
Abstract
The sequencing of large and complex genomes of crop species, facilitated by new sequencing technologies and bioinformatic approaches, has provided new opportunities for crop improvement. Current challenges include understanding how genetic variation translates into phenotypic performance in the field.
Similar content being viewed by others


Background
Genomics, the analysis of an organism's complete DNA sequence, has been one of the most transformative influences on biological studies. The genome sequences of organisms are fundamentally important for understanding the functions of individual genes and their networks, for defining evolutionary relationships and processes, and for revealing previously unknown regulatory mechanisms that coordinate the activities of genes. These genomics-based approaches are having a profound influence on both human disease diagnostics and treatment [1] and, equally importantly, on the improvement of crops for food and fuel production. In this review, we summarize progress in sequencing crop genomes, identify remaining technical challenges, and describe how genomics-based applications can aid crop improvement. We then assess the impact of genomics on plant breeding and crop improvement, showing how it is accelerating the improvement of staple and 'orphan' crops, and facilitating the utilization of untapped allelic variation. Finally, we speculate about the future impacts of genomics on plant biology and crop improvement by developing the concept of systems breeding, which integrates information on gene function, genome states, and regulatory networks across populations and species to create a predictive framework for estimating the contributions of genetic and epigenetic variation to phenotypes and field performance.
Progress in crop genome sequencing and analysis
Advances in sequencing crop genomes have mirrored the development of sequencing technologies (Table 1). Until 2010, Sanger sequencing of bacterial artificial chromosome (BAC)-based physical maps was the predominant approach used to access crop genomes such as rice, poplar and maize [1–3]. The rice genome comprises complete sequences of individual BACs assembled into physical maps that are anchored to genetic maps, whereas for maize, the sequences of individual BACs were not completely finished. For poplar, grapevine, sorghum and soybean [2, 4–6], whole genome shotgun (WGS) reads of libraries of randomly sheared fragments of different sizes and of BAC end sequences (BES) were assembled with powerful assembly algorithms such as ARACHNE [7, 8]. The trade-offs that shaped genome-sequencing strategies in the era before next-generation sequencing became available involved coverage, time and expense. Physical maps of BACs provide a good template for completing gaps and errors, but genome coverage of physical maps can be non-representative due to cloning bias. In addition, intensive hand-crafting is required to assess physical map integrity and to close gaps; this effort scales directly with genome size and complexity.
The sorghum genome [1–3, 5] was the first crop genome to be sequenced completely by the exclusive use of WGS sequence assemblies, which were then assessed for integrity using high-density genetic maps and physical maps. This pioneering analysis showed that scaffolds of Sanger sequence assemblies accurately span extensive repetitive DNA tracts and extend into telomeric and centromeric regions. The larger soybean genome was subsequently sequenced to similar high standards. The soybean genome is thought to be pseudo-diploid, derived from the diploidization of an allopolyploid in the past 50 million years [2, 4–6, 9], and this project successfully demonstrated that WGS assemblies are not confounded by large-scale genome duplication events.
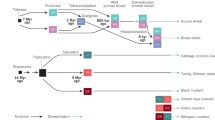
By 2010 to 2011, a mixture of sequencing technologies, all using WGS assembly methods, were being successfully applied to trees (apple, cacao and date palm), fruit (strawberry), vegetables (potato and Chinese cabbage) and forage crops (alfalfa relative) [10–16]. The Medicago and tomato [17] projects, which were initiated in the BAC-based Sanger sequencing era, were completed using next-generation sequencing. The contiguity of assemblies varied according to genome composition and size, with very high contiguity being achieved in potato and alfalfa by alignment to BAC sequences. The Brassica genomes are among the most challenging to sequence with respect to achieving large-scale assemblies because they have undergone three recent whole-genome duplications followed by partial diploidization [18]. Polyploidy has a centrally important role in plant genome evolution and in the formation of important crop genomes. Figure 1 illustrates three examples of polyploidy and how these events contribute to crop genetic diversity in different ways. In Brassica species, polyploidy has led to extensive structural heterogeneity and gene copy number variation when compared with their close relative Arabidopsis. The Brassica rapa genome sequence remains fragmentary, but alignments of Brassica chromosome segments to the Arabidopsis genome are exceptionally useful for advanced genetic analysis [19].

Diverse outcomes of polyploidy in crop species. Three examples of the consequences of allopolyploidy (in which hybrids have sets of chromosomes derived from different species) in important crop species are shown. (a) Oilseed rape (canola) is derived from a recent hybridization of Brassica rapa (Chinese cabbage, turnip) and Brassica oleraceae (broccoli, cauliflower, cabbage). The progenitor of these Brassica species was hexaploid (compared to Arabidopsis) after two rounds of whole-genome duplication. Extensive gene loss, possibly via deletion mechanisms [18], has occurred in these species. Upon hybridization to form allotetraploid Brassica napus, gene loss is accelerated, producing novel patterns of allelic diversity [19]. (b) Bread wheat is an allohexaploid derived from the relatively recent hybridization of allotetraploid durum (pasta) wheat and wild goat grass, Aeglilops tauschii. The Ph1 locus in the B genome [37] prevents pairing between the A, B and D genomes, leading to diploid meiosis and genome stability. This maintains the extensive genetic diversity from the three progenitor Triticeae genomes that underpins wheat crop productivity. (c) Sugarcane (Saccharum sp.) is a complex and unstable polyploid that is cultivated by cuttings. Hybrids between S. officinarum, which has high sugar content, and S. spontaneum, a vigorous wild relative, have variable chromosomal content from each parent. The genomes are closely related to the ancestral diploid Sorghum [42].
In its early stages, crop genomics relied on many small-scale science laboratories joining forces to generate the sequence data. However, this has changed radically with the emergence and leadership of large-scale genome sequencing centers, which focused their expertise and resources on important crop genomes. Two examples are the Joint Genome Institute (JGI) in the USA and the Beijing Genome Institute (BGI, Shenzen) in China, both of which provide exceptional expertise, capacity and levels of engagement with researchers. These centers and others are currently sequencing the genomes of many crucially important food and fuel crops, and are working in collaboration with science groups worldwide on improving our functional understanding of these genomes.
