
Abstract
The importance of RNA-protein interactions in controlling mRNA regulation and non-coding RNA function is increasingly appreciated. A variety of methods exist to comprehensively define RNA-protein interactions. We describe these methods and the considerations required for designing and interpreting these experiments.
Similar content being viewed by others


Introduction
Over the past decade there has been an increasing appreciation of the importance of RNA-protein interactions in controlling many aspects of gene regulation [1, 2]. The explosion in sequencing technologies has enabled exploration of the transcriptome at unprecedented depth [3]. This has led to a growing appreciation of the widespread role of alternative messenger RNA (mRNA) splicing [4–7], processing [8], editing [9–11] and methylation [12, 13] in generating diverse mRNAs and in controlling the stability and translation of mRNA. Furthermore, this has led to the identification of diverse classes of non-coding RNAs (ncRNAs), including many thousands of long non-coding RNAs (lncRNAs) that resemble mRNA but are not translated into proteins [14–17].
The central role of RNA-protein interactions in controlling mRNA processing [1, 2] and ncRNA function [18, 19] is now clear. Many proteins are known to be required for various aspects of mRNA processing [1]. These include the ubiquitously expressed serine-rich (SR) proteins [20] and heteronuclear ribonucleoproteins (hnRNPs) [21], as well as the cell type-specific Nova [22], Fox [23] and Muscleblind [24] proteins, which all play important roles in the regulation of alternative splicing in different cell types [2, 25, 26]. Yet, precisely how these proteins control the diversity of cell type-specific mRNA remains largely unclear [2, 27]. In addition, the proper cellular functions of virtually all ncRNAs - including those with catalytic roles [28, 29] - depend upon the formation of RNA-protein complexes [18, 19, 30]. These include classical examples such as ribosomal RNAs, small nuclear RNAs and small nucleolar RNAs that control translation, splicing and ribosomal biogenesis, as well as small ncRNAs such as microRNAs and piwi-associated RNAs that control mRNA stability and translation [31], and silencing of DNA repeats [32]. In addition, lncRNAs play key functional roles in controlling cellular regulation [18, 19, 33–37], likely through their interactions with diverse classes of proteins [18, 19]. To date, the full spectrum of proteins that interact with ncRNAs is still unknown [13, 14].
The past decade has seen a strong interplay between method development, exploration and discovery about RNA biology. The methods for exploring RNA-protein interactions can be split into two general categories: ‘protein-centric’ and ‘RNA-centric’ methods. The protein-centric methods generally rely on the ability to purify a protein [38–40], or class of proteins [41], followed by sequencing of the associated RNAs to map RNA-binding proteins (RBPs) across the transcriptome at high resolution. Conversely, the RNA-centric approaches generally capture a given RNA [42–44], or class of RNAs [45, 46], and identify the associated proteins using methods such as mass spectrometry (MS).
Protein-centric approaches have been widely used to generate binding maps of different RBPs across the transcriptome and have provided important insights into how mRNA processing is controlled in the cell [21, 23, 47, 48]. These methods have also been used to gain initial insights into some of the proteins that can interact with lncRNAs [49–51]. Because these methods require knowledge of the protein, they are of more limited utility for defining the proteins that associate with a given RNA transcript. The RNA-centric methods have been more generally used to determine the complexes associated with a specific ncRNA in the cell. Indeed, the protein compositions of several classical ncRNA complexes, including those of telomerase RNA [42], small nuclear RNA [43], 7SK RNA [44] and RNase P [52], have been identified using these approaches.
In this review, we discuss approaches for identifying RNA-protein interactions and the challenges associated with interpreting these data. We describe the various protein-centric methods, including native and crosslinking-based methods, and explore the caveats and considerations required for designing, performing and interpreting the results of these experiments. We describe approaches that have been developed to account for analytical biases that can arise in these data. Furthermore, we describe the various RNA-centric methods for the identification of unknown RNA-binding proteins, including the various RNA tags used, purification schemes and detection methods. While conceptually simple, the RNA-centric methods are still not as common as the protein-centric methods because they require an extraordinary amount of starting material to purify enough protein required for detection [53]. We describe the challenges associated with these methods and their interpretation. Finally, we discuss the future steps that will be needed to synthesize the results of these complementary approaches and enable the systematic application of such methods to new classes of ncRNAs.
Protein-centric methods to study RNA-protein interactions
The predominant methods for examining RNA-protein interactions are based on protein immunoprecipitation. These methods generally utilize antibodies to pull down the protein of interest and its associated RNA, which is reverse transcribed into cDNA, PCR amplified and sequenced [38, 54–59]. Bioinformatic analysis is then used to map reads back to their transcripts of origin and identify protein binding sites [60, 61].
There are several variants of these methods, which can be broken down into two main classes: native [39, 40, 51, 58, 62–64] and fully denaturing purifications [22, 55–57, 59, 65] (Figure 1a).

