
Abstract
Genome-wide association studies and comparative genomics have established major loci and specific polymorphisms affecting human skin, hair and eye color. Environmental changes have had an impact on selected pigmentation genes as populations have expanded into different regions of the globe.
Similar content being viewed by others
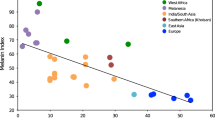
Introduction
Skin, hair and eye color vary dramatically among geographically and temporally separated human populations. It has long been speculated that this is due to adaptive changes, but the genetic causes and environmental selective pressures underlying this range of phenotypic variation have remained largely unknown. We now know of a large number of genes that impact on human pigmentation, especially in the melanosome biogenesis or the melanin biosynthetic pathways [1], and we are now in a position to characterize the genetic variation underlying the diversity seen in these pigmentation traits that has arisen during hominid evolution [2]. Recently, several locus-specific and genome-wide association studies (GWAS) searching for signatures of positive selection have highlighted relatively few and distinct loci in these pigmentation pathways, suggesting convergent evolution has occurred in different populations. Here we assess these population genetic findings in light of our current understanding of pigment biology.
The key pigment molecule melanin is an inert biopolymer produced by melanocyte cells present in the skin and hair follicles. It is transported within melanosome particles along thin cellular projections into the surrounding keratinocytes of the epidermis or the cortical region of the growing root sheath, but is retained in the iridial melanocytes of the eye [3]. Variations in genes within this pathway are therefore in a position to be pleiotropic in action, causing skin, hair and eye color to become correlated, for example, in Northern European populations with a high frequency of light hair, light skin and blue eyes or equatorial Africans with dark complexion, dark hair and brown eye color. However, since melanocytes located in these three compartments represent independent cellular populations [4] with alternative regulatory or signaling pathways [5], trait-specific variants also occur, producing assorted phenotypic combinations such as dark hair, light skin and blue eyes common in Europeans or the light hair, dark skin and brown eyes seen in Solomon Islanders. Another example of this is the sensitivity of follicular melanocytes to aging, gradually producing a silver-gray to white hair color, indicating a loss of cells from the bulb region over the years [6]. This happens in all humans, but age of onset varies both between and within ethnic groups. Notably, skin color has only been a target for natural selection in hominids following the development of hairlessness [7], and a genetic change allowing melanocytes to reside in the epidermis must therefore have occurred early during our evolution.
The biochemical pathway of melanogenesis, converting the amino acids phenylalanine, tyrosine and cysteine to melanin, is under complex genetic control involving the catalytic enzymes, structural matrix and ion-transport proteins of the melanosome (Figure 1) [8], trafficking molecules involved in melanosome maturation and export [9, 10] and ultimately degradation [11]. It also depends on regulatory pathways involving receptors, growth factors and transcription factors [12]. The discovery and characterization of human pigmentation genes (Table 1), often predicated on genes mapped to an animal coat color phenotype, and polymorphism of these genes within and between human populations, combined with functional studies, have provided the framework to understand normal variation in this physical trait and the imprints of the environment upon our genome.

Melanin formation in the melanosome. The conversion of phenylalanine to tyrosine by phenylalanine dehydroxylase (PAH) takes place in the cytoplasm of melanocytes and is necessary to maintain the supply of this substrate for melanogenesis to occur continuously, with its activity positively correlated with skin-type [140]. Active uptake of tyrosine by the melanosome is required, and is initiated by the process of oxidation by tyrosinase (TYR) and involves other enzymes such as DHI oxidase (TYRP1) and dopachrome tautomerase (DCT). Ion transport is critical to melanosome function, with TYR activity being pH-dependent and its absolute activity being critical for the rate of melanin production. The coupling of H+, Na+, Ca2+ and K+ transport by the V-ATP complex, with the involvement of SLC45A2, SLC24A5 and TPCN2 in the regulation of this process, is shown. Cystine as a negative regulator of melanogenesis is pumped out by CTNS. The SILV protein forms the matrix backbone through specific proteolysis of a precursor protein, upon which eumelanin is deposited. The melanosomes are then transported along thin cellular projections known as dendrites and deposited within keratinoctyes, which are the cells that take up the pigment and give the visible color.
Color genes and their variants
The earliest studies of pigmentation examined Mendelian inheritance, identifying genes of high penetrance. Mutations that eliminate melanin synthesis, causing the dilution of mouse coat colors, the human oculocutaneous albinisms (OCA1-4) [13] and related syndromic disorders of broader effect such as Hermansky-Pudlak syndrome (HPS1-8), initially allowed TYR, OCA2, TYRP1, SLC45A2 and HPS6 to be defined as human pigmentation-related genes.
After establishing the genetic basis of several pigmentation disease phenotypes, the next and more challenging step was to assign the major genes responsible for the normal spectrum of human coloration, which although a polygenic trait, was likely to be characterized by a limited number of major genes [14]. Using these disease genes as candidates and others established by the animal models, the search for alleles of major effect in humans was performed by genetic association tests. There are now numerous reports demonstrating association of general or population-specific polymorphisms within and flanking these loci with normal variation in skin, hair or eye color traits, while later analyses took the form of GWAS or examined identified candidate single nucleotide polymorphisms (SNPs) in large population groups [15–25], some of which have already been summarized [26]. These combined approaches have allowed identification of candidate SNPs, loci and genomic regions, with the genes identified summarized in Table 1. Each can then be tested for evidence of natural selection (Box 1).
Signs of selection
Once genes and variants apparently associated with color differences are identified it is possible to examine genome-wide SNP data using population genetic tests for selection. Such tools include decreased heterozygosity (LnRH), atypical levels of population differentiation of alleles (Fst) or decay of linkage disequilibrium (extended haplotype homozygosity) [27], or combinations of such tests [28]. Many of these approaches have found clear evidence for a selection of SNPs within pigmentation-related genes as being statistically significantly different when tested in different populations [28–37] (Table 2). One recent study of African, European and Asian samples has attempted to test and compare results of these methods, based on common SNPs on genotyping arrays to results from DNA resequencing of designated regions: OCA2, TYRP1, DCT and KITLG [38]. Surprisingly, the strong selection found by haplotype-based statistical tests was not replicated by neutrality tests based on the sequence data. Larger data sets of coding or exonic regions [39] may improve detection of population-specific selective sweeps [40].
Most studies searching for genes under selection in humans have accessed a limited population data set, such as the International HapMap Panel [41], but further data are continually becoming available from the 1000 Genomes Project [42].
Population genetic structure has been investigated by principal components analysis (PCA) of ancestry informative markers (AIMs) in large-scale genetic data obtained from thousands of individuals using the Human Genome Diversity Panel (HGDP) as a reference (1,043 individuals, 51 populations, 650,000 SNPs) [43]. In this fine-scale study using the AIM panel for differentiating individuals into the five broad geographic regions, the two top-scoring SNPs sit in a region no more than 30 kb from the SLC24A5 gene, with several other pigmentation genes also being strong markers. Although this is consistent with selection of these genes in the different groups, such population differentiation may equally have arisen due to the population bottlenecks that have occurred during human evolution (Box 1). Distinguishing between demographic forces and natural selection is extremely difficult [43], so additional evidence using other statistical approaches is required.
Hancock et al. [44] used allele-frequency data to test for adaptations to continuous climate variables at the genome-wide level and to identify genetic loci that underlie these adaptations in 61 populations worldwide. In addition to the HGDP dataset described earlier, this includes extra populations to expand information in Africa and Oceania. They gathered environmental data for nine continuous climate variables that have a strong impact on human physiology; however, these climate variables are simple proxies for selective pressures that are likely to be much more complex. These analyses did adjust for demographic history, and detected correlations between climate variables and a number of SNPs previously identified by GWAS as playing a prominent role in pigmentation and immune response phenotypes. The top candidate genes from the pigmentation and tanning pathways were SLC45A2 and OCA2, though notably MC1R, which is known to play such an important role in these processes, was not identified. The OCA2 gene was also found to be under selection in the European population using a model-based approach incorporating spatial ancestry analysis (SPA) of the HGDP dataset, in which large gradients in SNP allele frequencies in two- or three-dimensional geographical space are searched for [45].
Human pigmentation genes with signs of selection pressure
It is perhaps surprising that few genes of major influence in human pigmentation, and none with common polymorphism, have been described or characterized since our last review of the molecular genetics of human pigmentation [3]. Those under positive selection must directly alter the expression level of the gene transcript (OCA2, IRF4), be in linkage with functional SNPs that do so (ASIP, TYRP1, DCT, KITLG) or modify the biochemical activity of a protein (MC1R, TYR, SLC24A5, SLC45A2) [46]. The fact that there have been only a limited number of key genes identified implies any new discoveries influencing human pigmentation will represent a smaller and smaller proportion of phenotypic variation in these traits, or, in the case of CORIN [44], OPRM1 [47]NRG1 [48], BNC2 [49, 50] and CTNS [51], will be of as yet unknown influence, function or significance at the population level. This is in line with what is seen with other polygenic traits and human phenotypes, such as height and body weight [52].