Since 2012, analyses of the sequences of 12 crop genomes have been published, accounting for nearly half of the total published (Table 1) [20–28]. This explosion of data has been driven by cheaper and more effective sequencing technologies (primarily the Illumina [29] and Roche 454 [30] methods) coupled with increasingly sophisticated sequence and assembly strategies [31], which are generally delivered by large genome centers. Access to these technologies makes even a reasonably large crop genome project affordable and feasible within the period of a single research grant, and is having a major influence on strategies in crop genomics. For example, the role of multi-partner coordination has changed from raising and coordinating research funding and managing the distribution of research activities to a focus on data analyses, distribution and applications. These changes will further accelerate and greatly diversify the range of plant species and varieties sequenced.
The date palm genome [12] was sequenced using just paired end reads and remains fragmentary. Although this could be partly attributed to repeat composition, it is clear that the use of mate-pair libraries of different lengths, which provide accurately spaced pairs of sequence reads, substantially improves contiguity across medium-sized genomes of up to 1,000 Mb, as can be seen for citrus, diploid cotton, pigeonpea, chickpea and banana [21, 24, 25, 32, 33]. Contig and scaffold sizes were further increased in chickpea and pigeonpea by incorporating BES generated by Sanger sequence that have much longer read lengths paired over a 100 kb span. Increased lengths of Illumina reads, of up to 250 bases, are now available to users and should further improve contiguity. Using new assembly algorithms, the large genome of bamboo, a plant of major industrial and ecological significance, has recently been published [34].
Table 1 shows progress in sequencing two much larger Triticeae genomes, those of diploid barley (5,100 Mb) [27] and hexaploid bread wheat (17,000 Mb) [28]. Both the exceptional scale and high repeat content (approximately 80%) of these genomes provide significant challenges to straightforward WGS sequencing and assembly, with genes being separated by hundreds of kb of repeats such as nested retroelements [35]. In barley, a physical map of 67,000 BAC clones with a cumulative length of 4.98 Gb provided 304,523 BES reads as a framework for integration of 50X Illumina paired end and 2.5 kb mate pair reads. Contig median size was just 1.5 kb because the repeat content collapsed longer assemblies. Sequence assemblies were integrated with genetic and physical maps, and genic assemblies were assigned to chromosome arms. The chromosomal order of barley genes was then interpolated using synteny across multiple sequenced grass genomes and by ordering the genes according to the genetic or physical maps [36] (Figure 2).

The impact of whole genome sequencing on breeding. (a) Initial genetic maps consisted of few and sparse markers, many of which were anonymous markers (simple sequence repeats (SSR)) or markers based on restriction fragment length polymorphisms (RFLP). For example, if a phenotype of interest was affected by genetic variation within the SSR1-SSR2 interval, the complete region would be selected with little information about its gene content or allelic variation. (b) Whole genome sequencing of a closely related species enabled projection of gene content onto the target genetic map. This allowed breeders to postulate the presence of specific genes on the basis of conserved gene order across species (synteny), although this varies between species and regions. (c) Complete genome sequence in the target species provides breeders with an unprecedented wealth of information that allows them to access and identify variation that is useful for crop improvement. In addition to providing immediate access to gene content, putative gene function and precise genomic positions, the whole genome sequence facilitates the identification of both natural and induced (by TILLING) variation in germplasm collections and copy number variation between varieties. Promoter sequences allow epigenetic states to be surveyed, and expression levels can be monitored in different tissues or environments and in specific genetic backgrounds using RNAseq or microarrays. Integration of these layers of information can create gene networks, from which epistasis and target pathways can be identified. Furthermore, re-sequencing of varieties identifies a high density of SNP markers across genomic intervals, which enable genome-wide association studies (GWAS), genomic selection (GS) and more defined marker-assisted selection (MAS) strategies.
The bread wheat genome is a recent hexaploid composed of three related genomes (A, B and D), each the size of the barley genome, which do not pair and recombine, leading to their independent maintenance [37] (Figure 1). The challenge for wheat WGS strategies was to provide independent assemblies covering and representing genes from each homoeologous genome. The two closest diploid progenitors of the A and D genomes were sequenced to identify polymorphisms that could be used to assess WGS gene assemblies. Low coverage (5X) Roche 454 sequence was generated, and orthologous gene sequences from multiple grasses were used to guide assemblies. Approximately 94,000 genes were assembled and positively assigned to the A and D genomes using genome-specific single nucleotide polymorphisms (SNPs), with the remaining assemblies tentatively assigned to the B genome. Wheat gene assemblies, which are fragmentary compared to barley gene assemblies, were assigned to chromosomes using high-density genetic maps and conserved gene order.
The current wheat and barley gene-based assemblies are suitable for developing genetic markers [38] and for creating genetic maps for map-based cloning and marker-assisted breeding. To increase the gene coverage and contiguity of the barley genome, BACs in the physical map are being multiplex-sequenced using Illumina methods. This will result in chromosome assemblies with fewer gaps and more precisely ordered genes. This should establish barley as the pre-eminent genomic template and genetic reference for the Triticeae. On-going efforts in sequencing the bread wheat genome include sequencing purified flow-sorted chromosome arms to increase gene coverage and the complete assignment of homoeologous genes to the A, B, or D genome [39]. Constructing physical maps of BAC libraries made from purified chromosomes is also underway, with the chromosome 3B physical map [40] and BAC sequencing completed. Given sufficient funding and time, this strategy will provide the necessary high-quality reference genome. Since homoeologous genes can now be assembled and assigned to their genome, WGS can be used to improve the contiguity of wheat gene sequences by using long mate pair spans, in non-overlapping increments up to 40 kb using fosmid vectors [41], coupled to longer read lengths. New template preparation methods, such as Illumina Moleculo, which breaks assemblies down into separate 10 kb units, could be used to span large repeat units and to facilitate accurate assemblies covering large tracts of repeats. Although a colossal amount of sequencing is required, a whole-genome strategy for wheat, supplemented by the flow-sorted chromosome arm data, has the potential to provide users with a high-quality draft sequence relatively quickly and cheaply.