Protein-centric methods for detecting RNA-protein interactions. (a) Schematic of native and denaturing methods. RNA-protein crosslinks are represented by red Xs. Non-specific interactions in solution are labeled (NS) and represented by blue RNA fragments. (b) Computational considerations for identification of interaction sites. The top panel depicts two transcripts - one low-abundance and one high-abundance - that both contain a region that is twofold enriched in the immunoprecipitated (IP) sample over a control. Enrichment measurements in the low-abundance case suffer from high variance. The bottom panel shows simulated enrichment values in a low-abundance region and a high-abundance region, which both have a twofold enrichment in the IP sample. For the low abundance region, the observed log-fold changes are often far from the true underlying value while the abundant transcript shows a more consistent enrichment estimation. (c) A schematic of methods for mapping the precise protein binding sites on RNA. PAR-CLIP takes advantage of U → C transitions induced by UV crosslinking after 4SU incorporation. iCLIP uses the occasional arrest of reverse transcription at crosslink sites and tags and sequences these positions. CRAC relies on reverse transcription errors (deletions and substitutions) at crosslink sites to map sites. CRAC, cross-linking and analysis of cDNA; iCLIP, individual-nucleotide resolution cross-linking and immunoprecipitation; PAR-CLIP, photoactivatable-ribonucleoside-enhanced cross-linking and immunoprecipitation.
Native purifications
Native purification methods, often known simply as RIP (RNA immunoprecipitation), purify RNA-protein complexes under physiological conditions. The advantage to these methods is that they preserve the native complexes present in the cell. Yet, these methods also have several limitations. The first and perhaps best described is due to the non-physiological formation of RNA-protein interactions in solution. Indeed, it has been shown that purification of an RNA-binding protein can retrieve RNAs, even when the RNA and protein are not present in the same cell type and therefore could not be interacting in vivo[66]. Furthermore, the RNAs that are purified are generally very well correlated with the abundance of the RNA, with ribosomal RNAs being the largest contaminating RNA species in virtually all protein purifications [38]. As a consequence, specific interactions that occur with low abundance transcripts may be masked by non-specific interactions that occur with highly abundant transcripts [38].
Because of these issues, there has been some controversy about the nature of the interactions detected by these methods. For example, many lncRNA-protein interactions have been explored using native purifications of proteins such as those found in Polycomb repressive complex 2 (PRC2) [51, 58, 62]. In these studies, a very large percentage of lncRNAs, as well as mRNAs, were identified as interacting with PRC2 [58], with a recent study arguing that virtually all transcripts interact with PRC2 in the cell [62]. This has led to debate over the biological significance of lncRNA-PRC2 interactions, with some arguing that they are simply non-specific interactions [67]. However, it is clear that at least some lncRNAs interact with PRC2 [49, 50, 68] and that these interactions have clear functional roles [58, 69, 70]. While it is clear that both native and denaturing purification methods can identify a similar core set of functional interactions [71], the extent to which non-specific interactions are also identified by the native methods remains unclear. As such, interactions identified using native purification methods often require further experimental validation, such as through the integration of multiple distinct experimental approaches [49, 71, 72].
Denaturing methods for RNA-protein interactions
To account for these concerns, denaturing methods were introduced. By crosslinking RNA-protein complexes in the cell and purifying the complex under denaturing conditions, one can distinguish in vivo interactions that are crosslinked in the cell from interactions that form subsequently in solution.
The dominant method for crosslinking RNA-protein complexes is treatment of cells with short wavelength UV light to create a covalent linkage between physically interacting RNA and protein molecules in the cell, but not between interacting proteins [73]. Methods such as crosslinking and immunoprecipitation (CLIP) purify an RNA-protein complex using stringent wash conditions followed by denaturation of all complexes by heating in sodium dodecyl sulphate (SDS), running the samples on an SDS-polyacrylamide gel electrophoresis (PAGE) gel, and extracting the crosslinked RNA-protein complex, which will run at a size slightly larger than the protein itself, from the gel [74, 75]. The main limitation of this method is the low efficiency of UV crosslinking. To account for this, a variant that significantly increases crosslinking efficiency while retaining the main features of UV crosslinking was introduced: photoactivatable-ribonucleoside-enhanced (PAR)-CLIP [56]. This approach incorporates a nucleotide analog (such as 4′-thiouracil) into cells, followed by treatment of the cells with long-wavelength UV. The drawback to this approach is that it is only amenable to cells in culture and cannot be applied to primary tissues.
A significant concern with using UV crosslinking methods is that they may miss real RNA-protein interactions simply because they are not efficiently captured by UV crosslinking. Indeed, several RBP families that do not directly interact with nucleic acid bases but instead interact with other features, such as the sugar phosphate backbone, have been shown to have lower crosslinking efficiency with UV [76]. Because UV-induced crosslinking is still incompletely understood at the biophysical level [38], it is unclear which types of interactions might be missed or what frequency of real interactions may be missed. In addition, because UV only crosslinks direct RNA-protein interactions, it will not capture interactions that occur through a complex of multiple proteins. As an example, interactions with many chromatin regulatory proteins have proven challenging to identify by purification under denaturing conditions after UV crosslinking, likely because the precise protein that interacts directly with RNA is still unknown [33].
