1 genes, and both genes were epigenetically inactivated in sporadic RCC [68]. However, somatic inactivation (by mutation or methylation) of other genes associated with inherited kidney cancer, such as FLCN, FH and SDHB, is infrequent or absent (Table 1). Nevertheless, SPINT2 (HAI2), which encodes a secreted inhibitor of MET activity (activating mutations in the MET proto-oncogene are associated with familial pRCC, although somatic mutations are infrequent in sporadic pRCC [69, 70]), was found to be methylated in 30% of ccRCC and 45% of pRCC [71]. This observation demonstrates how TSG methylation can target familial RCC gene pathways. We note that several other epigenetically inactivated candidate TSGs, including members of the Wnt regulatory pathway [72], p16INK4a [73], CASP8 [74], GREM1 [75], RPRM [76], collagens [77], IGFBP1 [78], IGFBP3 [79] and PTGS2 [80], can be related to VHL-regulated pathways. However, genes involved in many other cellular processes have also been found to be epigenetically silenced in RCC (Table 1).
Identification of novel RCC TSGs by epigenetic analyses
Compared with the results of high-throughput sequencing studies of RCC [81], it seems that epigenetic studies have provided a much higher number of frequently inactivated candidate TSGs. Nevertheless, a combination of sequencing and epigenetic analysis provides the optimum strategy. Thus, although RASSF1A would not have been identified as an important RCC TSG by sequencing analysis alone, CDKN2A (which is mutated in approximately 10% of RCC and is the second most highly mutated gene in RCC [10]), is, on average, methylated in 11% of RCC [35, 36, 41, 43, 44, 55, 82], yielding a combined inactivation rate of about 21%. A wide variety of methodological approaches can be used to determine the promoter methylation status of candidate RCC TSGs and these have differing advantages and drawbacks (Tables 2 and 3). In addition to the detection of pathological promoter region methylation, it is important to demonstrate that this is associated with transcriptional silencing of the candidate TSG.
The functional epigenomics strategy uses 5-aza-2'-deoxycytidine treatment of cancer cell lines to identify genes whose expression is reactivated following demethylation. Although this strategy can provide an unbiased approach to identifying candidate epigenetically inactivated TSGs, only a minority of the re-expressed genes are ultimately proven to be silenced in primary tumors. Some techniques, such as methylation-specific PCR, can be very sensitive, and it is reassuring when results are available from a large number of tumors and multiple studies because the frequencies of methylation for individual genes can show considerable variation (Table 1). Such variation can reflect differences between cohorts of tumor samples or methylation detection methodologies, and only in a minority of cases are there data available from multiple studies and over 100 tumor samples. For less well studied genes the evidence for pathogenicity is strengthened by reports of frequent tumor-specific methylation (or mutations) in other tumor types; this is the case for BNC1 [83], PDLIM4 [84, 85], CST6 [86, 87]SLIT2 [88, 89], IGFBP3 [90, 91] and SPINT2 [92–94].
So far, epigenetic studies in RCC have concentrated on the methylation of CpG islands at or near to gene promoters. Recent studies in colorectal cancer have indicated that methylation extends well beyond discrete islands. Indeed, approximately 50% of these 'CpG island shores' were found more than 2 kb from the nearest annotated gene [95]. As with CpG island methylation, CpG shore methylation inversely correlates with gene expression. Further investigation of global genomic methylation patterns is necessary to elucidate the full role of epigenetic gene silencing (and oncogene activation) in RCC development. It is now accepted that in certain tumor types, colorectal being the best described, a subset of tumors show a CpG island methylator phenotype (CIMP+), which associates with specific lesions such as BRAF mutations and microsatellite instability [96]. However, the relevance of the CIMP+ phenotype to RCC has not yet been clearly defined [97]. The role of abnormal histone modification as an epigenetic factor in RCC development also remains to be investigated in depth. However, recent large-scale sequencing screens of RCC revealed mutations in the histone-modifying genes ubiquitously transcribed tetratricopeptide repeat gene on × chromosome (UTX), set domain-containing protein 2 (SETD2) and lysine-specific demethylase 5C (KDM5C, JARID1C), and that loss of these genes correlated with transcriptional deregulation [81, 98]. The interplay between erroneous histone modification and aberrant DNA methylation in the evolution of RCC merits further investigation.
Translational medicine and RCC epigenetics
Epigenetic biomarkers
Methylated TSGs provide attractive options for biomarkers for the detection and prognosis prediction of cancers, including RCC [99]. DNA-based assays are often more robust than RNA-based assays, and whereas the mutation spectrum causing TSG inactivation is usually diverse (which limits the utility of mutation-specific detection strategies for tumor screening programs), TSG inactivation by promoter hypermethylation provides a more homogeneous target for molecular screening strategies. So far, large-scale gene sequencing studies have demonstrated that, with the exception of VHL, there are no genes that are mutated very frequently, but a significant number of genes do show frequent tumor-specific methylation.