Several industrially important species, such as the conifers Norway spruce (Picea abies) [42] and loblolly pine (Pinus taeda), have very large genomes (approximately 20,000 to 24,000 Mb, respectively). They are being sequenced using WGS strategies involving fosmid pool sequencing and Illumina long-mate pair methods [43]. These tree species have particular characters that facilitate their genome analysis, including the absence of whole-genome duplication in their ancestry, relatively inactive retroelements and the presence of a large multicellular haploid gametophyte, the sequence of which does not exhibit heterozygosity.
Sugar cane, another important crop plant, is a hybrid between Saccharum officinarum and Saccharum spontaneum. These species are closely related to sorghum [44] and have haploid contents of 8 and 10 base chromosomes, respectively. Both S. officinarum and S. spontaneum have a monoploid genome size close to that of sorghum (760 Mb), but they are highly autopolyploid (2n = 80 and 2n = 40-128, respectively), resulting in a genome size of >15 Gb for hybrid sugar cane. Commercial cultivars are derived by backcrossing hybrids to S. officinarum, resulting in lines that have different chromosome contributions from each parental species [45]. The highly variable and heterozygous composition of commercial sugarcane genomes is a major challenge to genome sequencing. The sequencing of progenitor genomes, using WGS strategies and sorghum genes as templates, could create high-stringency orthologous gene assemblies. As in the analysis of the wheat draft genome, this strategy would generate information on ortholog copy number and identify sequence polymorphisms that could be used to genetically map desirable traits in the two progenitor species. Upon the development of commercial hybrids from sequenced progenitors, re-sequencing could identify desired genotypes and gene copy numbers.
A similar approach could be used for the biomass crop Miscanthus x giganteus, a sterile triploid derived from Miscanthus sinensis and tetraploid Miscanthus sacchariflorus. A recent genetic analysis has shown that M. sinensis has recently undergone whole-genome duplication [46] and a single dysploid chromosome fusion [47], neither of which occurred in the closely related sorghum genome [48]. The WGS strategy developed for wheat could be also applied to M. sinensis and its hybrids to determine gene copy numbers and to identify genetic variation in homoeologous gene copies.
Accessing and measuring sequence variation and the epigenome
It is reasonable to predict that within the next two years useful genome sequences will be available to support the genetic improvement of most of the important food and fuel crops. Crop improvement will depend, however, on the identification of useful genetic variation and its utilization by breeding and transformation. Such variation can be identified at a genome scale by comparison of multiple sequence reads to a single 'reference'. For example, in rice, low-coverage sequence of 1,083 Oryza sativa and 466 Oryza rufipogon (the progenitor species of cultivated rice) accessions [49] provided deep insights into the domestication of rice and the geographical distribution of variation, while providing material for quantitative trait loci (QTL) and genome-wide association studies (GWAS) [50]. The gene spaces of maize and wheat varieties are being re-sequenced using sequence capture methods that are based on the solution hybridization of sheared genomic DNA with biotinylated long overlapping oligos designed from gene sequences [51, 52]. The captured DNA is highly enriched in genic sequences, and its deep sequencing can distinguish closely related genes, including wheat homoeologs [53]. These approaches will facilitate the high-throughput sequencing of the gene space of multiple lines of crops, even those with very large polyploid genomes. These methods offer the ability to sequence rapidly the genomes or gene space of multiple accessions, wild relatives and even new species, which will undoubtedly accelerate the incorporation of unexplored and underutilized genetic variation into crops worldwide [54]. DNA sequence variation remains a primary focus, but extensive evidence from several crop species [55, 56] suggests that epigenetic changes are responsible for a range of stably heritable traits, and that epigenetic variation can be both induced and selected for during domestication [57]. The methylation status of captured DNA can be measured using bisulfite treatment followed by deep sequencing in a method called reduced representation bisulfite sequencing (RRBS) [58]. These important technological advances in sequence template preparation will permit the exceptionally detailed and cost-effective definition of variation in the sequences and epigenomes of multiple lines or species of crops, independently of their genome size and polyploid status [59].
Applying next-generation genomics to crop improvement
Accessing genome-wide sequence variation by re-sequencing significantly improves the availability of information that can be used to develop markers, thereby enhancing the genetic mapping of agronomic traits. For example, in wheat, fewer than 500 SNP markers were available in 2008 [60] with that number increasing to 1,536 in 2010 [61], 10,000 in 2011 and over 90,000 in 2012 [38]. This relatively high-density SNP information is proving extremely useful across different systems, including QTL mapping in bi-parental crosses and recombinant inbred lines, GWAS, and mapping QTL in advanced inter-cross lines such as those in multi-parent advanced generation inter-cross (MAGIC) [62] and nested association mapping (NAM) [63] populations. These approaches generally identify loci and causal genes for traits with relatively large phenotypic effects. The genomic segments that contain desired allelic variation can then be bred and combined in a single genetic background using markers to track the segments through marker-assisted selection (MAS).
Many important agricultural traits such as yield, however, result from relatively small effects across multiple loci. This implies that these loci might not be optimally identified through QTL or GWAS approaches and that their pyramiding through MAS will be inefficient. Therefore, breeders have begun to address these problems by developing a knowledge base of associations of polymorphic markers with phenotypes in breeding populations [64, 65]. These associations are used to develop a breeding model in which the frequency of desired marker alleles is optimized, thereby maximizing the estimated breeding value [66, 67]. Multiple cycles of selection are used to accumulate favorable alleles that are associated with desired phenotypes, although no causal relationship between a specific gene and a phenotype is established. This approach, termed genomic selection (GS) is incorporated into industrial-scale breeding processes that require very cheap high-throughput marker assays [68]. Next-generation sequencing of parental lines is influencing GS in several ways: by continuing to identify polymorphisms throughout the genome in both genic and inter-genic regions; by providing estimates of gene expression levels; and by providing information on the epigenetic states of genes (Figure 2). The first removes any limitations on marker density, while the latter two features are 'genomic features' that will surely have predictive power for complex traits. Speculatively, the encyclopedia of DNA elements (ENCODE) concept [69] of total genomic knowledge could eventually be incorporated into models for predicting performance from genomic information revealed by next-generation sequencing.