The polymorphisms present in several human pigmentation genes will now be discussed in specific detail to highlight the different types of changes occurring under natural selection. For some loci these changes occur in coding regions, while others are distal regulatory or intronic changes. In several cases the functional variant is yet to be identified, and there are strong associations with SNPs tagging the trait-associated haplotype.
MC1R (Ch16q24.3) and ASIP(Ch20q11.2)
The human melanocortin-1 receptor (MC1R) coding region is intronless and spans less than 1 kb [53] on chromosome 16; sequencing of this region has been used to test for selection for some years [54, 55]. In the case of MC1R, for example, multiple variants with the same effects on pigmentation appear at increased frequency with increasing latitude in multiple populations. In Europeans MC1R variant alleles are associated with pheomelanic red hair, fair skin and freckling, as well as skin cancer risk [56].
Savage et al. [57] showed that MC1R allelic diversity was low in Africa and high in Europe, and population differentiation was greatest between Asia and other populations [58]. Two tests for selection were applied. The Tajima D statistic was significantly negative in Africa and Southern Europe, and suggestive for Northern Europe, while Fu's Fs test was significantly negative in both Southern and Northern European samples. These results are consistent with the action of positive selection in the European populations.
Being relatively well studied, human pigmentation loci are a useful window into the distribution of mutation effect sizes. The usual theoretical model predicts an exponential distribution, with relatively more mutations of small effect. MC1R is especially interesting because it is a small intronless gene where polymorphism has repeatedly arisen quickly (5,000 to 10,000 years) under evolutionary pressure in vertebrates from fish to humans [59]. In humans, more than 90 nonsynonymous, frameshift or stop mutations are known to affect the activity of the protein, and eight of these are present at greater than 1% frequency in different populations. In European populations, compound MC1R heterozygotes are common (in an Australian twin sample, four times more common than homozygotes). Penetrance of compound heterozygotes is quite consistent with a multiplicative allelic model for hair color [60, 61]. This observation extends to epistatic effects of other pigmentation traits [62], with one mild exception being the interaction between MC1R and an ASIP rs4911442*C/T-tagged haplotype.
OCA2(Ch15q11.2)
The OCA2 gene, homologous to the mouse pink-eye dilute locus, is located on chromosome 15 and downstream of the HERC locus. The regulation of its expression has been proposed to exert the strongest influence on iris color [3], as well as having associations with skin and hair color [46] in European-derived populations. The OCA2 protein is thought to be a mature melanosomal membrane protein [63], with a potential role in trafficking other proteins to melanosomes [64]. The key determinant SNP rs12913832*T/C for the regulation of OCA2 expression is located in a short highly conserved region within intron 86 of the HERC2 gene, 21 kb upstream of the promoter of OCA2. The rs12913832*C-derived allele is highly associated with European blue eye color as a recessive trait. The ability of this conserved element to act as an enhancer regulating OCA2 transcription has recently been confirmed in experiments using melanocyte cultures carrying either rs12913832*T/T or rs12913832*C/C genotypes [65]. A long-range chromatin loop between this enhancer and the OCA2 promoter leads to elevated OCA2 expression in darkly pigmented rs12913832*T/T cells, but not in lightly pigmented rs12913832*C/C cells, concomitant with a reduction in transcription factor recruitment to this intronic region. This is the first validation that allele-dependent differences in chromatin loop formation result in differences in allelic gene expression effecting a common phenotypic trait, and serves as a paradigm for future studies for how common SNPs may direct changes in gene regulation.
The Asian-specific nonsynonymous SNP rs1800414*A/G in the OCA2 locus was first reported during the sequencing of the exonic regions of the human gene to establish its structure and to screen for mutations associated with albinism [66]. The rs1800414*G-derived allele is a His615Arg change in the protein encoded by exon 18, and was initially reported to be almost exclusively found in East and Southeast Asian populations [67, 68]. The influence of this allele on skin pigmentation was later demonstrated in an association study by measurement of melanin index in a sample of 122 individuals of East Asian ancestry and a replicate population with 207 Han Chinese [69]. This clearly demonstrated that the rs1800414*G/615Arg allele was present in those with lower melanin levels. An analysis of the rs1800414 SNP in a larger collection of samples from 72 populations [70] showed that this is essentially restricted to East Asia with frequencies as high as 0.76 for rs1800414*G and acting as a skin-lightening allele. Another hypofunctional OCA2 variant allele is rs74653330*G/A encoding Ala481Thr, with 70% of the function of the wild-type OCA2 protein [71]. This allele was first reported to be at high frequency in Northeast Asia [72], and a later study of 24 populations showed the highest frequency was 0.52 in the Oroquen population in China [73]. These reports provide evidence for lighter skin pigmentation evolving by means of selection acting at least partly genetically independently in Europeans and East Asians, that is, convergent evolution. However, in Europeans selection may have been predominantly for lighter eye color.
TYR(Ch11q14)
Tyrosinase (TYR) was the first human pigmentation gene to be identified and characterized, because many mutation reports show it to cause OCA1. Notably, TYR accounts for 46% of albinism cases in Caucasians [13], indicative of a frequency of mutant alleles approaching 1/100 in northern Europe [74]. Recent attention has been concentrated on two coding polymorphisms of TYR that appear at high frequency in Europeans and are largely absent in African populations: rs1042602*C/A Ser192Tyr, and rs1126809*G/A Arg402Gln. In a population of South Asian descent, the rs1042602*A/192Tyr allele was shown to be highly associated with lighter skin color [23]. Later work studied the frequency of rs1042602 in 1,871 normal Indian volunteers comprising 55 ethnic groups, and reported that the rs1042602*A/192Tyr allele was overrepresented in the Indo-Europeans [75]; however, no phenotypic associations with skin color were performed. Functional studies of the TYR protein accompanying this work revealed a tyrosinase enzyme activity for 192Tyr of only 60% of wild-type, possibly due to steric hindrance effects in the TYR protein copper-A catalytic site. The rs1126809*G/A Arg402Gln polymorphism also has reduced enzyme activity, with expression studies showing that it encodes a thermolabile variant protein with only 25% activity of wild type. The pathology of this mutation in Europeans has been controversial, with some reports questioning its association with albinism [13, 76]; this may be best reconciled by realizing the full spectrum of the albinism phenotype and the influence of the underlying skin complexion of the individual's ethnic background on this condition [77]. The relationship of these TYR coding variants to normal variation in pigmentation in Europeans has been studied by several groups [15, 25, 62, 78], with reported associations for lighter eye color, freckling and melanoma. Recently, both alleles have been shown to be protective against the development of vitiligo [79].
TYRP1 (Ch9p23) and DCT(Ch13q32)
While TYR catalyses the key initial step in melanin production, TYRP1 and DCT (TYRP2) act at subsequent steps, influencing the quantity and the quality of melanins and stabilizing the TYR enzyme. They also have some function in melanocyte survival after ultraviolet radiation (UVR) stress and in the maintenance of melanosomal structures. The TYRP gene family evolved by recurrent gene duplication from a common ancestral TYR early in evolution giving rise to TYRP1 and DCT [80]. In humans, mutations of TYRP1 causing OCA3 are frequently observed in South Africa [13]. Part of this phenotype is red bronze skin, ginger-red hair and blue irides. In a surprising finding, it was recently reported that a nonsynonymous amino acid change, Arg93Cys, in the TYRP1 protein is a major determinant of the blonde hair phenotype. This allele has a frequency of 0.26 on the Solomon Islands and is not seen outside of Oceania, so it represents a very recent evolutionary event, with the allele acting in a recessive manner [81]. Similarly, a very high frequency of a population-specific OCA2 mutation causing the expression of a full albinism phenotype has been found in the Polynesian Islands [82]. Both are cases of genetic drift resulting from population founder effects (Box 1). In the European population TYRP1 rs1408799*A has been associated with blue eye color [24, 25], though other SNPs are also associated with blue eyes [49, 83], so the causative SNP is yet to be recognized.
Recent work has shown that regulatory microRNAs also have a role in the regulation of TYRP1 function. Alleles that induce or disrupt miR-155 regulation have been demonstrated to be under different modes of selection among human populations, causing a strong negative correlation between the frequency of miR-155 regulation of TYRP1 in human populations and their latitude of residence. It has been proposed that local adaptation of microRNA regulation acts as a rheostat to optimize TYRP1 expression in response to differential UV radiation [84].
Differences in the eumelanic composition of human hair is dependent upon ancestry and age. This was determined by direct measurement of melanin content and type of eumelanin in a sample of African-American, East Asian and Caucasian individuals [85]. African and European hairs were found to have changes in the ratio of dihydroxyindole (DHI) to 5,6-dihydroxyindole-2-carboxylic acid (DHICA), favoring DHI more as people age. However, the Asian samples seemed to have low DHICA levels throughout life, and did not have graying to the extent of other populations [85]. Since a major role of dopachrome tautomerase (DCT) is the isomerization of dopachrome to DHICA, it is plausible that these ethnic differences reflect differences in DCT expression. Interestingly, Lao et al. [18] found a polymorphism in an intron of the DCT gene at high frequency in the East Asian population (Han Chinese) and that was suggestive of selection. This work was extended by Alonso et al. [29], who used a battery of different statistical tests to enhance the ability to detect selection, finding a signature of directional selection on DCT and TYRP1 in Asians. The later study of Edwards et al. [69] reported rs1407995*C/T and rs2031526*A/G as intronic polymorphisms of DCT showing very high frequency differences between East Asian and non-Asian populations (>0.6). In analysis of European haplotypes [86] of these two SNPs and in transmission-disequilibrium tests using rs1407995*C/T, there was an effect on eye color, but not skin or hair color.