Early diagnosis of RCC can be challenging. The classical clinical symptoms and signs of renal cancer are usually present only with late disease, when prognosis is poor; these symptoms - pain, palpable flank mass and hematuria - are present in only approximately 10% of patients [100]. The aim is to detect RCC early when the tumor is still confined, as this has a significant impact on long-term disease-free survival. Although an increasing number of RCCs are detected as incidental findings on abdominal imaging, distinguishing benign and malignant masses in such a situation can be difficult. However, DNA can be detected from cells sloughed from the tumor into urine or blood, and three studies [41–43] have successfully detected the presence of promoter methylation, by methylation-specific PCR, from DNA extracted from serum and urine of patients with RCC. Methylation of the Wnt antagonists SFRP1, SFRP2, SFRP4, SFRP5, DKK3 and WIF1 was detected in tumor DNA in the serum of patients in whom those genes were methylated in their tumor. Moreover, the frequency of methylation detection in serum correlated significantly with increased grade and stage, suggesting that detection of these methylation-specific PCR products may be useful as markers of tumor progression [63]. Using a panel of previously identified RCC-specific methylated genes, two of these studies [41, 43] have found a strong correlation between tumor methylation and methylated DNA obtained from patient urine. Methylation was not found in control, age-matched urine samples. The panels of genes used in these studies included VHL, RASSF1, MGMT, GSTP1, p16INK4, p14ARF, APC and TIMP3. The specificity for genes such as VHL and RASSF1, which are frequently methylated and believed to be inactivated at an early stage of tumor development, suggests that methylation-specific PCR-based hypermethylation panel arrays could have potential as an economically viable early detection screen for patients presenting non-specific symptoms and for distinguishing benign and malign renal masses.
Only a few genes that might have potential as prognostic biomarkers have been analyzed in urine or blood from RCC patients. However, the tumor methylation status of several TSGs has been correlated with prognosis. Two independent studies [63, 64] have reported an inverse correlation between SFRP1 promoter methylation and patient survival (in vitro and in vivo assays both suggested that SFRP1 had tumor suppressing activity in RCC [62, 64]). Methylation of COL14A1 and BNC1 was significantly associated with a poorer prognosis and this was a better prognostic indicator than tumor stage or grade [64]. JUP methylation was detected in a very high proportion of tumors tested (91%) and was reported to be an independent indicator of disease progression and patient survival [101]. Similarly, a significant correlation between methylation of the bone morphogenetic protein antagonist GREM1 and tumor grade and stage and poor prognosis was reported [102], and methylation of TU3A was significantly associated with advanced tumor stage (later than stage T2) and poor survival [50]. The methylation status of several TSGs has been correlated with tumor pathological characteristics but not prognosis. HOXB13 methylation, for example, was correlated with tumor grade, stage, size and microvessel invasion [103], whereas DKK1 methylation correlated with increased pathological grade [66] and DKK2 methylation correlated with both increased stage and grade [65]. However, most of these studies require replication and, although RASSF1 methylation was reported to correlate with stage [44, 46] and grade [44], the largest study so far found no correlation with grade [39].
Clearly it is important that there should be further studies of potential methylated biomarkers in tumor tissue and urine and/or blood with the ultimate aim of producing a panel of biomarkers that will enable non-invasive detection, molecular staging and prediction of prognosis. As the number of potential methylated TSG biomarkers increases, it will be of great importance to assay these in a standardized manner in prospective studies to establish their clinical utility.
Promoter methylation as a target for therapy
The identification of frequently methylated RCC TSGs highlights critical pathways that could potentially be targeted for novel therapeutic interventions in RCC and other cancer types. In addition, there are less gene-specific approaches to epigenetic therapy. Decitabine, the clinical form of the demethylating agent 5-aza-2'-deoxycytidine, has been investigated in several clinical trials for neoplasia, and promising responses have been reported in hematological malignancies (such as myelodysplastic syndrome [18, 104, 105]), although the response rates seem to be lower for common solid tumors. However, epigenetic therapy to alter cancer methylation or histone modification status is an area of increasing clinical trial activity. Clearly, strategies such as tumor methylation profiling, which could identify cancer patients most likely to respond to such therapies, would be a major advance.
Future prospects
Technological advances are accelerating the pace of methylation profiling for common human cancers. The advent of high-throughput hybridization-based assays can allow the methylation status of around 14,000 genes to be analyzed simultaneously (although only a few CpGs are interrogated for each gene) and strategies based on second generation massively parallel sequencing technologies will undoubtedly provide a more complete assessment of RCC epigenetics and elucidate novel RCC TSGs. One advantage of these approaches over the older 'candidate gene epigenetic status approach' is that the simultaneous analysis of many genes allows a better comparison of TSG methylation frequencies for specific genes and is likely to facilitate comparison between different studies. With increasing numbers of methylated TSGs in RCC identified, our knowledge of the molecular pathogenesis of RCC will increase and with it the potential for developing novel biomarkers and potential therapeutic interventions.
Abbreviations
- ccRCC:
-
clear cell renal cell carcinoma
- pRCC:
-
papillary RCC
- RCC:
-
renal cell carcinoma
- TSG:
-
tumor suppressor gene
References
Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, Boyle P: Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol. 2007, 18: 581-592.
Mancini V, Battaglia M, Ditonno P, Palazzo S, Lastilla G, Montironi R, Bettocchi C, Cavalcanti E, Ranieri E, Selvaggi FP: Current insights in renal cell cancer pathology. Urol Oncol. 2008, 26: 225-238.