Breeding uses natural allelic variation to improve crop performance. Sequence variation can be experimentally enhanced using, for example, ethyl methanesulphonate (EMS) to alkylate bases. TILLING (targeted induced local lesions in genomes) [70] is then used to screen for base changes in genes of interest to assess gene function and to create advantageous alleles for breeding. It is now feasible to use genome capture to sequence an entire mutant population, even in complex polyploid genomes such as wheat [52]. Here, polyploidy provides an advantage by buffering the influence of otherwise deleterious mutation loads.
Genetic manipulation using the Agrobacterium tumefaciens-mediated transfer of genes from any other organism is a mature technology that has been adapted for use in many of the crop species listed in Table 1. The precise modification of gene sequences using zinc-finger nucleases (ZFN) that can be engineered to recognize specific DNA sequences has been applied to a target locus in maize [71]. More recently a new type of precision tool for genome engineering has been developed from the prokaryotic clustered regularly interspaced short palindromic repeats (CRISPR) Cas9 immune system [72, 73]. The Cas9 nuclease is guided to specific target sequences for cleavage by an RNA molecule. Several types of genome editing are possible, such as the simultaneous editing of multiple sites, inducing deletions, and inserting new sequences by nick-mediated repair mechanisms.
Genomic features for future breeding
Genomics has radically altered the scope of genetics by providing a landscape of ordered genes and their epigenetic states, access to an enormous range of genetic variation, and the potential to measure gene expression directly with high precision and accuracy (Figure 2). This not only has important practical advantages for breeding but also facilitates systematic comparison of gene functions across sequenced genomes, bringing the wealth of knowledge of gene function and networks obtained in experimental species directly into the ambit of crop improvement. Given a suitable cyber-infrastructure, the integration of biological knowledge and models of networks across species, in a two-way flow from crops to experimental species and back again, will begin to generate new layers of knowledge that can be used for crop improvement. One layer is provided by ENCODE-level analyses [69]; although yet to start in plants, these analyses can guide the interpretation of gene function and variation, thus providing new information to inform the prediction of phenotype from genotype. Another information layer is provided by the systems-level integration of gene function into networks, such as those controlling flowering time in response to day-length and over-wintering (Figure 2). These networks have been identified in Arabidopsis and rice, with allelic variation in key 'hubs' strongly influencing network outputs. Evolutionary processes, such as gene duplication, and the possible footprints of domestication can be mapped to networks such as those controlling flowering time [74, 75]. Such 'systems breeding' approaches can use diverse genomic information to increase the precision with which phenotype can be predicted from genotype, thereby accelerating crop improvement and helping to address food security.
Abbreviations
- BAC:
-
bacterial artificial chromosome
- BES:
-
BAC end sequence
- CRISPR:
-
clustered regularly interspaced short palindromic repeats
- EMS:
-
ethyl methanesulphonate
- ENCODE:
-
Encyclopedia of DNA Elements
- GS:
-
genomic selection
- GWAS:
-
genome-wide association study
- MAGIC:
-
multi-parent advanced generation inter-cross
- MAS:
-
marker-assisted selection
- NAM:
-
nested association mapping
- QTL:
-
quantitative trait loci
- RRBS:
-
reduced representation bisulfite sequencing
- SNP:
-
single nucleotide polymorphism
- TILLING:
-
targeted induced local lesions in genomes
- WGS:
-
whole genome shotgun
- ZFN:
-
zinc-finger nucleases.
References
Sequencing Project IRG: The map-based sequence of the rice genome. Nature. 2005, 436: 793-800. 10.1038/nature03895.
Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen GL, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q, et al: The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science. 2006, 313: 1596-1604. 10.1126/science.1128691.
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, Minx P, Reily AD, Courtney L, Kruchowski SS, Tomlinson C, Strong C, Delehaunty K, Fronick C, Courtney B, Rock SM, Belter E, Du F, Kim K, Abbott RM, Cotton M, Levy A, Marchetto P, Ochoa K, Jackson SM, Gillam B, et al: The B73 maize genome: complexity, diversity, and dynamics. Science. 2009, 326: 1112-1115. 10.1126/science.1178534.
Jaillon O, Aury J-M, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyère C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, et al: The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007, 449: 463-467. 10.1038/nature06148.
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang H, Wang X, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang L, Carpita NC, et al: The sorghum bicolor genome and the diversification of grasses. Nature. 2009, 457: 551-556. 10.1038/nature07723.
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, et al: Genome sequence of the palaeopolyploid soybean. Nature. 2010, 463: 178-183. 10.1038/nature08670.
Hood L, Heath JR, Phelps ME, Lin B: Systems biology and new technologies enable predictive and preventative medicine. Science. 2004, 306: 640-643. 10.1126/science.1104635.
Batzoglou S: ARACHNE: a whole-genome shotgun assembler. Genome Res. 2002, 12: 177-189. 10.1101/gr.208902.
Gill N, Findley S, Walling JG, Hans C, Ma J, Doyle J, Stacey G, Jackson SA: Molecular and chromosomal evidence for allopolyploidy in soybean. Plant Physiol. 2009, 151: 1167-1174. 10.1104/pp.109.137935.
Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, Fontana P, Bhatnagar SK, Troggio M, Pruss D, Salvi S, Pindo M, Baldi P, Castelletti S, Cavaiuolo M, Coppola G, Costa F, Cova V, Dal Ri A, Goremykin V, Komjanc M, Longhi S, Magnago P, Malacarne G, Malnoy M, Micheletti D, Moretto M, Perazzolli M, Si-Ammour A, Vezzulli S, et al: The genome of the domesticated apple (Malus × domestica Borkh.). Nat Genet. 2010, 42: 833-839. 10.1038/ng.654.
Argout X, Salse J, Aury J-M, Guiltinan MJ, Droc G, Gouzy J, Allegre M, Chaparro C, Legavre T, Maximova SN, Abrouk M, Murat F, Fouet O, Poulain J, Ruiz M, Roguet Y, Rodier-Goud M, Barbosa-Neto JF, Sabot F, Kudrna D, Ammiraju JSS, Schuster SC, Carlson JE, Sallet E, Schiex T, Dievart A, Kramer M, Gelley L, Shi Z, Bérard A, et al: The genome of Theobroma cacao. Nat Genet. 2010, 43: 101-108.