Other crosslinking methods, such as formaldehyde, can eliminate the need to know the directly interacting protein, but alternative denaturing strategies are needed since purification from a denaturing SDS-PAGE gel would not resolve at the size of the protein. An alternative approach that leverages many of the conceptual features of the CLIP method is to utilize direct denaturing conditions rather than separation through an SDS-PAGE gel. These methods use affinity tags coupled to the protein of interest for capture by purification in denaturating conditions (that is, using urea or guanidine) [59–65]. The advantage to this approach is that it can be used with any crosslinking protocol, including formaldehyde crosslinking, which otherwise could not be separated on an SDS-PAGE gel [59]. Yet, this approach requires the ability to express a tagged version of the RBP of interest in the cell.
Analysis of protein-centric RNA-protein interaction data
There are two primary goals in the analysis of protein-centric experiments: defining which RNAs are bound by the specific protein and defining the specific protein-binding sites on these RNAs.
It is important to compare the sample to a negative control since observing reads from a specific RNA alone may not be indicative of a real interaction. One control is to normalize the coverage level of an RNA observed after purification to its abundance in total lysate. Yet, this control only accounts for issues due to RNA abundance: interactions can occur due to association with the purification resin or other features of the system. To account for this, other proteins can be used as negative controls. However, the negative control should be selected with care, as a non-RNA-binding protein is likely to have lower non-specific RNA binding. Indeed, simply mutating the RNA-binding domain of a protein has been shown to remove both specific and non-specific interactions formed by a protein [77]. The ideal control is to demonstrate that the interaction is not present in the absence of crosslinking [22, 38]. However, this control can only be used in conjunction with a fully denaturing protocol.
Furthermore, comparing the sample with a negative control requires proper statistical methods because the inherently low denominator for low abundance RNAs will lead to a higher variance in the enrichment measurement (Figure 1b). This challenge is similar to the problems faced when computing differential expression using RNA-Seq data [78], and many different statistical solutions, including parametric, non-parametric and permutation methods, have been proposed [79–81].
The second goal is to map protein-binding sites on RNA. A major consideration is the size of the RNA after digestion. While in theory the ideal size is that of the protein footprint itself, several considerations favor slightly larger sizes. One issue is the alignability of the sequencing reads, as very small fragments may not be able to be uniquely aligned to the transcriptome. Another concern is that overdigestion may lead to a loss of real binding sites by preferentially eliminating certain protein footprints [75].
Because UV-crosslinking is irreversible, reverse transcription can halt at the site of crosslinking even after protein removal [22, 25]. While this was originally considered a disadvantage of UV crosslinking, it has been successfully used by several methods, including the CLIP variant individual-nucleotide resolution CLIP (iCLIP), to identify protein-binding sites on RNA with improved resolution [55, 57]. In addition to RT stops, crosslink sites also show a higher rate of deletions and mismatches - these have also been used to identify binding sites [61] (Figure 1c). Yet, great care must be exercised when interpreting these RT-induced stop sites and errors, as RNA damage due to UV light is also known to inhibit reverse transcription [82].
RNA-centric methods
Protein-centric methods are of limited utility for identifying novel RBPs that interact with a specific RNA or for the characterization of novel classes of ncRNAs for which the identities of the RNA-binding proteins are still unknown. An alternative approach is to use an RNA-centric protein identification strategy. The general idea is simple: rather than using an antibody to capture a protein of interest and sequencing the associated RNA, these methods purify an RNA of interest and identify the associated protein complexes, using methods such as MS. We will explore the different variants of these methods below, focusing on those designed to comprehensively identify novel RNA-protein interactions.
RNA affinity capture methods
One general approach to capture RNA is to exploit naturally occurring interactions between RNA and protein - such as the bacteriophage MS2 viral coat protein, which binds tightly to an RNA stem loop structure [83]. In this approach, repeats of the MS2-binding RNA stem loop are appended to an RNA of interest and the tagged RNA complex is purified by coupling the MS2 protein to a solid support or resin [84–86]. These dual component interactions can be optimized to enable increased affinity and stability [44, 87]. As an example, a recent approach makes use of an engineered Csy4 protein, a component of the bacterial clustered regularly interspaced short palindromic repeats (CRISPR) system, to generate a tag with higher affinity than can be achieved for traditional RNA tags, including MS2 and PP7 [87]. Alternatively, artificially designed RNA aptamers can be developed and selected for binding to commonly used resin-conjugated proteins [43, 88]. An example of this is the S1 aptamer that binds to streptavidin [89, 90].