Clifford SC, Prowse AH, Affara NA, Buys CH, Maher ER: Inactivation of the von Hippel-Lindau (VHL) tumour suppressor gene and allelic losses at chromosome arm 3p in primary renal cell carcinoma: evidence for a VHL-independent pathway in clear cell renal tumourigenesis. Genes Chromosomes Cancer. 1998, 22: 200-209.
Foster K, Crossey PA, Cairns P, Hetherington JW, Richards FM, Jones MH, Bentley E, Affara NA, Ferguson-Smith MA, Maher ER: Molecular genetic investigation of sporadic renal cell carcinoma: analysis of allele loss on chromosomes 3p, 5q, 11p, 17 and 22. Br J Cancer. 1994, 69: 230-234.
Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM: Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA. 1994, 91: 9700-9704.
Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L: Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993, 260: 1317-1320.
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ: The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999, 399: 271-275.
Maher ER, Yates JR, Ferguson-Smith MA: Statistical analysis of the two stage mutation model in von Hippel-Lindau disease, and in sporadic cerebellar haemangioblastoma and renal cell carcinoma. J Med Genet. 1990, 27: 311-314.
Banks RE, Tirukonda P, Taylor C, Hornigold N, Astuti D, Cohen D, Maher ER, Stanley AJ, Harnden P, Joyce A, Knowles M, Selby PJ: Genetic and epigenetic analysis of von Hippel-Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer Res. 2006, 66: 2000-2011.
Catalogue of Somatic Mutations in Cancer. [http://www.sanger.ac.uk/genetics/CGP/cosmic/]
Bird A: DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16: 6-21.
Nguyen CT, Gonzales FA, Jones PA: Altered chromatin structure associated with methylation-induced gene silencing in cancer cells: correlation of accessibility, methylation, MeCP2 binding and acetylation. Nucleic Acids Res. 2001, 29: 4598-4606.
Fahrner JA, Eguchi S, Herman JG, Baylin SB: Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 2002, 62: 7213-7218.
Ballestar E, Paz MF, Valle L, Wei S, Fraga MF, Espada J, Cigudosa JC, Huang TH-M, Esteller M: Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. EMBO J. 2003, 22: 6335-6345.
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Pérez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M: Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005, 37: 391-400.
Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang S-HL, Watkins DN, Herman JG, Baylin SB: Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006, 2: e40-
Jones PA, Taylor SM: Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980, 20: 85-93.
Issa J-PJ, Kantarjian HM: Targeting DNA methylation. Clin Cancer Res. 2009, 15: 3938-3946.
Kikuchi R, Yagi S, Kusuhara H, Imai S, Sugiyama Y, Shiota K: Genome-wide analysis of epigenetic signatures for kidney-specific transporters. Kidney Int. 2010
Sørensen AL, Jacobsen BM, Reiner AH, Andersen IS, Collas P: Promoter DNA methylation patterns of differentiated cells are largely programmed at the progenitor stage. Mol Biol Cell. 2010, 21: 2066-2077.
Bodey B: Cancer-testis antigens: promising targets for antigen directed antineoplastic immunotherapy. Expert Opin Biol Ther. 2002, 2: 577-584.
Reik W, Lewis A: Co-evolution of X-chromosome inactivation and imprinting in mammals. Nat Rev Genet. 2005, 6: 403-410.
Herman JG, Baylin SB: Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003, 349: 2042-2054.
Feinberg AP, Tycko B: The history of cancer epigenetics. Nat Rev Cancer. 2004, 4: 143-153.
Egger G, Liang G, Aparicio A, Jones PA: Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004, 429: 457-463.
Esteller M: Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol. 2005, 45: 629-656.
Nakagawa Y, Uemura H, Shimizu K, Cho M, Yoshikawa M, Hirao Y, Yoshikawa K: [The role of MN/CA IX antigen in carcinogenesis and metastasis of renal cell carcinoma]. Hinyokika Kiyo. 2001, 47: 809-814.
Esteller M, Catasus L, Matias-Guiu X, Mutter GL, Prat J, Baylin SB, Herman JG: hMLH1 promoter hypermethylation is an early event in human endometrial tumorigenesis. Am J Pathol. 1999, 155: 1767-1772.
Prowse AH, Webster AR, Richards FM, Richard S, Olschwang S, Resche F, Affara NA, Maher ER: Somatic inactivation of the VHL gene in Von Hippel-Lindau disease tumors. Am J Hum Genet. 1997, 60: 765-771.
Ongenaert M, van Neste L, De Meyer T, Menschaert G, Bekaert S, Van Criekinge W: PubMeth: a cancer methylation database combining text-mining and expert annotation. Nucleic Acids Res. 2008, 36: D842-D846.
Kok K, Naylor SL, Buys CH: Deletions of the short arm of chromosome 3 in solid tumors and the search for suppressor genes. Adv Cancer Res. 1997, 71: 27-92.
Kovacs G, Frisch S: Clonal chromosome abnormalities in tumor cells from patients with sporadic renal cell carcinomas. Cancer Res. 1989, 49: 651-659.