SLC45A2(Ch5p14.3)
The most important polymorphism affecting skin and hair color is the rs16891982*G/C SNP on chromosome 5 in the SLC45A2 gene encoding the MATP protein change Leu374Phe [22]. In African and Asian populations, the ancestral rs16891982*G/374Leu allele dominates with a frequency of 0.99 to 1.0; in European populations it is present at a frequency of 0.02 to 0.3 with a strong north-south cline and with the rs16891982*G allele being strongly associated with olive skin and dark hair. The rs16891982*C/374Phe variant can be predicted to impair MATP function, and in cultured melanocytes 374Leu/Leu homozygotes have more MATP transcript and TYR protein than 374Phe/Phe homozygotes [46]. Reduction of MATP protein in melanocytes carrying an albinism mutation was shown to lead to mislocalization of TYR from melanosomes to the plasma membrane and incorporation of TYR into exosomes [87]. Another coding variant in SLC45A2, rs26722*G/A Glu272Lys, is also associated with dark hair, but is in strong disequilibrium with rs16891982*G/374Leu in European populations. Vierkotter et al. [88], however, have reported that the 272Lys protein variant is associated with increased presence of solar-induced lentigines in German (P = 0.13) and Japanese (P = 0.02) women, whereas all the Japanese samples were homozygous for 374Leu/Leu. There is also strong linkage disequilibrium between rs26722 and other intronic SNPs that predict coloring, rs28777 and rs35391 (r2 = 0.63), indicating an extended haplotype. Mutations of the SLC45A2 gene that cause OCA4 are highest in Asian populations [13].
SLC24A5(Ch15q21.1)
Lamason and coworkers [89] reported that a mutation in SLC24A5, encoding the NCKX5 protein, underlies the golden pigmentation phenotype in zebrafish, and found an important equivalent variant in humans. The key skin pigmentation SNP rs1426654*G/A, changing Thr111Ala, is fixed in European populations as the rs1426654*A/111Ala allele, while rs1426654*G/111Thr is close to fixation in African populations. In cultured human melanocytes, the TYR activity of the 111Ala/Ala and 111Ala/Thr genotypes are roughly twofold higher than for 111Thr/Thr homozygotes [46]. Quillen and coworkers [47] estimate the effect of rs1426654*C/T on melanin index, via admixture mapping, to be of the same magnitude as the SLC45A2 rs1426654*A/374Phe change, and their data seem consistent with an additive, rather than a dominant, allelic effect of the SLC24A5 rs1426654*A/111Ala allele on skin reflectance. Interestingly, in the indigenous American populations they studied, the rs1426654*A/111Ala allele is at intermediate frequencies, and shows no evidence of selection [47], while there is strong evidence for selection in Europeans. Mouse Slc24a5 null animals have normal coat color, but diminished RPE melanosomes (smaller and paler) and less pigmentation [90].
KITLG(Ch12q22)
The ligand for the c-KIT receptor (KITLG) is known to regulate the number of melanocytes during development, melanin distribution in the skin, and onset of familial progressive syndromes of both hyper- and hypo-pigmentation [91]. The first quantitative evidence that this gene was involved in skin color variation in humans was found using a parallel evolutionary approach based on genetic discoveries in marine and freshwater species of stickleback fish [92]. This study suggested that upstream cis-regulatory elements at the human KITLG locus were under selection in different natural environments, with major peaks of high-frequency derived alleles in the large intergenic flanking regions of KITLG found in Europeans and East Asians. Genotyping of the AIM SNP rs642742*T/C found a significant association with a higher melanin index in an African-American sample population. However, the causative changes in the locus remain unknown. Notably, a strong composite of multiple signals (CMS; indicative of selection) was reported for KITLG and narrowed down the candidate region [28], which included the rs12821256*T/C SNP associated with blonde hair in European populations [16, 25, 49], with SNPs in intron 1 also reported to be associated with light hair color [93].
IRF4(Ch6p25)
The interferon regulatory factor (IRF) family is a group of DNA-binding transcription factors that are involved with downstream regulation of interferon signaling, with the IRFs primarily associated with immune system development and response. The possible association of IRF4s with melanocytic biology has been used as a diagnostic marker for various melanoma subtypes [94]. Involvement with the melanin biosynthetic pathway was initially proposed when a GWAS identified a SNP in an intergenic region close to IRF4 displaying association with freckling [25]. Further genetic analysis has highlighted the rs12203592*C/T SNP, located in the fourth intron of the IRF4 gene and found to be strongly associated with hair color, eye color and skin tanning response to sunlight [16, 49, 95]. Chinese, Japanese and African populations are homozygous for the rs12203592*C allele, with only European populations possessing rs12203592*T. This SNP shows a north-south gradient across Europe, possibly indicative of a selective advantage [62, 96] and has been implicated in a strong genotype-by-age interaction on nevus count. Carriers of rs12203592*T possess higher nevi counts as adolescents, which reverses over age with adults possessing lower counts [97].
HBD3(Ch8p22)
The loci reviewed so far are known to be important determinants of pigmentation in humans and other species; however, there are several examples of genetic variants acting in nonhuman species only. The β-defensins are a family of small proteins mainly involved in innate mucosal and epidermal immunity. An in-frame glycine (Gly23del) deletion in the canine homolog of DEF103B (encoding β-defensin 3) is a major determinant of coat color in domestic dogs and wolves [98, 99]. The structure of β-defensin 3 is similar to ASIP protein, and it is now known to act as a high-affinity neutral agonist of MC1R, competing with melanocortin and ASIP. There are no reported human protein coding variants. The detection of equivalent variants in humans is complicated by the fact that the defensin gene cluster is in a 250 kbp segmental duplication, present as 1 to 12 copies in human and other primate populations [100]; there is also a dearth of SNPs in this region on standard genome-wide SNP arrays. The rs2737902*C/T SNP within the DEFB103 promoter is more common in East Asia than Africa [100]. The cline in rs2737902 allele frequency across HGDP populations is correlated with distance from the equator (Spearman r = 0.48), so it is possible that there is a causative relationship between skin color and genotype.
SILV (Ch12q13)
The SILV gene on chromosome 12 encodes the melanosomal Pmel17 protein in man and is important in several other species, but no common variants are associated with human skin or hair color differences. In the mouse gene knockout Pmel-/- [101] melanosomes appear spherical in contrast to the usual oblong shape. Complete inactivation of Pmel has only a mild effect on the coat color, despite a substantial reduction in the darker eumelanin content in hair, similar to that observed with the spontaneous silver mutation in the Pmel gene in mice. It is likely that alleles reported in other species with more striking effects on pigmentation are dominant-negative mutations [101].
Melanin and melanogenesis as a protective mechanism against sun exposure
The chemical contents, transfer and accumulation of melanosomes in keratinocytes determines hair and skin color, but establishing the biological function of melanin pigmentation can be enigmatic and dependent on environmental cues. While the number of melanocytes between different skin tones is relatively constant, dark skinned individuals have a higher density of large, singly dispersed melanosomes. These remain intact as they move upwards in the epidermis to form caps over the nuclei of keratinocytes, protecting against UVR DNA damage. Melanosomes in light-skinned Europeans are smaller and less dense, and aggregate into membrane-bound complexes and degrade rapidly [102]. It is assumed that the benefits of darker skin are to do with protection against UV light. Apart from protecting against the damaging effects of solar UVR, melanin has several other roles in the body, including scavenging reactive oxygen species, protecting nutrients from photodamage and possibly modulating the inflammatory response. Melanin can also provide camouflage, transport energy and bind drugs, and is involved in hearing, sight and regulation of body heat [103].
In addition, some of the precursors and intermediates of melanogenesis (Figure 2) also appear to be diffusible molecules involved in the photoprotective pathway as signaling or hormone-like regulators of melanocyte or keratinocyte functions [104]. In particular, the action of the DCT pigmentation gene and the DHICA metabolite it produces provides a new insight into the function of the melanogenic pathway that is distinct from the production of the final melanin polymer. A cell culture model treating human melanocytes with MC1R agonists has shown strong induction of the DCT protein in wild-type cells, but not in melanocytes homozygous for MC1R alleles associated with red hair color. This suggests that the ability to produce DHICA is compromised in Europeans carrying these variant alleles, along with an alteration in the type of melanin being synthesized [105]. Moreover, overexpression of DCT in WM35 amelanotic melanoma cells reduced their sensitivity to oxidative stress and their protection against DNA damage [106]. When Dct knockout mice were exposed to UVR, they had decreased levels of eumelanin and increased levels of reactive oxygen species (ROS), sunburn cells and apoptotic cells. DHICA-derived melanin showed a strong hydroxyl radical scavenging ability [107] and inhibited lipid peroxidation [108].