Al-Dous EK, George B, Al-Mahmoud ME, Al-Jaber MY, Wang H, Salameh YM, Al-Azwani EK, Chaluvadi S, Pontaroli AC, DeBarry J, Arondel V, Ohlrogge J, Saie IJ, Suliman-Elmeer KM, Bennetzen JL, Kruegger RR, Malek JA: De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat Biotechnol. 2011, 29: 521-527. 10.1038/nbt.1860.
Shulaev V, Sargent DJ, Crowhurst RN, Mockler TC, Folkerts O, Delcher AL, Jaiswal P, Mockaitis K, Liston A, Mane SP, Burns P, Davis TM, Slovin JP, Bassil N, Hellens RP, Evans C, Harkins T, Kodira C, Desany B, Crasta OR, Jensen RV, Allan AC, Michael TP, Setubal JC, Celton J-M, Rees DJG, Williams KP, Holt SH, Rojas JJR, Chatterjee M, et al: The genome of woodland strawberry (Fragaria vesca). Nat Genet. 2010, 43: 109-116.
Xu X, Pan S, Cheng S, Zhang B, Mu D, Ni P, Zhang G, Yang S, Li R, Wang J, Orjeda G, Guzman F, Torres M, Lozano R, Ponce O, Martinez D, la Cruz De G, Chakrabarti SK, Patil VU, Skryabin KG, Kuznetsov BB, Ravin NV, Kolganova TV, Beletsky AV, Mardanov AV, Di Genova A, Bolser DM, Martin DMA, Li G, Yang Y, et al: Genome sequence and analysis of the tuber crop potato. Nature. 2011, 475: 189-195. 10.1038/nature10158.
Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, Bai Y, Mun J-H, Bancroft I, Cheng F, Huang S, Li X, Hua W, Wang J, Wang X, Freeling M, Pires JC, Paterson AH, Chalhoub B, Wang B, Hayward A, Sharpe AG, Park B-S, Weisshaar B, Liu B, Li B, Liu B, Tong C, Song C, Duran C, et al: The genome of the mesopolyploid crop species Brassica rapa. Nat Genet. 2011, 43: 1035-1039. 10.1038/ng.919.
Young ND, Debellé F, Oldroyd GED, Geurts R, Cannon SB, Udvardi MK, Benedito VA, Mayer KFX, Gouzy J, Schoof H, Van de Peer Y, Proost S, Cook DR, Meyers BC, Spannagl M, Cheung F, De Mita S, Krishnakumar V, Gundlach H, Zhou S, Mudge J, Bharti AK, Murray JD, Naoumkina MA, Rosen B, Silverstein KAT, Tang H, Rombauts S, Zhao PX, Zhou P, et al: The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 2011, 480: 520-524.
Sato S, Tabata S, Hirakawa H, Asamizu E, Shirasawa K, Isobe S, Kaneko T, Nakamura Y, Shibata D, Aoki K, Egholm M, Knight J, Bogden R, Li C, Shuang Y, Xu X, Pan S, Cheng S, Liu X, Ren Y, Wang J, Albiero A, Dal Pero F, Todesco S, Van Eck J, Buels RM, Bombarely A, Gosselin JR, Huang M, Leto JA, et al: The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012, 485: 635-641. 10.1038/nature11119.
Town CD, Cheung F, Maiti R, Crabtree J, Haas BJ, Wortman JR, Hine EE, Althoff R, Arbogast TS, Tallon LJ, Vigouroux M, Trick M, Bancroft I: Comparative genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss, fragmentation, and dispersal after polyploidy. Plant Cell. 2006, 18: 1348-1359. 10.1105/tpc.106.041665.
Bancroft I, Morgan C, Fraser F, Higgins J, Wells R, Clissold L, Baker D, Long Y, Meng J, Wang X, Liu S, Trick M: Dissecting the genome of the polyploid crop oilseed rape by transcriptome sequencing. Nat Biotechnol. 2011, 29: 762-766. 10.1038/nbt.1926.
Prochnik S, Marri PR, Desany B, Rabinowicz PD, Kodira C, Mohiuddin M, Rodriguez F, Fauquet C, Tohme J, Harkins T, Rokhsar DS, Rounsley S: The cassava genome: current progress, future directions. Tropical Plant Biol. 2012, 5: 88-94. 10.1007/s12042-011-9088-z.
Varshney RK, Chen W, Li Y, Bharti AK, Saxena RK, Schlueter JA, Donoghue MTA, Azam S, Fan G, Whaley AM, Farmer AD, Sheridan J, Iwata A, Tuteja R, Penmetsa RV, Wu W, Upadhyaya HD, Yang S-P, Shah T, Saxena KB, Michael T, McCombie WR, Yang B, Zhang G, Yang H, Wang J, Spillane C, Cook DR, May GD, Xu X, et al: Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotechnol. 2011, 30: 83-89. 10.1038/nbt.2022.
Bennetzen JL, Schmutz J, Wang H, Percifield R, Hawkins J, Pontaroli AC, Estep M, Feng L, Vaughn JN, Grimwood J, Jenkins J, Barry K, Lindquist E, Hellsten U, Deshpande S, Wang X, Wu X, Mitros T, Triplett J, Yang X, Ye C-Y, Mauro-Herrera M, Wang L, Li P, Sharma M, Sharma R, Ronald PC, Panaud O, Kellogg EA, Brutnell TP, et al: Reference genome sequence of the model plant Setaria. Nat Biotechnol. 2012, 30: 555-561. 10.1038/nbt.2196.
Garcia-Mas J, Benjak A, Sanseverino W, Bourgeois M, Mir G, González VM, Hénaff E, Camara F, Cozzuto L, Lowy E, Alioto T, Capella-Gutiérrez S, Blanca J, Cañizares J, Ziarsolo P, Gonzalez-Ibeas D, Rodríguez-Moreno L, Droege M, Du L, Alvarez-Tejado M, Lorente-Galdos B, Melé M, Yang L, Weng Y, Navarro A, Marques-Bonet T, Aranda MA, Nuez F, Picó B, Gabaldón T, et al: The genome of melon (Cucumis melo L.). Proc Natl Acad Sci USA. 2012, 109: 11872-11877. 10.1073/pnas.1205415109.