Beroukhim R, Brunet J-P, Di Napoli A, Mertz KD, Seeley A, Pires MM, Linhart D, Worrell RA, Moch H, Rubin MA, Sellers WR, Meyerson M, Linehan WM, Kaelin WG, Signoretti S: Patterns of gene expression and copy-number alterations in von-Hippel Lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009, 69: 4674-4681.
Young AC, Craven RA, Cohen D, Taylor C, Booth C, Harnden P, Cairns DA, Astuti D, Gregory W, Maher ER, Knowles MA, Joyce A, Selby PJ, Banks RE: Analysis of VHL gene alterations and their relationship to clinical parameters in sporadic conventional renal cell carcinoma. Clin Cancer Res. 2009, 15: 7582-7592.
Dulaimi E, Ibanez de Caceres I, Uzzo RG, Al-Saleem T, Greenberg RE, Polascik TJ, Babb JS, Grizzle WE, Cairns P: Promoter hypermethylation profile of kidney cancer. Clin Cancer Res. 2004, 10: 3972-3979.
Hori Y, Oda Y, Kiyoshima K, Yamada Y, Nakashima Y, Naito S, Tsuneyoshi M: Oxidative stress and DNA hypermethylation status in renal cell carcinoma arising in patients on dialysis. J Pathol. 2007, 212: 218-226.
Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, Navratilova M, Szeszenia-Dabrowska N, Mates D, Mukeria A, Holcatova I, Schmidt LS, Toro JR, Karami S, Hung R, Gerard GF, Linehan WM, Merino M, Zbar B, Boffetta P, Brennan P, Rothman N, Chow WH, Waldman FM, Moore LE: Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008, 14: 4726-4734.
Hesson LB, Cooper WN, Latif F: Evaluation of the 3p21.3 tumour-suppressor gene cluster. Oncogene. 2007, 26: 7283-7301.
Morrissey C, Martinez A, Zatyka M, Agathanggelou A, Honorio S, Astuti D, Morgan NV, Moch H, Richards FM, Kishida T, Yao M, Schraml P, Latif F, Maher ER: Epigenetic inactivation of the RASSF1A 3p21.3 tumor suppressor gene in both clear cell and papillary renal cell carcinoma. Cancer Res. 2001, 61: 7277-7281.
Lusher ME, Lindsey JC, Latif F, Pearson ADJ, Ellison DW, Clifford SC: Biallelic epigenetic inactivation of the RASSF1A tumor suppressor gene in medulloblastoma development. Cancer Res. 2002, 62: 5906-5911.
Battagli C, Uzzo RG, Dulaimi E, Ibanez de Caceres I, Krassenstein R, Al-Saleem T, Greenberg RE, Cairns P: Promoter hypermethylation of tumor suppressor genes in urine from kidney cancer patients. Cancer Res. 2003, 63: 8695-8699.
Onay H, Pehlivan S, Koyuncuoglu M, Kirkali Z, Ozkinay F: Multigene methylation analysis of conventional renal cell carcinoma. Urol Int. 2009, 83: 107-112.
Hoque MO, Begum S, Topaloglu O, Jeronimo C, Mambo E, Westra WH, Califano JA, Sidransky D: Quantitative detection of promoter hypermethylation of multiple genes in the tumor, urine, and serum DNA of patients with renal cancer. Cancer Res. 2004, 64: 5511-5517.
Costa VL, Henrique R, Ribeiro FR, Pinto M, Oliveira J, Lobo F, Teixeira MR, Jerónimo C: Quantitative promoter methylation analysis of multiple cancer-related genes in renal cell tumors. BMC Cancer. 2007, 7: 133-
Dreijerink K, Braga E, Kuzmin I, Geil L, Duh FM, Angeloni D, Zbar B, Lerman MI, Stanbridge EJ, Minna JD, Protopopov A, Li J, Kashuba V, Klein G, Zabarovsky ER: The candidate tumor suppressor gene, RASSF1A, from human chromosome 3p21.3 is involved in kidney tumorigenesis. Proc Natl Acad Sci USA. 2001, 98: 7504-7509.
Gonzalgo ML, Yegnasubramanian S, Yan G, Rogers CG, Nicol TL, Nelson WG, Pavlovich CP: Molecular profiling and classification of sporadic renal cell carcinoma by quantitative methylation analysis. Clin Cancer Res. 2004, 10: 7276-7283.
Yoon JH, Dammann R, Pfeifer GP: Hypermethylation of the CpG island of the RASSF1A gene in ovarian and renal cell carcinomas. Int J Cancer. 2001, 94: 212-217.
Vanaja DK, Ballman KV, Morlan BW, Cheville JC, Neumann RM, Lieber MM, Tindall DJ, Young CYF: PDLIM4 repression by hypermethylation as a potential biomarker for prostate cancer. Clin Cancer Res. 2006, 12: 1128-1136.
van den Boom J, Wolter M, Blaschke B, Knobbe CB, Reifenberger G: Identification of novel genes associated with astrocytoma progression using suppression subtractive hybridization and real-time reverse transcription-polymerase chain reaction. Int J Cancer. 2006, 119: 2330-2338.
Awakura Y, Nakamura E, Ito N, Kamoto T, Ogawa O: Methylation-associated silencing of TU3A in human cancers. Int J Oncol. 2008, 33: 893-899.