The differences between these methods can be exploited when trying to elute their respective RNA-protein complexes. In general, protein complexes are eluted from a support resin by boiling in SDS [87]. This approach will dissociate bound material from the resin, including complexes bound specifically through the tag and those bound non-specifically directly to the resin. For several of these affinity tags, complexes can be eluted more specifically. For example, in the case of the S1 aptamer, the weaker affinity of the S1-streptavidin interaction compared with the biotin-streptavidin interaction can be exploited to enable specific elution of the RNA through a competitive approach using high concentrations of biotin [91]. In the CRISPR system, because of the nature of the Csy4 mutant used, one can specifically cleave the complex through the addition of imidazole. Indeed, the specificity of elution dramatically increases the specificity of the purified complexes and can improve detection sensitivity [87].
Purification of RNA and associated protein complexes
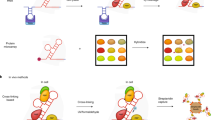
RNA-centric approaches can be grouped into one of two major classes: in vitro and in vivo purification methods (Figure 2a). The in vitro approaches generally employ a synthetic RNA bait to capture and identify proteins from cellular extracts [43, 88, 90]. In contrast, the in vivo approaches capture the RNA-protein complexes present in the cell [45, 46, 85, 92]. While the in vivo methods preserve the context of true RNA-protein interactions, they are more technically challenging, especially if the target RNA is of low abundance in the cell.

RNA-centric methods for the purification and identification of RNA-binding proteins. (a) Examples of purification schemes for RNA-binding proteins using in vitro and in vivo approaches. For in vitro approaches, a tagged RNA construct is generated and bound to a solid support. In this example the MS2 protein-RNA interaction tagging method is shown with the target RNA (red), MS2-binding motif (purple) and MS2 protein (gray). Cell lysate is prepared and proteins from lysate are captured using the tagged RNA in vitro. For in vivo approaches, the target RNA is crosslinked to specific interacting RNA-binding proteins in living cells using UV, formaldehyde or other cross-linkers. Cells are lysed and the RNA-protein complexes captured from solution. In both scenarios, the complex is washed to remove non-specific interactions (green proteins). Finally the bound proteins are eluted. (b) MS is commonly used to identify the RBPs in a purified sample. In non-quantitative MS approaches, RBPs are purified from unlabeled cell material using either an RNA of interest or a control construct. After separation by one-dimensional gel electrophoresis, specific protein bands from the sample are selected, excised and identified by MS analysis. In quantitative MS approaches, proteins are differentially labeled based on their initial cell populations. Experimental and control purifications are performed on these labeled populations and the purified RBPs are pooled to create a single sample. MS analysis allows direct comparison of labeled peptides, which can then be quantified to determine specific proteins in the sample compared with the control. SILAC, stable isotope labeling by amino acids in cell culture.
Similar to the protein-centric methods, purification of RNA under native conditions can lead to re-association or formation of non-specific RNA-protein interactions in solution. Studies using in vitro approaches or performing purifications in native conditions have generally found association between the RNA of interest and highly abundant proteins in the cell, such as hnRNPs [85, 91, 92]. Whether these represent real biological interactions or non-specific associations is unclear because only a handful of RNAs have been purified to date. To address this, a recent study made use of UV crosslinking and purified RNA complexes under fully denaturing conditions (using 8 M urea), which will only capture in vivo crosslinked complexes [85]. Using this approach, there were clear differences in the proteins identified after purifications performed in native and denaturing conditions. DNA-binding proteins and other abundant nucleic acid-binding proteins were present only in the native purification, but not in the denaturing purification, suggesting that at least some of these purified proteins might be due to non-specific association in solution. Other approaches use stringent, high-salt wash conditions to reduce non-specific interactions during RNA-protein complex purification [45, 93, 94].
The challenge with denaturing approaches is that they require complexes to be crosslinked in the cell, which is not efficient. In addition, several crosslinking strategies, such as formaldehyde crosslinking, may have additional technical challenges associated with the identification of crosslinked peptides by MS [95].
Defining the proteins that associate with an RNA
We will focus on MS methods for identification of RNA-binding proteins. There are two principal ways that have been used to comprehensively identify these protein complexes by MS: non-quantitative and quantitative MS (Figure 2b).
In the non-quantitative methods, purified proteins from the RNA sample of interest and a control are separated by gel electrophoresis and stained for total protein. Protein bands that are present only in the sample of interest but not the control are extracted and the proteins identified by MS [84]. Alternatively, the total proteome can be analyzed by MS to detect all proteins purified in a sample [87, 96]. The advantage to the latter approach is that all proteins can be identified in the sample, including those that are not visible on the gel. In this approach, the control can also be analyzed to identify non-specific proteins for exclusion. However, it is difficult to directly compare the quantities of proteins identified in the sample and control, due to variations in the relative intensity of identified peptides in independent runs [53].
To overcome this limitation, one can use quantitative MS to simultaneously compare the proteins in the sample and control. There are several ways to do this (reviewed in [53]). In one popular method used for RNA-protein analysis, cells are metabolically labeled to generate differentially tagged protein pools for MS analysis, in which the isotopes of the proteins are compared to provide direct quantification [97]. The advantage to this approach is that the ratios of peptides from the experimental and control samples can be directly compared to allow discrimination of true binding partners from non-specific interactors. This method can account for some of the issues associated with abundant protein association. As an example, in quantitative MS experiments, most of the abundant proteins, such as hnRNPs, show equal abundance in both experimental and control samples, suggesting that these interactions are not specific to the RNA of interest [91].