Protection pathways in the skin against ultraviolet radiation (UVR). UVR induces DNA damage, which leads to activation of p53 and the formation of POMC and MC1R activation factors. MC1R action can be blocked by ASIP. Upon receptor activation of cAMP, dopachrome tautomerase (DCT) activity is upregulated and this leads to the generation of 5,6-dihydroxyindole-2-carboxylic acid (DHICA). MC1R also activates melanosome maturation and transfer to the keratinocyte. DHICA removes reactive oxygen species (ROS), and activates catalase (CAT) and peroxisome proliferator activated receptor (PPAR) in keratinocytes. Finally, DCT provides antagonistic feedback to p53 in melanocytes [112].
As DHICA is a diffusible molecule, it may enter keratinocytes directly to induce increased UVR resistance [109]. When primary keratinocyte cultures were treated with DHICA a range of cellular changes were seen, including expression and activity of the antioxidant enzymes superoxide dismutase (SOD) and catalase. This led to decreased cell damage and apoptosis after UVA exposure. The regulation of this DHICA-mediated differentiation of the keratinocytes was found to involve peroxisome proliferator activated receptors (PPARs) [109]. In summary, these experiments indicate that DCT, and the subsequent action of its product DHICA, are intrinsically linked to protection of skin cells against cell death and ROS after UVR exposure, as well as their role in the generation of protective eumelanin pigment.
Skin color differences through environmental selection - but why?
It is now apparent that we have identified human pigmentation genes that have been under evolutionary selective pressure. Specific alleles have reached high geographic/population-specific frequencies and in some cases have reached near fixation, as for example the SLC24A5 and SLC45A2 genes in Europeans. The reasons for this selection are likely to be diverse, not all based simply on the actions of the melanin pigment itself, and possibly even as varied as the alleles themselves. The involvement of other signaling pathways must now be considered, such as MC1R in the DNA damage response [110, 111], and DCT as a regulator of melanogenic intermediates [108] and p53 function [112]. Others will no doubt become apparent as our biological understanding of the role of melanogenesis and melanocyte cell physiology grows.
Some possible selective pressures acting on skin pigmentation in high and low UVR environments include the need for vitamin D synthesis and protection from photolysis of folate [113], the requirement for the skin to be both a permeability barrier against multiple forms of stress causing water loss and the first line of the innate immune system [114], and protection from skin cancer [115] and disease [116].
Overwhelming epidemiological evidence points to permanent dark skin pigmentation having been selected as a protective measure against the deleterious effects of solar UVR exposure in tropical climates, including Australia. As vitamin D3 is synthesized only when UVR penetrates the skin, the dose received not only varies according to the season, length of day, latitude, altitude and occupation of the individual, but it is also critically dependent upon the physical characteristics of skin thickness and melanin content, and, importantly, a person's age. Vitamin D is available from the diet in low quantities but varies according to the food source. A lack of the vitamin can have serious consequences for evolutionary fitness. This understanding initially led to the vitamin D hypothesis: that lighter skin types were selected in the northern hemisphere due to low environmental UVR exposure and deficiencies in dietary sources [117]. However, the influence of vitamin D on the selection of lighter skin color has been disputed [118]. Nessvi et al. [119] found that ethnicity rather than skin color better predicted 25-hydroxyvitamin D3 levels in New Zealanders. This is congruent with the observation that no pigmentation gene variants have been demonstrated to affect serum vitamin D levels in GWAS [120, 121], the exception being an association with OCA2 that was lost after adjusting for lifestyle [122]. Perhaps more surprisingly, pigmentation genes were not associated with blood folate concentrations in a GWAS [123], consistent with earlier clinical studies of UVA exposure effects on folate levels [124]. However, the behavior of melanin pigment itself after UVR exposure in undergoing the immediate pigment darkening response may participate in protection against folate photodegradation [125].
The ability to produce melanin appears across a wide range of species and within tissues not exposed to UVR, suggesting other functions of the pigment, but its contribution to the barrier function of skin and as a driver of human epidermal pigmentation have only recently been hypothesized [126]. This is partly based on the finding that UVR damages the barrier, so selection for increased pigmentation in darker skin would improve this barrier function. Darkly pigmented skin does display more rapid barrier recovery after acute damage than lightly pigmented skin types [127]. Thus, these authors have proposed that pigmentation in hominids evolved in response to the combined stress of UVR exposure and low humidity. Moreover, there is an intimate link between maintaining the permeability barrier of the skin and antimicrobial defenses, and some researchers suggest that darkly pigmented melanocytes enhance this defense by acidifying the outer epidermis [126, 127]. The cellular basis for this epidermal acidification of dark skin is suggested to be the melanosomes themselves [127], involving polymorphisms in the ion-transporter proteins of the melanosome [114] that are central to skin color differences, as discussed earlier. However, there is inconsistency here with the neutralization of the melanosome that occurs with an increase in melanogenesis [128–130], undermining in part the barrier hypothesis involving melanosome pH and immune defenses. Evidence for pathogen-driven and immune selection in different populations has not appeared in the pigmentation gene set [131].
Eye color
Eye color as a trait may be under multiple selection pressures, including sexual- [132] or personality-related factors [133], but there may also have been strong co-selection for lightening of pigmentary traits in multiple human populations. The selection pressure on the OCA2-HERC region associated with blue eye color in Europeans is so strong [70] that other elements may have been in play, and we have proposed that this could have been in response to an environmental factor, lack of sunlight, to overcome seasonal affective disorder (SAD) [3]. SAD is a major depressive illness that has been linked with eye color; perhaps those with blue eyes were able to withstand the dark, depressing days of the Neolithic European winters better than those with brown eye color. Notably, the eye has specialized neurons in the retina (retinal ganglion cells) that can detect blue light as a non-visual response to help regulate circadian rhythms [134]: a missense allele of the photopigment melanopsin in retinal ganglion cells may predispose carriers to SAD [135], supporting this as a possible connection for the selective pressure on the OCA2 rs12913832*T/C SNP.
Conclusions
Each of our genomes are unique, but in comparison to other species the human population in general has a low level of genetic heterogeneity, with few regions under selection [136]. The genome differs in very specific ways, acting under the constraint of the environment that individuals find themselves in. The pigmentation gene set has responded in different ways at different periods of our history [137], with light skin developing in parallel in Europe and Asia by at least two mechanisms.
We can discern evidence for complete or near-complete selective sweeps for SLC24A5, TYR SLC45A2, and for incomplete sweeps for multiple other loci such as MC1R (Table 2). An interpretation of the patterns of allelic diversity at different pigmentation loci is that once the alleles of large effect came close to fixation under 'hard' selection, then selective pressure was reduced with 'soft' selection on other loci, encouraging intermediate frequency polymorphism of functional alleles, most notably in MC1R. We presume the functional variants segregating in OCA2 and TYRP1 in Oceania reflect weak purifying selection and drift, given that dark skin color in the Solomon Islands means baseline protection against UV damage is high. The puzzling observation that there are large ethnic differences in MC1R and IRF4 allele frequencies, but few long-associated haplotypes, might be seen as an effect of a combination of softened selection plus strong selection on linked loci. In the case of IRF4, there are multiple nearby SNPs in the same gene affecting lymphocyte maturation; for MC1R, loci such as FANCA and CDK10 are close, and these may be under selection themselves [138]. A final point is whether there is unequivocal evidence for selection on pigmentation traits other than skin color. We have suggested that the blue eye colors variants in OCA2 are the strongest candidates for this.
While it is largely believed that human pigmentation has faced adaptive selection and been shaped by the environment and climate, the genetic causes and selective pressures responsible have been difficult to detect and resolve. With the flood of information coming from human genome projects there has been substantial progression of the field, as has been highlighted with the pigmentation loci and polymorphisms discussed in this article (Table 2). By studying compound haplotype systems within these genes, an attempt has been made to calculate the age of the alleles associated with lightening of European skin color. It is proposed that changes within KITLG appeared 30,000 years ago, and within TYRP1, SLC24A5 and SLC45A2 at 11,000-19,000 years ago, respectively [139]. So in the future, even the timing of when these selective sweeps occurred may become known.
Box 1. How do genetic differences arise in human populations?
Human genomic diversity is a consequence of our history and behavior, due to continual population expansion, contraction, movement, family structure, environmental (diet and shelter) and cultural differences, which all come together to influence the demographic forces that underlie evolution. These forces are genetic drift, which are stochastic changes of allele frequencies in the population, and natural selection. A serial founder effectto our populations has been found to be an important model for humans, and has generated patterns of diversity among small populations that have dispersed over long distances from our first origin in Africa. Not all differences in allele frequencies arise from adaptive evolutionor positive selectionfor alleles; most are consistent with neutralevolution. Balancing selectionis where multiple alleles at a locus are maintained in a population: specific types of balancing selection include heterozygote advantageand frequency-dependent selection. Purifying selectionor negative selectionresults in the removal of deleterious alleles from a population. Background selectionoccurs in the case where purifying selection against deleterious mutations also removes variation at linked neutral sites. In genetic hitchhiking, positive selection for a beneficial mutation increases frequency of neutral variants on the same haplotype. Such a change in allele and haplotype frequencies might be large, due to a highly advantageous mutation (completeor hard sweep; or partialor soft sweepif it is more gradual). Population bottleneckshave occurred commonly in our evolution and result in a reduction in population size that increases the effects of drift and can skew the frequency of gene polymorphisms. The effect of a bottleneck on patterns of genetic variation depends on how severe the decrease in population size is and the duration of the bottleneck. All of these processes need to be considered when looking for human pigmentation gene allele frequency differences between populations, so as to work out which are signals of adaptive evolution or stochastic gene frequency changes.