Druck T, Hadaczek P, Fu TB, Ohta M, Siprashvili Z, Baffa R, Negrini M, Kastury K, Veronese ML, Rosen D, Rothstein J, McCue P, Cotticelli MG, Inoue H, Croce CM, Huebner K: Structure and expression of the human FHIT gene in normal and tumor cells. Cancer Res. 1997, 57: 504-512.
Sozzi G, Pastorino U, Moiraghi L, Tagliabue E, Pezzella F, Ghirelli C, Tornielli S, Sard L, Huebner K, Pierotti MA, Croce CM, Pilotti S: Loss of FHIT function in lung cancer and preinvasive bronchial lesions. Cancer Res. 1998, 58: 5032-5037.
Dumon KR, Ishii H, Fong LY, Zanesi N, Fidanza V, Mancini R, Vecchione A, Baffa R, Trapasso F, During MJ, Huebner K, Croce CM: FHIT gene therapy prevents tumor development in Fhit-deficient mice. Proc Natl Acad Sci USA. 2001, 98: 3346-3351.
Kvasha S, Gordiyuk V, Kondratov A, Ugryn D, Zgonnyk YM, Rynditch AV, Vozianov AF: Hypermethylation of the 5'CpG island of the FHIT gene in clear cell renal carcinomas. Cancer Lett. 2008, 265: 250-257.
Morris MR, Hesson LB, Wagner KJ, Morgan NV, Astuti D, Lees RD, Cooper WN, Lee J, Gentle D, Macdonald F, Kishida T, Grundy R, Yao M, Latif F, Maher ER: Multigene methylation analysis of Wilms' tumour and adult renal cell carcinoma. Oncogene. 2003, 22: 6794-6801.
Vincan E: Frizzled/WNT signalling: the insidious promoter of tumour growth and progression. Front Biosci. 2004, 9: 1023-1034.
Widelitz R: Wnt signaling through canonical and non-canonical pathways: recent progress. Growth Factors. 2005, 23: 111-116.
Kawano Y, Kypta R: Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003, 116: 2627-2634.
Niehrs C: Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 2006, 25: 7469-7481.
Awakura Y, Nakamura E, Ito N, Kamoto T, Ogawa O: Methylation-associated silencing of SFRP1 in renal cell carcinoma. Oncol Rep. 2008, 20: 1257-1263.
Dahl E, Wiesmann F, Woenckhaus M, Stoehr R, Wild PJ, Veeck J, Knüchel R, Klopocki E, Sauter G, Simon R, Wieland WF, Walter B, Denzinger S, Hartmann A, Hammerschmied CG: Frequent loss of SFRP1 expression in multiple human solid tumours: association with aberrant promoter methylation in renal cell carcinoma. Oncogene. 2007, 26: 5680-5691.
Gumz ML, Zou H, Kreinest PA, Childs AC, Belmonte LS, LeGrand SN, Wu KJ, Luxon BA, Sinha M, Parker AS, Sun LZ, Ahlquist DA, Wood CG, Copland JA: Secreted frizzled-related protein 1 loss contributes to tumor phenotype of clear cell renal cell carcinoma. Clin Cancer Res. 2007, 13: 4740-4749.
Urakami S, Shiina H, Enokida H, Hirata H, Kawamoto K, Kawakami T, Kikuno N, Tanaka Y, Majid S, Nakagawa M, Igawa M, Dahiya R: Wnt antagonist family genes as biomarkers for diagnosis, staging, and prognosis of renal cell carcinoma using tumor and serum DNA. Clin Cancer Res. 2006, 12: 6989-6997.
Morris MR, Ricketts C, Gentle D, Abdulrahman M, Clarke N, Brown M, Kishida T, Yao M, Latif F, Maher ER: Identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma. Oncogene. 2010, 29: 2104-2117.
Hirata H, Hinoda Y, Nakajima K, Kawamoto K, Kikuno N, Kawakami K, Yamamura S, Ueno K, Majid S, Saini S, Ishii N, Dahiya R: Wnt antagonist gene DKK2 is epigenetically silenced and inhibits renal cancer progression through apoptotic and cell cycle pathways. Clin Cancer Res. 2009, 15: 5678-5687.
Hirata H, Hinoda Y, Nakajima K, Kawamoto K, Kikuno N, Ueno K, Yamamura S, Zaman MS, Khatri G, Chen Y, Saini S, Majid S, Deng G, Ishii N, Dahiya R: Wnt antagonist DKK1 acts as a tumor suppressor gene that induces apoptosis and inhibits proliferation in human renal cell carcinoma. Int J Cancer. 2010, 10.1002/ijc.25507.
Saini S, Liu J, Yamamura S, Majid S, Kawakami K, Hirata H, Dahiya R: Functional significance of secreted Frizzled-related protein 1 in metastatic renal cell carcinomas. Cancer Res. 2009, 69: 6815-6822.
Chen J, Lui W-O, Vos MD, Clark GJ, Takahashi M, Schoumans J, Khoo SK, Petillo D, Lavery T, Sugimura J, Astuti D, Zhang C, Kagawa S, Maher ER, Larsson C, Alberts AS, Kanayama HO, Teh BT: The t(1;3) breakpoint-spanning genes LSAMP and NORE1 are involved in clear cell renal cell carcinomas. Cancer Cell. 2003, 4: 405-413.