Xu Q, Chen L-L, Ruan X, Chen D, Zhu A, Chen C, Bertrand D, Jiao W-B, Hao B-H, Lyon MP, Chen J, Gao S, Xing F, Lan H, Chang J-W, Ge X, Lei Y, Hu Q, Miao Y, Wang L, Xiao S, Biswas MK, Zeng W, Guo F, Cao H, Yang X, Xu X-W, Cheng Y-J, Xu J, Liu J-H, et al: The draft genome of sweet orange (Citrus sinensis). Nat Genet. 2012, 45: 59-66. 10.1038/ng.2472.
Wang K, Wang Z, Li F, Ye W, Wang J, Song G, Yue Z, Cong L, Shang H, Zhu S, Zou C, Li Q, Yuan Y, Lu C, Wei H, Gou C, Zheng Z, Yin Y, Zhang X, Liu K, Wang B, Song C, Shi N, Kohel RJ, Percy RG, Yu JZ, Zhu Y-X, Wang J, Yu S: The draft genome of a diploid cotton Gossypium raimondii. Nat Genet. 2012, 44: 1098-1103. 10.1038/ng.2371.
Paterson AH, Wendel JF, Gundlach H, Guo H, Jenkins J, Jin D, Llewellyn D, Showmaker KC, Shu S, Udall J, Yoo M-J, Byers R, Chen W, Doron-Faigenboim A, Duke MV, Gong L, Grimwood J, Grover C, Grupp K, Hu G, Lee T-H, Li J, Lin L, Liu T, Marler BS, Page JT, Roberts AW, Romanel E, Sanders WS, Szadkowski E, et al: Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 2012, 492: 423-427. 10.1038/nature11798.
Mayer KFX, Waugh R, Langridge P, Close TJ, Wise RP, Graner A, Matsumoto T, Sato K, Schulman A, Muehlbauer GJ, Stein N, Ariyadasa R, Schulte D, Poursarebani N, Zhou R, Steuernagel B, Mascher M, Scholz U, Shi B, Langridge P, Madishetty K, Svensson JT, Bhat P, Moscou M, Resnik J, Close TJ, Muehlbauer GJ, Hedley P, Liu H, Morris J, et al: A physical, genetic and functional sequence assembly of the barley genome. Nature. 2012, 491: 711-716.
Brenchley R, Spannagl M, Pfeifer M, Barker GLA, D'Amore R, Allen AM, McKenzie N, Kramer M, Kerhornou A, Bolser D, Kay S, Waite D, Trick M, Bancroft I, Gu Y, Huo N, Luo M-C, Sehgal S, Gill B, Kianian S, Anderson O, Kersey P, Dvorak J, McCombie WR, Hall A, Mayer KFX, Edwards KJ, Bevan MW, Hall N: Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature. 2012, 491: 705-710. 10.1038/nature11650.
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, et al: Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008, 456: 53-59. 10.1038/nature07517.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen Y-J, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim J-B, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, et al: Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2006, 441: 120-
Maccallum I, Przybylski D, Gnerre S, Burton J, Shlyakhter I, Gnirke A, Malek J, McKernan K, Ranade S, Shea TP, Williams L, Young S, Nusbaum C, Jaffe DB: ALLPATHS 2: small genomes assembled accurately and with high continuity from short paired reads. Genome Biol. 2009, 10: R103-10.1186/gb-2009-10-10-r103.
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Tar'an B, Millan T, Zhang X, Ramsay LD, Iwata A, Wang Y, Nelson W, Farmer AD, Gaur PM, Soderlund C, Penmetsa RV, Xu C, Bharti AK, He W, Winter P, Zhao S, Hane JK, Carrasquilla-Garcia N, Condie JA, Upadhyaya HD, Luo M-C, et al: Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol. 2013, 31: 240-246. 10.1038/nbt.2491.
D'Hont A, Denoeud F, Aury J-M, Baurens F-C, Carreel F, Garsmeur O, Noel B, Bocs S, Droc G, Rouard M, Da Silva C, Jabbari K, Cardi C, Poulain J, Souquet M, Labadie K, Jourda C, Lengellé J, Rodier-Goud M, Alberti A, Bernard M, Correa M, Ayyampalayam S, Mckain MR, Leebens-Mack J, Burgess D, Freeling M, Mbéguié-A-Mbéguié D, Chabannes M, Wicker T, et al: The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature. 2012, 488: 213-217. 10.1038/nature11241.
Peng Z, Lu Y, Li L, Zhao Q, Feng Q, Gao Z, Lu H, Hu T, Yao N, Liu K, Li Y, Fan D, Guo Y, Li W, Lu Y, Weng Q, Zhou C, Zhang L, Huang T, Zhao Y, Zhu C, Liu X, Yang X, Wang T, Miao K, Zhuang C, Cao X, Tang W, Liu G, Liu Y, et al: The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat Genet. 2013, 45: 456-461. 10.1038/ng.2569.
Choulet F, Wicker T, Rustenholz C, Paux E, Salse J, Leroy P, Schlub S, Le Paslier M-C, Magdelenat G, Gonthier C, Couloux A, Budak H, Breen J, Pumphrey M, Liu S, Kong X, Jia J, Gut M, Brunel D, Anderson JA, Gill BS, Appels R, Keller B, Feuillet C: Megabase level sequencing reveals contrasted organization and evolution patterns of the wheat gene and transposable element spaces. Plant Cell. 2010, 22: 1686-1701. 10.1105/tpc.110.074187.
Mayer KFX, Martis M, Hedley PE, Simková H, Liu H, Morris JA, Steuernagel B, Taudien S, Roessner S, Gundlach H, Kubaláková M, Suchánková P, Murat F, Felder M, Nussbaumer T, Graner A, Salse J, Endo T, Sakai H, Tanaka T, Itoh T, Sato K, Platzer M, Matsumoto T, Scholz U, Dolezel J, Waugh R, Stein N: Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell. 2011, 23: 1249-1263. 10.1105/tpc.110.082537.
Griffiths S, Sharp R, Foote TN, Bertin I, Wanous M, Reader S, Colas I, Moore G: Molecular characterization of Ph1 as a major chromosome pairing locus in polyploid wheat. Nature. 2006, 439: 749-752. 10.1038/nature04434.