Evidence that a particular locus has experienced selection can be inferred [27] from inter-population differences in allele frequencies (Fst-based tests) that are larger than expected based on: the (inferred) population demographic histories; correlations between population allele frequencies and directly or indirectly measured agents of selection correlated with geography, such as UV exposure, temperature, precipitation, diet, pathogen exposure; the diversity and spectrum of alleles at the locus within and between populations, usually called neutrality tests(HKA test; Tajima D, Fu and Li F, Fay and Wu H); the extent of linkage disequilibrium surrounding the locus (expected haplotype homozygosity EHH, PX-EHH, iHs); and variation in the degree of population admixture. Tests will differ in their power to detect selection at mutations and events of different ages and types. Some neutrality tests are less applicable to data from current GWAS SNP arrays where common SNPs are overrepresented.
Abbreviations
- AIM:
-
ancestry informative marker
- DCT:
-
dopachrome tautomerase
- DHI:
-
dihydroxyindole
- DHICA:
-
5,6-dihyroxyindole-2-carboxylic acid
- GWAS:
-
genome-wide association study
- HGDP:
-
Human Genome Diversity Panel
- IRF:
-
interferon regulatory factor
- OCA:
-
oculocutaneous albinism
- PCA:
-
principal components analysis
- POMC:
-
pro-opiomelanocortin
- PPAR:
-
peroxisome proliferator activated receptor
- ROS:
-
reactive oxygen species
- SAD:
-
seasonal affective disorder
- SNP:
-
single nucleotide polymerization
- TYR:
-
tyrosinase
- UVA:
-
ultraviolet-A radiation
- UVR:
-
ultraviolet radiation.
References
Sturm RA: Molecular genetics of human pigmentation diversity. Hum Mol Gen. 2009, 18: R9-17. 10.1093/hmg/ddp003.
Westerhof W: Evolutionary, biologic, and social aspects of skin color. Dermatol Clinics. 2007, 25: 293-302. 10.1016/j.det.2007.05.001. vii
Sturm RA, Larsson M: Genetics of human iris colour and patterns. Pigment Cell Melanoma Res. 2009, 22: 544-562. 10.1111/j.1755-148X.2009.00606.x.
Tobin DJ: The cell biology of human hair follicle pigmentation. Pigment Cell Melanoma Res. 2011, 24: 75-88. 10.1111/j.1755-148X.2010.00803.x.
Van Raamsdonk CD, Barsh GS, Wakamatsu K, Ito S: Independent regulation of hair and skin color by two G protein-coupled pathways. Pigment Cell Melanoma Res. 2009, 22: 819-826. 10.1111/j.1755-148X.2009.00609.x.
Commo S, Gaillard O, Bernard BA: Human hair greying is linked to a specific depletion of hair follicle melanocytes affecting both the bulb and the outer root sheath. Br J Dermatol. 2004, 150: 435-443. 10.1046/j.1365-2133.2004.05787.x.
Rogers AR, Iltis D, Wooding S: Genetic variation at the MC1R locus and the time since loss of human body hair. Curr Anthropol. 2004, 45: 105-108. 10.1086/381006.
Ito S, Wakamatsu K: Human hair melanins: what we have learned and have not learned from mouse coat color pigmentation. Pigment Cell Melanoma Res. 2011, 24: 63-74. 10.1111/j.1755-148X.2010.00755.x.
Ando H, Niki Y, Ito M, Akiyama K, Matsui MS, Yarosh DB, Ichihashi M: Melanosomes are transferred from melanocytes to keratinocytes through the processes of packaging, release, uptake, and dispersion. J Invest Dermatol. 2012, 132: 1222-1229. 10.1038/jid.2011.413.
Singh SK, Kurfurst R, Nizard C, Schnebert S, Perrier E, Tobin DJ: Melanin transfer in human skin cells is mediated by filopodia - a model for homotypic and heterotypic lysosome-related organelle transfer. FASEB J. 2010, 24: 3756-3769. 10.1096/fj.10-159046.
Ebanks JP, Koshoffer A, Wickett RR, Schwemberger S, Babcock G, Hakozaki T, Boissy RE: Epidermal keratinocytes from light vs. dark skin exhibit differential degradation of melanosomes. J Invest Dermatol. 2011, 131: 1226-1233. 10.1038/jid.2011.22.
Lin JY, Fisher DE: Melanocyte biology and skin pigmentation. Nature. 2007, 445: 843-850. 10.1038/nature05660.
Rooryck C, Morice F, Didier L, Taieb A, Arveiler B: Genetic basis of oculocutaneous albinism. Expert Rev Dermatol. 2009, 4: 611-622. 10.1586/edm.09.53.
Barsh GS: What controls variation in human skin color?. PLoS Biol. 2003, 1: E27-
Gudbjartsson DF, Sulem P, Stacey SN, Goldstein AM, Rafnar T, Sigurgeirsson B, Benediktsdottir KR, Thorisdottir K, Ragnarsson R, Sveinsdottir SG, Magnusson V, Lindblom A, Kostulas K, Botella-Estrada R, Soriano V, Juberías P, Grasa M, Saez B, Andres R, Scherer D, Rudnai P, Gurzau E, Koppova K, Kiemeney LA, Jakobsdottir M, Steinberg S, Helgason A, Gretarsdottir S, Tucker MA, Mayordomo JI, et al: ASIP and TYR pigmentation variants associate with cutaneous melanoma and basal cell carcinoma. Nat Genet. 2008, 40: 886-891. 10.1038/ng.161.
Han J, Kraft P, Nan H, Guo Q, Chen C, Qureshi A, Hankinson SE, Hu FB, Duffy DL, Zhao ZZ, Martin NG, Montgomery GW, Hayward NK, Thomas G, Hoover RN, Chanock S, Hunter DJ: A genome-wide association study identifies novel alleles associated with hair color and skin pigmentation. PLoS Genet. 2008, 4: e1000074-10.1371/journal.pgen.1000074.
Izagirre N, Garcia I, Junquera C, de la Rua C, Alonso S: A scan for signatures of positive selection in candidate loci for skin pigmentation in humans. Mol Biol Evol. 2006, 23: 1697-1706. 10.1093/molbev/msl030.
Lao O, de Gruijter JM, van Duijn K, Navarro A, Kayser M: Signatures of positive selection in genes associated with human skin pigmentation as revealed from analyses of single nucleotide polymorphisms. Ann Hum Genet. 2007, 71: 354-369. 10.1111/j.1469-1809.2006.00341.x.
McEvoy B, Beleza S, Shriver MD: The genetic architecture of normal variation in human pigmentation: an evolutionary perspective and model. Hum Mol Genet. 2006, 15 (Spec No 2): R176-181.
Myles S, Somel M, Tang K, Kelso J, Stoneking M: Identifying genes underlying skin pigmentation differences among human populations. Hum Genet. 2007, 120: 613-621.
Norton HL, Kittles RA, Parra E, McKeigue P, Mao X, Cheng K, Canfield VA, Bradley DG, McEvoy B, Shriver MD: Genetic evidence for the convergent evolution of light skin in Europeans and East Asians. Mol Biol Evol. 2007, 24: 710-722.
Soejima M, Tachida H, Ishida T, Sano A, Koda Y: Evidence for recent positive selection at the human AIM1 locus in a European population. Mol Biol Evol. 2006, 23: 179-188.
Stokowski RP, Pant PV, Dadd T, Fereday A, Hinds DA, Jarman C, Filsell W, Ginger RS, Green MR, van der Ouderaa FJ, Cox DR: A genomewide association study of skin pigmentation in a South Asian population. Am J Hum Genet. 2007, 81: 1119-1132. 10.1086/522235.
Sulem P, Gudbjartsson DF, Stacey SN, Helgason A, Rafnar T, Jakobsdottir M, Steinberg S, Gudjonsson SA, Palsson A, Thorleifsson G, Pálsson S, Sigurgeirsson B, Thorisdottir K, Ragnarsson R, Benediktsdottir KR, Aben KK, Vermeulen SH, Goldstein AM, Tucker MA, Kiemeney LA, Olafsson JH, Gulcher J, Kong A, Thorsteinsdottir U, Stefansson K: Two newly identified genetic determinants of pigmentation in Europeans. Nat Genet. 2008, 40: 835-837. 10.1038/ng.160.