The choice of which MS approach to use for the identification of RBPs depends on the nature of the upstream purification. When utilizing a protocol where the resulting protein purification yields little background in the control sample, a non-quantitative approach may work well. The CRISPR-Csy4 system, for instance, was previously shown to enable very high stringency and specific elution, and because of this a non-quantitative approach provided reliable results [87]. Similarly, when employing crosslinking followed by a denaturing purification strategy, non-quantitative MS might provide a good approach. In contrast, when using a system with higher background, a quantitative MS approach can provide increased ability to discriminate between specific and non-specific binders.
Analytical challenges with RBP MS analysis
There are several analytical challenges for identifying proteins associated with an RNA by MS. Similar to the protein-centric methods, great care must be taken to select informative negative controls for the RNA-centric methods. Controls that are often used include a different cellular RNA [92], sequences lacking known protein-binding structures [85–91], tag-only controls [44], antisense RNA [71, 98] or non-specific RNA sequences [99]. In these cases, any non-specific protein interactions due to abundance, nucleic acid binding or the tag itself should be identical for the target RNA and controls. However, the ideal negative control is not clearly established because there may be some specific features of the RNA of interest that bind non-specifically to certain proteins. In cases where protein-RNA crosslinking is employed, the ideal control would be a non-crosslinked sample because it represents the identical purification of the same RNA but without any in vivo crosslinked complexes [96]. However, this approach requires the use of in vivo crosslinking followed by a denaturing purification and therefore is not applicable to all purification methods. In the absence of this, several different negative controls should be included to ensure robustness of the results identified.
A significant challenge in the identification of unknown RBPs is the generation of sufficient material for MS, particularly for low abundance RNA-protein complexes. Unlike sequencing methods that enable nucleic acid amplification, the amount of protein purified in these experiments cannot be amplified. For this reason, RNA-centric methods have mostly been applied to highly abundant RNAs, such as 7SK [44], snRNPs [100], Let-7 [99] and IRES [85]. More recently, these approaches have been used to define proteins associated with all mRNA by UV crosslinking RNA-protein complexes, capturing polyadenylated transcripts using oligo-dT coupled magnetic beads, and detecting associated proteins by quantitative MS [45, 46, 94]. Yet, application of this approach to identify binding partners of individual mRNAs, lncRNAs or other low abundance RNAs is still a significant challenge.
Future directions
While much work has been done to develop methods to identify and examine RNA-protein interactions, there are still significant challenges that need to be addressed. To date, we still do not know the protein complexes that interact with most RNAs in the cell - including mRNAs, classical ncRNAs and lncRNAs. For lncRNAs in particular, we know little about the diversity of proteins that they may interact with. Many of the protein complexes that have been identified to interact with lncRNAs do not fall into traditional RNA-binding protein classes, making it difficult to generate accurate predictions of what these complexes may look like. Understanding the protein complexes that interact with lncRNAs will be an important first step toward understanding their various biological functions and mechanisms. The major challenge with defining these proteins is that the RNA-centric methods are still not well suited for exploring low abundance transcripts. Future work will be needed to address this challenge and to define the protein complexes that interact with a given lncRNA or individual mRNA.
Although the development of the protein-centric and RNA-centric approaches has mostly proceeded independently, we can now begin to combine the results of these complementary approaches to build a complete picture of the repertoire of RBPs in a cell and define their roles in binding and modulating the functions of various classes of RNA [101]. Several recent studies have begun to examine protein binding at a transcriptome-wide scale [45, 94, 102–106]. In these studies, RBPs [45, 94, 102, 104] and/or their binding sites [45, 94, 102–106] have been identified by MS or high-throughput sequencing, respectively. By exploring the different components of RNA-protein complexes, we will be able to identify new RBPs, as well as discriminate the timing of the binding of a set of given RBPs to an individual RNA [107]. This will ultimately provide a more complete understanding of the function of RNA-protein complexes, including how these complexes assemble and how they modulate cellular function.
Abbreviations
- CLIP:
-
Crosslinking and immunoprecipitation
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- hnRNP:
-
Heterogenous ribonucleoprotein
- lncRNA:
-
Large non-coding RNA
- MS:
-
Mass spectrometry
- ncRNA:
-
Non-coding RNA
- PAR:
-
Photoactivatable-ribonucleoside-enhanced
- PCR2:
-
Polycomb repressive complex 2
- RBP:
-
RNA-binding protein.
References
Chen M, Manley JL: Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009, 10: 741-754.
Licatalosi DD, Darnell RB: RNA processing and its regulation: global insights into biological networks. Nat Rev Genet. 2010, 11: 75-87.
Wang Z, Gerstein M, Snyder M: RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009, 10: 57-63.