Schmidt L, Junker K, Nakaigawa N, Kinjerski T, Weirich G, Miller M, Lubensky I, Neumann HP, Brauch H, Decker J, Vocke C, Brown JA, Jenkins R, Richard S, Bergerheim U, Gerrard B, Dean M, Linehan WM, Zbar B: Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene. 1999, 18: 2343-2350.
Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, Scherer SW, Zhuang Z, Lubensky I, Dean M, Allikmets R, Chidambaram A, Bergerheim UR, Feltis JT, Casadevall C, Zamarron A, Bernues M, Richard S, Lips CJ, Walther MM, Tsui LC, Geil L, Orcutt ML, Stackhouse T, Lipan J, Slife L, Brauch H, Decker J, Niehans G, Hughson MD, et al: Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997, 16: 68-73.
Morris MR, Gentle D, Abdulrahman M, Maina EN, Gupta K, Banks RE, Wiesener MS, Kishida T, Yao M, Teh B, Latif F, Maher ER: Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer Res. 2005, 65: 4598-4606.
Chitalia VC, Foy RL, Bachschmid MM, Zeng L, Panchenko MV, Zhou MI, Bharti A, Seldin DC, Lecker SH, Dominguez I, Cohen HT: Jade-1 inhibits Wnt signalling by ubiquitylating beta-catenin and mediates Wnt pathway inhibition by pVHL. Nat Cell Biol. 2008, 10: 1208-1216.
Zatyka M, da Silva NF, Clifford SC, Morris MR, Wiesener MS, Eckardt K-U, Houlston RS, Richards FM, Latif F, Maher ER: Identification of cyclin D1 and other novel targets for the von Hippel-Lindau tumor suppressor gene by expression array analysis and investigation of cyclin D1 genotype as a modifier in von Hippel-Lindau disease. Cancer Res. 2002, 62: 3803-3811.
Qi H, Ohh M: The von Hippel-Lindau tumor suppressor protein sensitizes renal cell carcinoma cells to tumor necrosis factor-induced cytotoxicity by suppressing the nuclear factor-kappaB-dependent antiapoptotic pathway. Cancer Res. 2003, 63: 7076-7080.
Genetos DC, Toupadakis CA, Raheja LF, Wong A, Papanicolaou SE, Fyhrie DP, Loots GG, Yellowley CE: Hypoxia decreases sclerostin expression and increases Wnt signaling in osteoblasts. J Cell Biochem. 2010, 110: 457-467.
Roe J-S, Youn H-D: The positive regulation of p53 by the tumor suppressor VHL. Cell Cycle. 2006, 5: 2054-2056.
Bishop T, Lau KW, Epstein ACR, Kim SK, Jiang M, O'Rourke D, Pugh CW, Gleadle JM, Taylor MS, Hodgkin J, Ratcliffe PJ: Genetic analysis of pathways regulated by the von Hippel-Lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol. 2004, 2: e289-
Kajimura S, Aida K, Duan C: Understanding hypoxia-induced gene expression in early development: in vitro and in vivo analysis of hypoxia-inducible factor 1-regulated zebra fish insulin-like growth factor binding protein 1 gene expression. Mol Cell Biol. 2006, 26: 1142-1155.
Slomiany MG, Rosenzweig SA: IGF-1-induced VEGF and IGFBP-3 secretion correlates with increased HIF-1 alpha expression and activity in retinal pigment epithelial cell line D407. Invest Ophthalmol Vis Sci. 2004, 45: 2838-2847.
Yang S, Kim J, Ryu J-H, Oh H, Chun C-H, Kim BJ, Min BH, Chun J-S: Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med. 2010, 16: 687-693.
Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, Teague J, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Forbes S, Jia M, Jones D, Knott H, Kok CY, Lau KW, Leroy C, Lin ML, McBride DJ, Maddison M, Maguire S, McLay K, Menzies A, Mironenko T, et al: Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010, 463: 360-363.
Sanz-Casla MT, Maestro ML, del Barco V, Zanna I, Moreno J, Vidaurreta M, Almansa I, Fernández C, Blanco J, Maestro C, Resel L: Loss of heterozygosity and methylation of p16 in renal cell carcinoma. Urol Res. 2003, 31: 159-162.
Tong WG, Wierda WG, Lin E, Kuang SQ, Bekele BN, Estrov Z, Wei Y, Yang H, Keating MJ, Garcia-Manero G: Genome-wide DNA methylation profiling of chronic lymphocytic leukemia allows identification of epigenetically repressed molecular pathways with clinical impact. Epigenetics. 2010, 5: 499-508.
Boumber YA, Kondo Y, Chen X, Shen L, Gharibyan V, Konishi K, Estey E, Kantarjian H, Garcia-Manero G, Issa J-PJ: RIL, a LIM gene on 5q31, is silenced by methylation in cancer and sensitizes cancer cells to apoptosis. Cancer Res. 2007, 67: 1997-2005.
Tellez CS, Shen L, Estécio MRH, Jelinek J, Gershenwald JE, Issa J-PJ: CpG island methylation profiling in human melanoma cell lines. Melanoma Res. 2009, 19: 146-155.