Sulem P, Gudbjartsson DF, Stacey SN, Helgason A, Rafnar T, Magnusson KP, Manolescu A, Karason A, Palsson A, Thorleifsson G, Jakobsdottir M, Steinberg S, Pálsson S, Jonasson F, Sigurgeirsson B, Thorisdottir K, Ragnarsson R, Benediktsdottir KR, Aben KK, Kiemeney LA, Olafsson JH, Gulcher J, Kong A, Thorsteinsdottir U, Stefansson K: Genetic determinants of hair, eye and skin pigmentation in Europeans. Nat Genet. 2007, 39: 1443-1452. 10.1038/ng.2007.13.
Gerstenblith MR, Shi J, Landi MT: Genome-wide association studies of pigmentation and skin cancer: a review and meta-analysis. Pigment Cell Melanoma Res. 2010, 23: 587-606. 10.1111/j.1755-148X.2010.00730.x.
Oleksyk TK, Smith MW, O'Brien SJ: Genome-wide scans for footprints of natural selection. Philos Trans R Soc Lond B Biol Sci. 2010, 365: 185-205. 10.1098/rstb.2009.0219.
Grossman SR, Shlyakhter I, Karlsson EK, Byrne EH, Morales S, Frieden G, Hostetter E, Angelino E, Garber M, Zuk O, Lander ES, Schaffner SF, Sabeti PC: A composite of multiple signals distinguishes causal variants in regions of positive selection. Science. 2010, 327: 883-886. 10.1126/science.1183863.
Alonso S, Izagirre N, Smith-Zubiaga I, Gardeazabal J, Diaz-Ramon JL, Diaz-Perez JL, Zelenika D, Boyano MD, Smit N, de la Rua C: Complex signatures of selection for the melanogenic loci TYR, TYRP1 and DCT in humans. BMC Evol Biol. 2008, 8: 74-10.1186/1471-2148-8-74.
Casto AM, Feldman MW: Genome-wide association study SNPs in the human genome diversity project populations: does selection affect unlinked SNPs with shared trait associations?. PLoS Genet. 2011, 7: e1001266-10.1371/journal.pgen.1001266.
Kimura R, Fujimoto A, Tokunaga K, Ohashi J: A practical genome scan for population-specific strong selective sweeps that have reached fixation. PloS One. 2007, 2: e286-10.1371/journal.pone.0000286.
Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, Absher D, Srinivasan BS, Barsh GS, Myers RM, Feldman MW, Pritchard JK: Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009, 19: 826-837. 10.1101/gr.087577.108.
Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, Schaffner SF, Lander ES, International HapMap Consortium, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, et al: Genome-wide detection and characterization of positive selection in human populations. Nature. 2007, 449: 913-918. 10.1038/nature06250.
Tang K, Thornton KR, Stoneking M: A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 2007, 5: e171-10.1371/journal.pbio.0050171.
Tennessen JA, Akey JM: Parallel adaptive divergence among geographically diverse human populations. PLoS Genet. 2011, 7: e1002127-10.1371/journal.pgen.1002127.
Voight BF, Kudaravalli S, Wen X, Pritchard JK: A map of recent positive selection in the human genome. PLoS Biol. 2006, 4: e72-10.1371/journal.pbio.0040072.
Williamson SH, Hubisz MJ, Clark AG, Payseur BA, Bustamante CD, Nielsen R: Localizing recent adaptive evolution in the human genome. PLoS Genet. 2007, 3: e90-10.1371/journal.pgen.0030090.
de Gruijter JM, Lao O, Vermeulen M, Xue Y, Woodwark C, Gillson CJ, Coffey AJ, Ayub Q, Mehdi SQ, Kayser M, Tyler-Smith C: Contrasting signals of positive selection in genes involved in human skin-color variation from tests based on SNP scans and resequencing. Invest Genet. 2011, 2: 24-10.1186/2041-2223-2-24.
Tennessen JA, Madeoy J, Akey JM: Signatures of positive selection apparent in a small sample of human exomes. Genome Res. 2010, 20: 1327-1334. 10.1101/gr.106161.110.
Tennessen JA, O'Connor TD, Bamshad MJ, Akey JM: The promise and limitations of population exomics for human evolution studies. Genome Biol. 2011, 12: 127-10.1186/gb-2011-12-9-127.
International HapMap 3 Consortium, Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Peltonen L, Dermitzakis E, Bonnen PE, Altshuler DM, Gibbs RA, de Bakker PI, Deloukas P, Gabriel SB, Gwilliam R, Hunt S, Inouye M, Jia X, Palotie A, Parkin M, Whittaker P, Yu F, Chang K, Hawes A, Lewis LR, Ren Y, et al: Integrating common and rare genetic variation in diverse human populations. Nature. 2010, 467: 52-58. 10.1038/nature09298.
1000 Genomes Project Consortium: A map of human genome variation from population-scale sequencing. Nature. 2010, 467: 1061-1073. 10.1038/nature09534.
Paschou P, Lewis J, Javed A, Drineas P: Ancestry informative markers for fine-scale individual assignment to worldwide populations. J Med Genet. 2010, 47: 835-847. 10.1136/jmg.2010.078212.
Hancock AM, Witonsky DB, Alkorta-Aranburu G, Beall CM, Gebremedhin A, Sukernik R, Utermann G, Pritchard JK, Coop G, Di Rienzo A: Adaptations to climate-mediated selective pressures in humans. PLoS Genet. 2011, 7: e1001375-10.1371/journal.pgen.1001375.
Yang WY, Novembre J, Eskin E, Halperin E: A model-based approach for analysis of spatial structure in genetic data. Nat Genet. 2012, 44: 725-731. 10.1038/ng.2285.
Cook AL, Chen W, Thurber AE, Smit DJ, Smith AG, Bladen TG, Brown DL, Duffy DL, Pastorino L, Bianchi-Scarra G, Leonard JH, Stow JL, Sturm RA: Analysis of cultured human melanocytes based on polymorphisms within the SLC45A2/MATP, SLC24A5/NCKX5, and OCA2/P loci. J Invest Dermatol. 2009, 129: 392-405. 10.1038/jid.2008.211.
Quillen EE, Bauchet M, Bigham AW, Delgado-Burbano ME, Faust FX, Klimentidis YC, Mao X, Stoneking M, Shriver MD: OPRM1 and EGFR contribute to skin pigmentation differences between indigenous Americans and Europeans. Hum Genet. 2012, 131: 1073-1080. 10.1007/s00439-011-1135-1.
Choi W, Wolber R, Gerwat W, Mann T, Batzer J, Smuda C, Liu H, Kolbe L, Hearing VJ: The fibroblast-derived paracrine factor neuregulin-1 has a novel role in regulating the constitutive color and melanocyte function in human skin. J Cell Sci. 2010, 123: 3102-3111. 10.1242/jcs.064774.
Eriksson N, Macpherson JM, Tung JY, Hon LS, Naughton B, Saxonov S, Avey L, Wojcicki A, Pe'er I, Mountain J: Web-based, participant-driven studies yield novel genetic associations for common traits. PLoS Genet. 2010, 6: e1000993-10.1371/journal.pgen.1000993.
Lang MR, Patterson LB, Gordon TN, Johnson SL, Parichy DM: Basonuclin-2 requirements for zebrafish adult pigment pattern development and female fertility. PLoS Genet. 2009, 5: e1000744-10.1371/journal.pgen.1000744.
Chiaverini C, Sillard L, Flori E, Ito S, Briganti S, Wakamatsu K, Fontas E, Berard E, Cailliez M, Cochat P, Foulard M, Guest G, Niaudet P, Picardo M, Bernard FX, Antignac C, Ortonne JP, Ballotti R: Cystinosin is a melanosomal protein that regulates melanin synthesis. FASEB J. 2012, 26: 3779-3789. 10.1096/fj.11-201376.
Kemper KE, Visscher PM, Goddard ME: Genetic architecture of body size in mammals. Genome Biol. 2012, 13: 244-
Smith AG, Box NF, Marks LH, Chen W, Smit DJ, Wyeth JR, Huttley GA, Easteal S, Sturm RA: The human melanocortin-1 receptor locus: analysis of transcription unit, locus polymorphism and haplotype evolution. Gene. 2001, 281: 81-94. 10.1016/S0378-1119(01)00791-0.
Harding RM, Healy E, Ray AJ, Ellis NS, Flanagan N, Todd C, Dixon C, Sajantila A, Jackson IJ, Birch-Machin MA, Rees JL: Evidence for variable selective pressures at MC1R. Am J Hum Genet. 2000, 66: 1351-1361. 10.1086/302863.
Makova KD, Ramsay M, Jenkins T, Li WH: Human DNA sequence variation in a 6.6-kb region containing the melanocortin 1 receptor promoter. Genetics. 2001, 158: 1253-1268.
Beaumont KA, Liu YY, Sturm RA: The melanocortin-1 receptor gene polymorphism and association with human skin cancer. Prog Mol Biol Trans Sci. 2009, 88: 85-153.
Savage SA, Gerstenblith MR, Goldstein AM, Mirabello L, Fargnoli MC, Peris K, Landi MT: Nucleotide diversity and population differentiation of the melanocortin 1 receptor gene, MC1R. BMC Genet. 2008, 9: 31-
Nakayama K, Soemantri A, Jin F, Dashnyam B, Ohtsuka R, Duanchang P, Isa MN, Settheetham-Ishida W, Harihara S, Ishida T: Identification of novel functional variants of the melanocortin 1 receptor gene originated from Asians. Hum Genet. 2006, 119: 322-330. 10.1007/s00439-006-0141-1.