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB: Alternative isoform regulation in human tissue transcriptomes. Nature. 2008, 456: 470-476.
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ: Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008, 40: 1413-1415.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B: Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008, 5: 621-628.
Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, Schmidt D, O'Keeffe S, Haas S, Vingron M, Lehrach H, Yaspo ML: A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008, 321: 956-960.
Ulitsky I, Shkumatava A, Jan CH, Subtelny AO, Koppstein D, Bell GW, Sive H, Bartel DP: Extensive alternative polyadenylation during zebrafish development. Genome Res. 2012, 22: 2054-2066.
Bahn JH, Lee JH, Li G, Greer C, Peng G, Xiao X: Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res. 2012, 22: 142-150.
Peng Z, Cheng Y, Tan BC, Kang L, Tian Z, Zhu Y, Zhang W, Liang Y, Hu X, Tan X, Guo J, Dong Z, Liang Y, Bao L, Wang J: Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol. 2012, 30: 253-260.
Danecek P, Nellåker C, McIntyre RE, Buendia-Buendia JE, Bumpstead S, Ponting CP, Flint J, Durbin R, Keane TM, Adams DJ: High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome Biol. 2012, 13: 26-
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G: Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012, 485: 201-206.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR: Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. 2012, 149: 1635-1646.
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES: Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009, 458: 223-227.
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL: Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25: 1915-1927.
Bánfai B, Jia H, Khatun J, Wood E, Risk B, Gundling WE, Kundaje A, Gunawardena HP, Yu Y, Xie L, Krajewski K, Strahl BD, Chen X, Bickel P, Giddings MC, Brown JB, Lipovich L: Long noncoding RNAs are rarely translated in two human cell lines. Genome Res. 2012, 22: 1646-1657.
Guttman M, Russell P, Ingolia NT, Weissman JS, Lander ES: Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013, 154: 240-251.
Guttman M, Rinn JL: Modular regulatory principles of large non-coding RNAs. Nature. 2012, 482: 339-346.
Rinn JL, Chang HY: Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012, 81: 145-166.
Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, Qiu J, Huang J, Yeo GW, Ares M, Fu XD: Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell. 2013, 50: 223-235.
Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, Yan BY, Donohue JP, Shiue L, Hoon S, Brenner S, Ares M, Yeo GW: Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012, 1: 167-178.
Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB: CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003, 302: 1212-1215.
Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH: An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol. 2009, 16: 130-137.
Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, Lécuyer E, Burge CB: Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012, 150: 710-724.
Ge H, Manley JL: A protein factor, ASF, controls cell-specific alternative splicing of SV40 early pre-mRNA in vitro. Cell. 1990, 62: 25-34.
Caceres JF, Stamm S, Helfman DM, Krainer AR: Regulation of alternative splicing in vivo by overexpression of antagonistic splicing factors. Science. 1994, 265: 1706-1709.
Darnell JE: Reflections on the history of pre-mRNA processing and highlights of current knowledge: a unified picture. RNA. 2013, 19: 443-460.
Fica SM, Tuttle N, Novak T, Li NS, Lu J, Koodathingal P, Dai Q, Staley JP, Piccirilli JA: RNA catalyses nuclear pre-mRNA splicing. Nature. 2013, 503: 229-234.
Leung EK, Suslov N, Tuttle N, Sengupta R, Piccirilli JA: The mechanism of peptidyl transfer catalysis by the ribosome. Annu Rev Biochem. 2011, 80: 527-555.
Eddy SR: Non-coding RNA genes and the modern RNA world. Nat Rev Genet. 2001, 2: 919-929.
Bartel DP: MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004, 116: 281-297.
Siomi MC, Sato K, Pezic D, Aravin AA: PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol. 2011, 12: 246-258.
Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, Yang X, Amit I, Meissner A, Regev A, Rinn JL, Root DE, Lander ES: lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011, 477: 295-300.
Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R: Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010, 143: 46-58.
Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, Wysocka J, Lei M, Dekker J, Helms JA, Chang HY: A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011, 472: 120-124.
Ponting CP, Oliver PL, Reik W: Evolution and functions of long noncoding RNAs. Cell. 2009, 136: 629-641.
Ulitsky I, Bartel DP: lincRNAs: genomics, evolution, and mechanisms. Cell. 2013, 154: 26-46.
Darnell RB: HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip Rev RNA. 2010, 1: 266-286.
Hogan DJ, Riordan DP, Gerber AP, Herschlag D, Brown PO: Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLOS Biol. 2008, 6: e255-
Gerber AP, Herschlag D, Brown PO: Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLOS Biol. 2004, 2: E79-
Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS: Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009, 324: 218-223.
Lingner J, Cech TR: Purification of telomerase from Euplotes aediculatus: requirement of a primer 3' overhang. Proc Natl Acad Sci U S A. 1996, 93: 10712-10717.