Veena MS, Lee G, Keppler D, Mendonca MS, Redpath JL, Stanbridge EJ, Wilczynski SP, Srivatsan ES: Inactivation of the cystatin E/M tumor suppressor gene in cervical cancer. Genes Chromosomes Cancer. 2008, 47: 740-754.
Rivenbark AG, Livasy CA, Boyd CE, Keppler D, Coleman WB: Methylation-dependent silencing of CST6 in primary human breast tumors and metastatic lesions. Exp Mol Pathol. 2007, 83: 188-197.
Dallol A, Krex D, Hesson L, Eng C, Maher ER, Latif F: Frequent epigenetic inactivation of the SLIT2 gene in gliomas. Oncogene. 2003, 22: 4611-4616.
Dunwell TL, Dickinson RE, Stankovic T, Dallol A, Weston V, Austen B, Catchpoole D, Maher ER, Latif F: Frequent epigenetic inactivation of the SLIT2 gene in chronic and acute lymphocytic leukemia. Epigenetics. 2009, 4: 265-269.
Hanafusa T, Yumoto Y, Nouso K, Nakatsukasa H, Onishi T, Fujikawa T, Taniyama M, Nakamura S, Uemura M, Takuma Y, Yumoto E, Higashi T, Tsuji T: Reduced expression of insulin-like growth factor binding protein-3 and its promoter hypermethylation in human hepatocellular carcinoma. Cancer Lett. 2002, 176: 149-158.
Wiley A, Katsaros D, Fracchioli S, Yu H: Methylation of the insulin-like growth factor binding protein-3 gene and prognosis of epithelial ovarian cancer. Int J Gynecol Cancer. 2006, 16: 210-218.
Kongkham PN, Northcott PA, Ra YS, Nakahara Y, Mainprize TG, Croul SE, Smith CA, Taylor MD, Rutka JT: An epigenetic genome-wide screen identifies SPINT2 as a novel tumor suppressor gene in pediatric medulloblastoma. Cancer Res. 2008, 68: 9945-9953.
Tung EK-K, Wong C-M, Yau T-O, Lee JM-F, Ching Y-P, Ng IO-L: HAI-2 is epigenetically downregulated in human hepatocellular carcinoma, and its Kunitz domain type 1 is critical for anti-invasive functions. Int J Cancer. 2009, 124: 1811-1819.
Dong W, Chen X, Xie J, Sun P, Wu Y: Epigenetic inactivation and tumor suppressor activity of HAI-2/SPINT2 in gastric cancer. Int J Cancer. 2010, 127: 1526-1534.
Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP: The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009, 41: 178-186.
Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, Leggett B, Levine J, Kim M, French AJ, Thibodeau SN, Jass J, Haile R, Laird PW: CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006, 38: 787-793.
McRonald FE, Morris MR, Gentle D, Winchester L, Baban D, Ragoussis J, Clarke NW, Brown MD, Kishida T, Yao M, Latif F, Maher ER: CpG methylation profiling in VHL related and VHL unrelated renal cell carcinoma. Mol Cancer. 2009, 8: 31-
van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, Edkins S, Hardy C, O'Meara S, Teague J, Butler A, Hinton J, Latimer C, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Cole J, Forbes S, Jia M, Jones D, Kok CY, Leroy C, Lin ML, McBride DJ, Maddison M, Maquire S, McLay K, Menzies A, et al: Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009, 41: 521-523.
Cairns P: Gene methylation and early detection of genitourinary cancer: the road ahead. Nat Rev Cancer. 2007, 7: 531-543.
Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP: Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998, 72: 141-196.
Breault JE, Shiina H, Igawa M, Ribeiro-Filho LA, Deguchi M, Enokida H, Urakami S, Terashima M, Nakagawa M, Kane CJ, Carroll PR, Dahiya R: Methylation of the gamma-catenin gene is associated with poor prognosis of renal cell carcinoma. Clin Cancer Res. 2005, 11: 557-564.
van Vlodrop IJH, Baldewijns MML, Smits KM, Schouten LJ, van Neste L, Van Criekinge W, van Poppel H, Lerut E, Schuebel KE, Ahuja N, Herman JG, de Bruïne AP, van Engeland M: Prognostic significance of Gremlin1 (GREM1) promoter CpG island hypermethylation in clear cell renal cell carcinoma. Am J Pathol. 2010, 176: 575-584.
Okuda H, Toyota M, Ishida W, Furihata M, Tsuchiya M, Kamada M, Tokino T, Shuin T: Epigenetic inactivation of the candidate tumor suppressor gene HOXB13 in human renal cell carcinoma. Oncogene. 2006, 25: 1733-1742.
Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, Stone RM, Nelson D, Powell BL, DeCastro CM, Ellerton J, Larson RA, Schiffer CA, Holland JF: Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002, 20: 2429-2440.
Oki Y, Jelinek J, Shen L, Kantarjian HM, Issa JP: Induction of hypomethylation and molecular response after decitabine therapy in patients with chronic myelomonocytic leukemia. Blood. 2008, 111: 2382-2384.
Ibanez de Caceres I, Dulaimi E, Hoffman AM, Al-Saleem T, Uzzo RG, Cairns P: Identification of novel target genes by an epigenetic reactivation screen of renal cancer. Cancer Res. 2006, 66: 5021-5028.