Hofreiter M, Schoneberg T: The genetic and evolutionary basis of colour variation in vertebrates. Cell Mol Life Sci. 2010, 67: 2591-2603. 10.1007/s00018-010-0333-7.
Sturm RA, Duffy DL, Box NF, Newton RA, Shepherd AG, Chen W, Marks LH, Leonard JH, Martin NG: Genetic association and cellular function of MC1R variant alleles in human pigmentation. Ann N Y Acad Sci. 2003, 994: 348-358. 10.1111/j.1749-6632.2003.tb03199.x.
Duffy DL, Box NF, Chen W, Palmer JS, Montgomery GW, James MR, Hayward NK, Martin NG, Sturm RA: Interactive effects of MC1R and OCA2 on melanoma risk phenotypes. Hum Mol Gen. 2004, 13: 447-461.
Duffy DL, Zhao ZZ, Sturm RA, Hayward NK, Martin NG, Montgomery GW: Multiple pigmentation gene polymorphisms account for a substantial proportion of risk of cutaneous malignant melanoma. J Invest Dermatol. 2010, 130: 520-528. 10.1038/jid.2009.258.
Sitaram A, Piccirillo R, Palmisano I, Harper DC, Dell'Angelica EC, Schiaffino MV, Marks MS: Localization to mature melanosomes by virtue of cytoplasmic dileucine motifs is required for human OCA2 function. Mol Biol Cell. 2009, 20: 1464-1477. 10.1091/mbc.E08-07-0710.
Hoyle DJ, Rodriguez-Fernandez IA, Dell'angelica EC: Functional interactions between OCA2 and the protein complexes BLOC-1, BLOC-2, and AP-3 inferred from epistatic analyses of mouse coat pigmentation. Pigment Cell Melanoma Res. 2011, 24: 275-281. 10.1111/j.1755-148X.2010.00815.x.
Visser M, Kayser M, Palstra RJ: HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome Res. 2012, 22: 446-455. 10.1101/gr.128652.111.
Lee ST, Nicholls RD, Jong MT, Fukai K, Spritz RA: Organization and sequence of the human P gene and identification of a new family of transport proteins. Genomics. 1995, 26: 354-363. 10.1016/0888-7543(95)80220-G.
Anno S, Abe T, Yamamoto T: Interactions between SNP alleles at multiple loci contribute to skin color differences between caucasoid and mongoloid subjects. Int Journal Biol Sci. 2008, 4: 81-86.
Yuasa I, Umetsu K, Harihara S, Kido A, Miyoshi A, Saitou N, Dashnyam B, Jin F, Lucotte G, Chattopadhyay PK, Henke L, Henke J: Distribution of two Asian-related coding SNPs in the MC1R and OCA2 genes. Biochem Genet. 2007, 45: 535-542. 10.1007/s10528-007-9095-9.
Edwards M, Bigham A, Tan J, Li S, Gozdzik A, Ross K, Jin L, Parra EJ: Association of the OCA2 polymorphism His615Arg with melanin content in east Asian populations: further evidence of convergent evolution of skin pigmentation. PLoS Genet. 2010, 6: e1000867-10.1371/journal.pgen.1000867.
Donnelly MP, Paschou P, Grigorenko E, Gurwitz D, Barta C, Lu RB, Zhukova OV, Kim JJ, Siniscalco M, New M, Li H, Kajuna SL, Manolopoulos VG, Speed WC, Pakstis AJ, Kidd JR, Kidd KK: A global view of the OCA2-HERC2 region and pigmentation. Hum Genet. 2012, 131: 683-696. 10.1007/s00439-011-1110-x.
Sviderskaya EV, Bennett DC, Ho L, Bailin T, Lee ST, Spritz RA: Complementation of hypopigmentation in p-mutant (pink-eyed dilution) mouse melanocytes by normal human P cDNA, and defective complementation by OCA2 mutant sequences. J Invest Dermatol. 1997, 108: 30-34. 10.1111/1523-1747.ep12285621.
Yuasa I, Umetsu K, Harihara S, Miyoshi A, Saitou N, Park KS, Dashnyam B, Jin F, Lucotte G, Chattopadhyay PK, Henke L, Henke J: OCA2 481Thr, a hypofunctional allele in pigmentation, is characteristic of northeastern Asian populations. J Hum Genet. 2007, 52: 690-693. 10.1007/s10038-007-0167-9.
Yuasa I, Harihara S, Jin F, Nishimukai H, Fujihara J, Fukumori Y, Takeshita H, Umetsu K, Saitou N: Distribution of OCA2*481Thr and OCA2*615Arg, associated with hypopigmentation, in several additional populations. Leg Med (Tokyo). 2011, 13: 215-217. 10.1016/j.legalmed.2011.04.003.
King RA, Pietsch J, Fryer JP, Savage S, Brott MJ, Russell-Eggitt I, Summers CG, Oetting WS: Tyrosinase gene mutations in oculocutaneous albinism 1 (OCA1): definition of the phenotype. Hum Genet. 2003, 113: 502-513. 10.1007/s00439-003-0998-1.
Chaki M, Sengupta M, Mondal M, Bhattacharya A, Mallick S, Bhadra R, Ray K: Molecular and functional studies of tyrosinase variants among Indian oculocutaneous albinism type 1 patients. J Invest Dermatol. 2011, 131: 260-262. 10.1038/jid.2010.274.
Oetting WS, Pietsch J, Brott MJ, Savage S, Fryer JP, Summers CG, King RA: The R402Q tyrosinase variant does not cause autosomal recessive ocular albinism. Am J Med Genet Part A. 2009, 149A: 466-469. 10.1002/ajmg.a.32654.
Chiang PW, Spector E, Tsai AC: Oculocutaneous albinism spectrum. Am J Med Genet Part A. 2009, 149A: 1590-1591. 10.1002/ajmg.a.32939.
Hu HH, Guedj M, Descamps V, Jouary T, Bourillon A, Ezzedine K, Taieb A, Bagot M, Bensussan A, Saiag P, Grandchamp B, Basset-Seguin N, Soufir N: Assessment of tyrosinase variants and skin cancer risk in a large cohort of French subjects. J Dermatol Sci. 2011, 64: 127-133. 10.1016/j.jdermsci.2011.07.003.
Jin Y, Ferrara T, Gowan K, Holcomb C, Rastrou M, Erlich HA, Fain PR, Spritz RA: Next-generation DNA re-sequencing identifies common variants of TYR and HLA-A that modulate the risk of generalized vitiligo via antigen presentation. J Invest Dermatol. 2012, 132: 1730-1733. 10.1038/jid.2012.37.
Esposito R, D'Aniello S, Squarzoni P, Pezzotti MR, Ristoratore F, Spagnuolo A: New insights into the evolution of metazoan tyrosinase gene family. PloS One. 2012, 7: e35731-10.1371/journal.pone.0035731.
Kenny EE, Timpson NJ, Sikora M, Yee MC, Moreno-Estrada A, Eng C, Huntsman S, Burchard EG, Stoneking M, Bustamante CD, Myles S: Melanesian blond hair is caused by an amino acid change in TYRP1. Science. 2012, 336: 554-10.1126/science.1217849.
Johanson HC, Chen W, Wicking C, Sturm RA: Inheritance of a novel mutated allele of the OCA2 gene associated with high incidence of oculocutaneous albinism in a Polynesian community. J Hum Genet. 2010, 55: 103-111. 10.1038/jhg.2009.130.
Liu F, van Duijn K, Vingerling JR, Hofman A, Uitterlinden AG, Janssens AC, Kayser M: Eye color and the prediction of complex phenotypes from genotypes. Curr Biol. 2009, 19: R192-193. 10.1016/j.cub.2009.01.027.
Li J, Liu Y, Xin X, Kim TS, Cabeza EA, Ren J, Nielsen R, Wrana JL, Zhang Z: Evidence for positive selection on a number of microRNA regulatory interactions during recent human evolution. PLoS Genet. 2012, 8: e1002578-10.1371/journal.pgen.1002578.
Commo S, Wakamatsu K, Lozano I, Panhard S, Loussouarn G, Bernard BA, Ito S: Age-dependent changes in eumelanin composition in hairs of various ethnic origins. Int J Cosmet Sci. 2012, 34: 102-107. 10.1111/j.1468-2494.2011.00691.x.
Zhu G, Montgomery GW, James MR, Trent JM, Hayward NK, Martin NG, Duffy DL: A genome-wide scan for naevus count: linkage to CDKN2A and to other chromosome regions. Eur J Hum Genet. 2007, 15: 94-102. 10.1038/sj.ejhg.5201729.
Cullinane AR, Vilboux T, O'Brien K, Curry JA, Maynard DM, Carlson-Donohoe H, Ciccone C, Markello TC, Gunay-Aygun M, Huizing M, Gahl WA: Homozygosity mapping and whole-exome sequencing to detect SLC45A2 and G6PC3 mutations in a single patient with oculocutaneous albinism and neutropenia. J Invest Dermatol. 2011, 131: 2017-2025. 10.1038/jid.2011.157.