Hartmuth K, Urlaub H, Vornlocher HP, Will CL, Gentzel M, Wilm M, Lührmann R: Protein composition of human prespliceosomes isolated by a tobramycin affinity-selection method. Proc Natl Acad Sci U S A. 2002, 99: 16719-16724.
Hogg JR, Collins K: RNA-based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA. 2007, 13: 868-880.
Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, Krijgsveld J, Hentze MW: Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012, 149: 1393-1406.
Kwon SC, Yi H, Eichelbaum K, Föhr S, Fischer B, You KT, Castello A, Krijgsveld J, Hentze MW, Kim VN: The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol. 2013, 20: 1122-1130.
Xue Y, Zhou Y, Wu T, Zhu T, Ji X, Kwon YS, Zhang C, Yeo G, Black DL, Sun H, Fu XD, Zhang Y: Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell. 2009, 36: 996-1006.
Zhang C, Frias MA, Mele A, Ruggiu M, Eom T, Marney CB, Wang H, Licatalosi DD, Fak JJ, Darnell RB: Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science. 2010, 329: 439-443.
Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT: Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008, 322: 750-756.
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY: Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007, 129: 1311-1323.
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL: Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009, 106: 11667-11672.
Li Y, Altman S: Partial reconstitution of human RNase P in HeLa cells between its RNA subunit with an affinity tag and the intact protein components. Nucleic Acids Res. 2002, 30: 3706-3711.
Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B: Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem. 2007, 389: 1017-1031.
Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, Darnell JC, Darnell RB: HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008, 456: 464-469.
König J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J: iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010, 17: 909-915.
Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T: Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010, 141: 129-141.
Granneman S, Kudla G, Petfalski E, Tollervey D: Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc Natl Acad Sci U S A. 2009, 106: 9613-9618.
Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT: Genome-wide identification of Polycomb-associated RNAs by RIP-seq. Mol Cell. 2010, 40: 939-953.
Singh G, Ricci EP, Moore MJ: RIPiT-Seq: A high-throughput approach for footprinting RNA:protein complexes. Methods. 2013, doi:10.1016/j.ymeth.2013.09.013
Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M: A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods. 2011, 8: 559-564.
Zhang C, Darnell RB: Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol. 2011, 29: 607-614.
Davidovich C, Zheng L, Goodrich KJ, Cech TR: Promiscuous RNA binding by Polycomb repressive complex 2. Nat Struct Mol Biol. 2013, 20: 1250-1257.
Keene JD, Komisarow JM, Friedersdorf MB: RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc. 2006, 1: 302-307.
Oeffinger M, Wei KE, Rogers R, DeGrasse JA, Chait BT, Aitchison JD, Rout MP: Comprehensive analysis of diverse ribonucleoprotein complexes. Nat Methods. 2007, 4: 951-956.
Creamer TJ, Darby MM, Jamonnak N, Schaughency P, Hao H, Wheelan SJ, Corden JL: Transcriptome-wide binding sites for components of the Saccharomyces cerevisiae non-poly(A) termination pathway: Nrd1, Nab3, and Sen1. PLoS Genet. 2001, 7: e1002329-
Mili S, Steitz JA: Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA. 2004, 10: 1692-1694.
Brockdorff N: Noncoding RNA and Polycomb recruitment. RNA. 2013, 19: 429-442.
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY: Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010, 329: 689-693.
Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES, Plath K, Guttman M: The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013, 341: 1237973-
Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y: Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003, 300: 131-135.
Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M, Tuschl T, Ohler U, Keene JD: Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011, 43: 327-339.
Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, Attardi LD, Regev A, Lander ES, Jacks T, Rinn JL: A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010, 142: 409-419.
Brimacombe R, Stiege W, Kyriatsoulis A, Maly P: Intra-RNA and RNA-protein cross-linking techniques in Escherichia coli ribosomes. Methods Enzymol. 1988, 164: 287-309.
Ule J, Jensen K, Mele A, Darnell RB: CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods. 2005, 37: 376-386.
Wang Z, Tollervey J, Briese M, Turner D, Ule J: CLIP: construction of cDNA libraries for high-throughput sequencing from RNAs cross-linked to proteins in vivo. Methods. 2009, 48: 287-293.
Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ: The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell. 2012, 151: 750-764.
Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D: Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010, 24: 2615-2620.
Garber M, Grabherr MG, Guttman M, Trapnell C: Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011, 8: 469-477.
Anders S, Huber W: Differential expression analysis for sequence count data. Genome Biol. 2010, 11: R106-
Rapaport F, Khanin R, Liang Y, Pirun M, Krek A, Zumbo P, Mason CE, Socci ND, Betel D: Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013, 14: R95-
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L: Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012, 7: 562-578.
Kladwang W, Hum J, Das R: Ultraviolet shadowing of RNA can cause significant chemical damage in seconds. Sci Rep. 2012, 2: 517-
Carey J, Cameron V, de Haseth PL, Uhlenbeck OC: Sequence-specific interaction of R17 coat protein with its ribonucleic acid binding site. Biochemistry. 1983, 22: 2601-2610.