Christoph F, Weikert S, Kempkensteffen C, Krause H, Schostak M, Köllermann J, Miller K, Schrader M: Promoter hypermethylation profile of kidney cancer with new proapoptotic p53 target genes and clinical implications. Clin Cancer Res. 2006, 12: 5040-5046.
Majid S, Dar AA, Ahmad AE, Hirata H, Kawakami K, Shahryari V, Saini S, Tanaka Y, Dahiya AV, Khatri G, Dahiya R: BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis. 2009, 30: 662-670.
Morris MR, Gentle D, Abdulrahman M, Clarke N, Brown M, Kishida T, Yao M, Teh BT, Latif F, Maher ER: Functional epigenomics approach to identify methylated candidate tumour suppressor genes in renal cell carcinoma. Br J Cancer. 2008, 98: 496-501.
Yamada D, Kikuchi S, Williams YN, Sakurai-Yageta M, Masuda M, Maruyama T, Tomita K, Gutmann DH, Kakizoe T, Kitamura T, Kanai Y, Murakami Y: Promoter hypermethylation of the potential tumor suppressor DAL-1/4.1B gene in renal clear cell carcinoma. Int J Cancer. 2006, 118: 916-923.
Christoph F, Kempkensteffen C, Weikert S, Köllermann J, Krause H, Miller K, Schostak M, Schrader M: Methylation of tumour suppressor genes APAF-1 and DAPK-1 and in vitro effects of demethylating agents in bladder and kidney cancer. Br J Cancer. 2006, 95: 1701-1707.
Zhang Q, Ying J, Zhang K, Li H, Ng KM, Zhao Y, He Q, Yang X, Xin D, Liao S-K, Tao Q, Jin J: Aberrant methylation of the 8p22 tumor suppressor gene DLC1 in renal cell carcinoma. Cancer Lett. 2007, 249: 220-226.
Khoo SK, Kahnoski K, Sugimura J, Petillo D, Chen J, Shockley K, Ludlow J, Knapp R, Giraud S, Richard S, Nordenskjöld M, Teh BT: Inactivation of BHD in sporadic renal tumors. Cancer Res. 2003, 63: 4583-4587.
da Silva NF, Gentle D, Hesson LB, Morton DG, Latif F, Maher ER: Analysis of the Birt-Hogg-Dubé (BHD) tumour suppressor gene in sporadic renal cell carcinoma and colorectal cancer. J Med Genet. 2003, 40: 820-824.
Gad S, Lefèvre SH, Khoo SK, Giraud S, Vieillefond A, Vasiliu V, Ferlicot S, Molinié V, Denoux Y, Thiounn N, Chrétien Y, Méjean A, Zerbib M, Benoît G, Hervé JM, Allègre G, Bressac-de Paillerets B, Teh BT, Richard S: Mutations in BHD and TP53 genes, but not in HNF1beta gene, in a large series of sporadic chromophobe renal cell carcinoma. Br J Cancer. 2007, 96: 336-340.
Seliger B, Handke D, Schabel E, Bukur J, Lichtenfels R, Dammann R: Epigenetic control of the ubiquitin carboxyl terminal hydrolase 1 in renal cell carcinoma. J Transl Med. 2009, 7: 90-
Dallol A, Forgacs E, Martinez A, Sekido Y, Walker R, Kishida T, Rabbitts P, Maher ER, Minna JD, Latif F: Tumour specific promoter region methylation of the human homologue of the Drosophila Roundabout gene DUTT1 (ROBO1) in human cancers. Oncogene. 2002, 21: 3020-3028.
Astuti D, Da Silva NF, Dallol A, Gentle D, Martinsson T, Kogner P, Grundy R, Kishida T, Yao M, Latif F, Maher ER: SLIT2 promoter methylation analysis in neuroblastoma, Wilms' tumour and renal cell carcinoma. Br J Cancer. 2004, 90: 515-521.
Bachman KE, Herman JG, Corn PG, Merlo A, Costello JF, Cavenee WK, Baylin SB, Graff JR: Methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene suggest a suppressor role in kidney, brain, and other human cancers. Cancer Res. 1999, 59: 798-802.
Kempkensteffen C, Hinz S, Schrader M, Christoph F, Magheli A, Krause H, Schostak M, Miller K, Weikert S: Gene expression and promoter methylation of the XIAP-associated Factor 1 in renal cell carcinomas: correlations with pathology and outcome. Cancer Lett. 2007, 254: 227-235.
Lee M-G, Huh J-S, Chung S-K, Lee J-H, Byun D-S, Ryu B-K, Kang M-J, Chae K-S, Lee S-J, Lee C-H, Kim JI, Chang SG, Chi SG: Promoter CpG hypermethylation and downregulation of XAF1 expression in human urogenital malignancies: implication for attenuated p53 response to apoptotic stresses. Oncogene. 2006, 25: 5807-5822.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Both authors contributed to manuscript preparation and editing.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Morris, M.R., Maher, E.R. Epigenetics of renal cell carcinoma: the path towards new diagnostics and therapeutics. Genome Med 2, 59 (2010). https://doi.org/10.1186/gm180
Published:
DOI: https://doi.org/10.1186/gm180