Allen AM, Barker GLA, Berry ST, Coghill JA, Gwilliam R, Kirby S, Robinson P, Brenchley RC, D'Amore R, McKenzie N, Waite D, Hall A, Bevan M, Hall N, Edwards KJ: Transcript-specific, single-nucleotide polymorphism discovery and linkage analysis in hexaploid bread wheat (Triticum aestivum L.). Plant Biotechnol J. 2011, 9: 1086-1099. 10.1111/j.1467-7652.2011.00628.x.
Berkman PJ, Skarshewski A, Lorenc MT, Lai K, Duran C, Ling EYS, Stiller J, Smits L, Imelfort M, Manoli S, McKenzie M, Kubaláková M, Simková H, Batley J, Fleury D, Dolezel J, Edwards D: Sequencing and assembly of low copy and genic regions of isolated Triticum aestivum chromosome arm 7DS. Plant Biotechnol J. 2011, 9: 768-775. 10.1111/j.1467-7652.2010.00587.x.
Paux E, Sourdille P, Salse J, Saintenac C, Choulet F, Leroy P, Korol A, Michalak M, Kianian S, Spielmeyer W, Lagudah E, Somers D, Kilian A, Alaux M, Vautrin S, Bergès H, Eversole K, Appels R, Safár J, Simková H, Dolezel J, Bernard M, Feuillet C: A physical map of the 1-gigabase bread wheat chromosome 3B. Science. 2008, 322: 101-104. 10.1126/science.1161847.
Williams LJS, Tabbaa DG, Li N, Berlin AM, Shea TP, MacCallum I, Lawrence MS, Drier Y, Getz G, Young SK, Jaffe DB, Nusbaum C, Gnirke A: Paired-end sequencing of Fosmid libraries by Illumina. Genome Res. 2012, 22: 2241-2249. 10.1101/gr.138925.112.
Nystedt B, Street NR, Wetterbom A, Zuccolo A, Lin Y-C, Scofield DG, Vezzi F, Delhomme N, Giacomello S, Alexeyenko A, Vicedomini R, Sahlin K, Sherwood E, Elfstrand M, Gramzow L, Holmberg K, Hällman J, Keech O, Klasson L, Koriabine M, Kucukoglu M, Käller M, Luthman J, Lysholm F, Niittylä T, Olson A, Rilakovic N, Ritland C, Rosselló JA, Sena J, et al: The Norway spruce genome sequence and conifer genome evolution. Nature. 2013, 497: 579-584. 10.1038/nature12211.
Dendrome. [http://dendrome.ucdavis.edu/]
Jannoo N, Grivet L, Chantret N, Garsmeur O, Glaszmann J-C, Arruda P, D'Hont A: Orthologous comparison in a gene-rich region among grasses reveals stability in the sugarcane polyploid genome. Plant J. 2007, 50: 574-585. 10.1111/j.1365-313X.2007.03082.x.
Grivet L, Arruda P: Sugarcane genomics: depicting the complex genome of an important tropical crop. Curr Opin Plant Biol. 2002, 5: 122-127. 10.1016/S1369-5266(02)00234-0.
Swaminathan K, Chae WB, Mitros T, Varala K, Xie L, Barling A, Glowacka K, Hall M, Jezowski S, Ming R, Hudson M, Juvik JA, Rokhsar DS, Moose SP: A framework genetic map for Miscanthus sinensis from RNAseq-based markers shows recent tetraploidy. BMC Genomics. 2012, 13: 142-10.1186/1471-2164-13-142.
Luo MC, Deal KR, Akhunov ED, Akhunova AR, Anderson OD, Anderson JA, Blake N, Clegg MT, Coleman-Derr D, Conley EJ, Crossman CC, Dubcovsky J, Gill BS, Gu YQ, Hadam J, Heo HY, Huo N, Lazo G, Ma Y, Matthews DE, McGuire PE, Morrell PL, Qualset CO, Renfro J, Tabanao D, Talbert LE, Tian C, Toleno DM, Warburton ML, You FM, et al: Genome comparisons reveal a dominant mechanism of chromosome number reduction in grasses and accelerated genome evolution in Triticeae. Proc Natl Acad Sci USA. 2009, 106: 15780-15785. 10.1073/pnas.0908195106.
Ming R, Liu SC, Lin YR, da Silva J, Wilson W, Braga D, van Deynze A, Wenslaff TF, Wu KK, Moore PH, Burnquist W, Sorrells ME, Irvine JE, Paterson AH: Detailed alignment of saccharum and sorghum chromosomes: comparative organization of closely related diploid and polyploid genomes. Genetics. 1998, 150: 1663-1682.
Huang X, Kurata N, Wei X, Wang Z-X, Wang A, Zhao Q, Zhao Y, Liu K, Lu H, Li W, Guo Y, Lu Y, Zhou C, Fan D, Weng Q, Zhu C, Huang T, Zhang L, Wang Y, Feng L, Furuumi H, Kubo T, Miyabayashi T, Yuan X, Xu Q, Dong G, Zhan Q, Li C, Fujiyama A, Toyoda A, et al: A map of rice genome variation reveals the origin of cultivated rice. Nature. 2012, 490: 497-501. 10.1038/nature11532.
Huang X, Zhao Y, Wei X, Li C, Wang A, Zhao Q, Li W, Guo Y, Deng L, Zhu C, Fan D, Lu Y, Weng Q, Liu K, Zhou T, Jing Y, Si L, Dong G, Huang T, Lu T, Feng Q, Qian Q, Li J, Han B: Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet. 2011, 44: 32-39. 10.1038/ng.1018.
Lai J, Li R, Xu X, Jin W, Xu M, Zhao H, Xiang Z, Song W, Ying K, Zhang M, Jiao Y, Ni P, Zhang J, Li D, Guo X, Ye K, Jian M, Wang B, Zheng H, Liang H, Zhang X, Wang S, Chen S, Li J, Fu Y, Springer NM, Yang H, Wang J, Dai J, Schnable PS, et al: Genome-wide patterns of genetic variation among elite maize inbred lines. Nat Genet. 2010, 42: 1027-1030. 10.1038/ng.684.