Slobodin B, Gerst JE: A novel mRNA affinity purification technique for the identification of interacting proteins and transcripts in ribonucleoprotein complexes. RNA. 2010, 16: 2277-2290.
Tsai BP, Wang X, Huang L, Waterman ML: Quantitative profiling of in vivo-assembled RNA-protein complexes using a novel integrated proteomic approach. Mol Cell Proteomics. 2011, 10: M110.007385-
Bardwell VJ, Wickens M: Purification of RNA and RNA-protein complexes by an R17 coat protein affinity method. Nucleic Acids Res. 1990, 18: 6587-6594.
Lee HY, Haurwitz RE, Apffel A, Zhou K, Smart B, Wenger CD, Laderman S, Bruhn L, Doudna JA: RNA-protein analysis using a conditional CRISPR nuclease. Proc Natl Acad Sci U S A. 2013, 110: 5416-5421.
Bachler M, Schroeder R, von Ahsen U: StreptoTag: a novel method for the isolation of RNA-binding proteins. RNA. 1999, 5: 1509-1516.
Leppek K, Stoecklin G: An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res. 2013, doi:10.1093/nar/gkt956
Srisawat C, Engelke DR: Streptavidin aptamers: affinity tags for the study of RNAs and ribonucleoproteins. RNA. 2001, 7: 632-6341.
Butter F, Scheibe M, Morl M, Mann M: Unbiased RNA-protein interaction screen by quantitative proteomics. Proc Natl Acad Sci U S A. 2009, 106: 10626-10631.
Zielinski J, Kilk K, Peritz T, Kannanayakal T, Miyashiro KY, Eiríksdóttir E, Jochems J, Langel U, Eberwine J: In vivo identification of ribonucleoprotein-RNA interactions. Proc Natl Acad Sci U S A. 2006, 103: 1557-1562.
Zeng F, Peritz T, Kannanayakal TJ, Kilk K, Eiríksdóttir E, Langel U, Eberwine J: A protocol for PAIR: PNA-assisted identification of RNA binding proteins in living cells. Nat Protoc. 2006, 1: 920-927.
Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, Wyler E, Bonneau R, Selbach M, Dieterich C, Landthaler M: The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012, 46: 674-690.
Sutherland BW, Toews J, Kast J: Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. J Mass Spectrom. 2008, 43: 699-715.
Mitchell SF, Jain S, She M, Parker R: Global analysis of yeast mRNPs. Nat Struct Mol Biol. 2013, 20: 127-133.
Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M: Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002, 1: 376-386.
Arun G, Akhade VS, Donakonda S, Rao MR: mrhl RNA, a long noncoding RNA, negatively regulates Wnt signaling through its protein partner Ddx5/p68 in mouse spermatogonial cells. Mol Cell Biol. 2012, 32: 3140-3152.
Newman MA, Thomson JM, Hammond SM: Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA. 2008, 14: 1539-1549.
Zhou Z, Licklider LJ, Gygi SP, Reed R: Comprehensive proteomic analysis of the human spliceosome. Nature. 2002, 419: 182-185.
Scheibe M, Butter F, Hafner M, Tuschl T, Mann M: Quantitative mass spectrometry and PAR-CLIP to identify RNA-protein interactions. Nucleic Acids Res. 2012, 40: 9897-9902.
Castello A, Horos R, Strein C, Fischer B, Eichelbaum K, Steinmetz LM, Krijgsveld J, Hentze MW: System-wide identification of RNA-binding proteins by interactome capture. Nat Protoc. 2013, 8: 491-500.
Freeberg MA, Han T, Moresco JJ, Kong A, Yang YC, Lu ZJ, Yates JR, Kim JK: Pervasive and dynamic protein binding sites of the mRNA transcriptome in Saccharomyces cerevisiae. Genome Biol. 2013, 14: R13-
Kwon SC, Yi H, Eichelbaum K, Föhr S, Fischer B, You KT, Castello A, Krijgsveld J, Hentze MW, Kim VN: The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol. 2013, 20: 1122-1130.
Silverman IM, Li F, Alexander A, Goff L, Trapnell C, Rinn JL, Gregory BD: RNase-mediated protein footprint sequencing reveals protein-binding sites throughout the human transcriptome. Genome Biol. 2014, 15: R3-
Schueler M, Munschauer M, Gregersen LH, Finzel A, Loewer A, Chen W, Landthaler M, Dieterich C: Differential protein occupancy profiling of the mRNA transcriptome. Genome Biol. 2014, 15: RX-
Klass DM, Scheibe M, Butter F, Hogan GJ, Mann M, Brown PO: Quantitative proteomic analysis reveals concurrent RNA-protein interactions and identifies new RNA-binding proteins in Saccharomyces cerevisiae. Genome Res. 2013, 23: 1028-1038.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
McHugh, C.A., Russell, P. & Guttman, M. Methods for comprehensive experimental identification of RNA-protein interactions. Genome Biol 15, 203 (2014). https://doi.org/10.1186/gb4152
Published:
DOI: https://doi.org/10.1186/gb4152