Winfield MO, Wilkinson PA, Allen AM, Barker GLA, Coghill JA, Burridge A, Hall A, Brenchley RC, D'Amore R, Hall N, Bevan MW, Richmond T, Gerhardt DJ, Jeddeloh JA, Edwards KJ: Targeted re-sequencing of the allohexaploid wheat exome. Plant Biotechnol J. 2012, 10: 733-742. 10.1111/j.1467-7652.2012.00713.x.
Saintenac C, Jiang D, Akhunov ED: Targeted analysis of nucleotide and copy number variation by exon capture in allotetraploid wheat genome. Genome Biol. 2011, 12: R88-10.1186/gb-2011-12-9-r88.
Varshney R, Graner A, Sorrells M: Genomics-assisted breeding for crop improvement. Trends Plant Sci. 2005, 10: 621-630. 10.1016/j.tplants.2005.10.004.
Kashkush K, Feldman M, Levy AA: Gene loss, silencing and activation in a newly synthesized wheat allotetraploid. Genetics. 2002, 160: 1651-1659.
Shivaprasad PV, Dunn RM, Santos BA, Bassett A, Baulcombe DC: Extraordinary transgressive phenotypes of hybrid tomato are influenced by epigenetics and small silencing RNAs. EMBO J. 2012, 31: 257-266.
Feldman M, Levy AA: Genome evolution in allopolyploid wheat - a revolutionary reprogramming followed by gradual changes. J Genet Genomics. 2009, 36: 511-518. 10.1016/S1673-8527(08)60142-3.
Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R: Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33: 5868-5877. 10.1093/nar/gki901.
Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O, Alix A, McCosh RB, Chen H, Schork NJ, Ecker JR: Patterns of population epigenomic diversity. Nature. 2013, 495: 193-198. 10.1038/nature11968.
Chao S, Zhang W, Akhunov E, Sherman J, Ma Y, Luo M-C, Dubcovsky J: Analysis of gene-derived SNP marker polymorphism in US wheat (Triticum aestivum L.) cultivars. Mol Breeding. 2008, 23: 23-33.
Chao S, Dubcovsky J, Dvorak J, Luo M-C, Baenziger SP, Matnyazov R, Clark DR, Talbert LE, Anderson JA, Dreisigacker S, Glover K, Chen J, Campbell K, Bruckner PL, Rudd JC, Haley S, Carver BF, Perry S, Sorrells ME, Akhunov ED: Population- and genome-specific patterns of linkage disequilibrium and SNP variation in spring and winter wheat (Triticum aestivum L.). BMC Genomics. 2010, 11: 727-10.1186/1471-2164-11-727.
Kover PX, Valdar W, Trakalo J, Scarcelli N, Ehrenreich IM, Purugganan MD, Durrant C, Mott R: A multiparent advanced generation inter-cross to fine-map quantitative traits in Arabidopsis thaliana. PLoS Genet. 2009, 5: e1000551-10.1371/journal.pgen.1000551.
McMullen MD, Kresovich S, Villeda HS, Bradbury P, Li H, Sun Q, Flint-Garcia S, Thornsberry J, Acharya C, Bottoms C, Brown P, Browne C, Eller M, Guill K, Harjes C, Kroon D, Lepak N, Mitchell SE, Peterson B, Pressoir G, Romero S, Oropeza Rosas M, Salvo S, Yates H, Hanson M, Jones E, Smith S, Glaubitz JC, Goodman M, Ware D, et al: Genetic properties of the maize nested association mapping population. Science. 2009, 325: 737-740. 10.1126/science.1174320.
Morrell PL, Buckler ES, Ross-Ibarra J: Crop genomics: advances and applications. Nat Rev Genet. 2011, 13: 85-96.
Meuwissen TH, Hayes BJ, Goddard ME: Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001, 157: 1819-1829.
Xu SS: Estimating polygenic effects using markers of the entire genome. Genetics. 2003, 163: 789-801.
Heffner EL, Sorrells ME, Jannink J-L: Genomic selection for crop improvement. Crop Sci. 2009, 49: 1-10.2135/cropsci2008.08.0512.
Eathington SR, Crosbie TM, Edwards MD, Reiter RS, Bull JK: Molecular markers in a commercial breeding program. Crop Sci. 2007, 47: S154-S163.
The ENCODE Project Consortium: The ENCODE (ENCyclopedia Of DNA Elements) project. Science. 2004, 306: 636-640.
Uauy C, Paraiso F, Colasuonno P, Tran RK, Tsai H, Berardi S, Comai L, Dubcovsky J: A modified TILLING approach to detect induced mutations in tetraploid and hexaploid wheat. BMC Plant Biol. 2009, 9: 115-10.1186/1471-2229-9-115.
Shukla VK, Doyon Y, Miller JC, DeKelver RC, Moehle EA, Worden SE, Mitchell JC, Arnold NL, Gopalan S, Meng X, Choi VM, Rock JM, Wu Y-Y, Katibah GE, Zhifang G, McCaskill D, Simpson MA, Blakeslee B, Greenwalt SA, Butler HJ, Hinkley SJ, Zhang L, Rebar EJ, Gregory PD, Urnov FD: Precise genome modification in the crop species Zea mays using zinc-finger nucleases. Nature. 2009, 459: 437-441. 10.1038/nature07992.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E: A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012, 337: 816-821. 10.1126/science.1225829.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM: RNA-guided human genome engineering via Cas9. Science. 2013, 339: 823-826. 10.1126/science.1232033.
Higgins JA, Bailey PC, Laurie DA: Comparative genomics of flowering time pathways using Brachypodium distachyon as a model for the temperate grasses. PLoS ONE. 2010, 5: e10065-10.1371/journal.pone.0010065.
Yan L, Fu D, Li C, Blechl A, Tranquilli G, Bonafede M, Sanchez A, Valarik M, Yasuda S, Dubcovsky J: The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc Natl Acad Sci USA. 2006, 103: 19581-19586. 10.1073/pnas.0607142103.
Acknowledgements
This work was supported by grants BB/J004588/1, BB/J004596/1 and BB/J003557/1 from the UK Biotechnology and Biological Sciences Research Council (BBSRC) and the John Innes Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Bevan, M.W., Uauy, C. Genomics reveals new landscapes for crop improvement. Genome Biol 14, 206 (2013). https://doi.org/10.1186/gb-2013-14-6-206
Published:
DOI: https://doi.org/10.1186/gb-2013-14-6-206