Vierkotter A, Kramer U, Sugiri D, Morita A, Yamamoto A, Kaneko N, Matsui M, Krutmann J: Development of lentigines in German and Japanese women correlates with variants in the SLC45A2 gene. J Invest Dermatol. 2012, 132: 733-736. 10.1038/jid.2011.350.
Lamason RL, Mohideen MA, Mest JR, Wong AC, Norton HL, Aros MC, Jurynec MJ, Mao X, Humphreville VR, Humbert JE, Sinha S, Moore JL, Jagadeeswaran P, Zhao W, Ning G, Makalowska I, McKeigue PM, O'Donnell D, Kittles R, Parra EJ, Mangini NJ, Grunwald DJ, Shriver MD, Canfield VA, Cheng KC: SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science. 2005, 310: 1782-1786. 10.1126/science.1116238.
Vogel P, Read RW, Vance RB, Platt KA, Troughton K, Rice DS: Ocular albinism and hypopigmentation defects in Slc24a5-/- mice. Vet Pathol. 2008, 45: 264-279. 10.1354/vp.45-2-264.
Picardo M, Cardinali G: The genetic determination of skin pigmentation: KITLG and the KITLG/c-Kit pathway as key players in the onset of human familial pigmentary diseases. J Invest Dermatol. 2011, 131: 1182-1185. 10.1038/jid.2011.67.
Miller CT, Beleza S, Pollen AA, Schluter D, Kittles RA, Shriver MD, Kingsley DM: cis-Regulatory changes in KIT ligand expression and parallel evolution of pigmentation in sticklebacks and humans. Cell. 2007, 131: 1179-1189. 10.1016/j.cell.2007.10.055.
Mengel-From J, Wong TH, Morling N, Rees JL, Jackson IJ: Genetic determinants of hair and eye colours in the Scottish and Danish populations. BMC Genet. 2009, 10: 88-
Sundram U, Harvell JD, Rouse RV, Natkunam Y: Expression of the B-cell proliferation marker MUM1 by melanocytic lesions and comparison with S100, gp100 (HMB45), and MelanA. Mod Pathol. 2003, 16: 802-810. 10.1097/01.MP.0000081726.49886.CF.
Nan H, Kraft P, Qureshi AA, Guo Q, Chen C, Hankinson SE, Hu FB, Thomas G, Hoover RN, Chanock S, Hunter DJ, Han J: Genome-wide association study of tanning phenotype in a population of European ancestry. J Invest Dermatol. 2009, 129: 2250-2257. 10.1038/jid.2009.62.
Moskvina V, Smith M, Ivanov D, Blackwood D, Stclair D, Hultman C, Toncheva D, Gill M, Corvin A, O'Dushlaine C, Morris DW, Wray NR, Sullivan P, Pato C, Pato MT, Sklar P, Purcell S, Holmans P, O'Donovan MC, Owen MJ, Kirov G: Genetic differences between five European populations. Human Hered. 2010, 70: 141-149. 10.1159/000313854.
Duffy DL, Iles MM, Glass D, Zhu G, Barrett JH, Höiom V, Zhao ZZ, Sturm RA, Soranzo N, Hammond C, Kvaskoff M, Whiteman DC, Mangino M, Hansson J, Newton-Bishop JA, GenoMEL, Bataille V, Hayward NK, Martin NG, Bishop DT, Spector TD, Montgomery GW: IRF4 variants have age-specific effects on nevus count and predispose to melanoma. Am J Hum Genet. 2010, 87: 6-16. 10.1016/j.ajhg.2010.05.017.
Anderson TM, vonHoldt BM, Candille SI, Musiani M, Greco C, Stahler DR, Smith DW, Padhukasahasram B, Randi E, Leonard JA, Bustamante CD, Ostrander EA, Tang H, Wayne RK, Barsh GS: Molecular and evolutionary history of melanism in North American gray wolves. Science. 2009, 323: 1339-1343. 10.1126/science.1165448.
Candille SI, Kaelin CB, Cattanach BM, Yu B, Thompson DA, Nix MA, Kerns JA, Schmutz SM, Millhauser GL, Barsh GS: A-defensin mutation causes black coat color in domestic dogs. Science. 2007, 318: 1418-1423. 10.1126/science.1147880.
Hardwick RJ, Machado LR, Zuccherato LW, Antolinos S, Xue Y, Shawa N, Gilman RH, Cabrera L, Berg DE, Tyler-Smith C, Kelly P, Tarazona-Santos E, Hollox EJ: A worldwide analysis of beta-defensin copy number variation suggests recent selection of a high-expressing DEFB103 gene copy in East Asia. Hum Mutat. 2011, 32: 743-750. 10.1002/humu.21491.
Hellström AR, Watt B, Fard SS, Tenza D, Mannström P, Narfström K, Ekesten B, Ito S, Wakamatsu K, Larsson J, Ulfendahl M, Kullander K, Raposo G, Kerje S, Hallböök F, Marks MS, Andersson L: Inactivation of Pmel alters melanosome shape but has only a subtle effect on visible pigmentation. PLoS Genet. 2011, 7: e1002285-10.1371/journal.pgen.1002285.
Brenner M, Hearing VJ: The protective role of melanin against UV damage in human skin. Photochem Photobiol. 2008, 84: 539-549. 10.1111/j.1751-1097.2007.00226.x.
Plonka PM, Passeron T, Brenner M, Tobin DJ, Shibahara S, Thomas A, Slominski A, Kadekaro AL, Hershkovitz D, Peters E, Nordlund JJ, Abdel-Malek Z, Takeda K, Paus R, Ortonne JP, Hearing VJ, Schallreuter KU: What are melanocytes really doing all day long...?. Exp Dermatol. 2009, 18: 799-819. 10.1111/j.1600-0625.2009.00912.x.
Slominski A, Zmijewski MA, Pawelek J: l-tyrosine and l-dihydroxyphenylalanine as hormone-like regulators of melanocyte functions. Pigment Cell Melanoma Res. 2012, 25: 14-27. 10.1111/j.1755-148X.2011.00898.x.
Roberts DW, Newton RA, Leonard JH, Sturm RA: Melanocytes expressing MC1R polymorphisms associated with red hair color have altered MSH-ligand activated pigmentary responses in coculture with keratinocytes. J Cell Physiol. 2008, 215: 344-355. 10.1002/jcp.21318.
Michard Q, Commo S, Belaidi JP, Alleaume AM, Michelet JF, Daronnat E, Eilstein J, Duche D, Marrot L, Bernard BA: TRP-2 specifically decreases WM35 cell sensitivity to oxidative stress. Free Rad Biol Med. 2008, 44: 1023-1031. 10.1016/j.freeradbiomed.2007.11.021.
Jiang S, Liu XM, Dai X, Zhou Q, Lei TC, Beermann F, Wakamatsu K, Xu SZ: Regulation of DHICA-mediated antioxidation by dopachrome tautomerase: implication for skin photoprotection against UVA radiation. Free Rad Biol Med. 2010, 48: 1144-1151. 10.1016/j.freeradbiomed.2010.01.033.
Panzella L, Napolitano A, d'Ischia M: Is DHICA the key to dopachrome tautomerase and melanocyte functions?. Pigment Cell Melanoma Res. 2011, 24: 248-249. 10.1111/j.1755-148X.2010.00771.x.
Kovacs D, Flori E, Maresca V, Ottaviani M, Aspite N, Dell'anna ML, Panzella L, Napolitano A, Picardo M, d'Ischia M: The eumelanin intermediate 5,6-dihydroxyindole-2-carboxylic acid is a messenger in the cross-talk among epidermal cells. J Invest Dermatol. 2012, 132: 1196-1205. 10.1038/jid.2011.457.
Kadekaro AL, Leachman S, Kavanagh RJ, Swope V, Cassidy P, Supp D, Sartor M, Schwemberger S, Babcock G, Wakamatsu K, Ito S, Koshoffer A, Boissy RE, Manga P, Sturm RA, Abdel-Malek ZA: Melanocortin 1 receptor genotype: an important determinant of the damage response of melanocytes to ultraviolet radiation. FASEB J. 2010, 24: 3850-3860. 10.1096/fj.10-158485.
Wong SS, Ainger SA, Leonard JH, Sturm RA: MC1R variant allele effects on UVR-induced phosphorylation of p38, p53, and DDB2 repair protein responses in melanocytic cells in culture. J Invest Dermatol. 2012, 132: 1452-1461. 10.1038/jid.2011.473.
Sendoel A, Kohler I, Fellmann C, Lowe SW, Hengartner MO: HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature. 2010, 465: 577-583. 10.1038/nature09141.
Jablonski NG, Chaplin G: Colloquium paper: human skin pigmentation as an adaptation to UV radiation. Proc Nat Acad Sci USA. 2010, 107 (Suppl 2): 8962-8968.
Elias PM, Menon G, Wetzel BK, Williams JJ: Barrier requirements as the evolutionary "driver" of epidermal pigmentation in humans. Am J Hum Biol. 2010, 22: 526-537. 10.1002/ajhb.21043.
Jablonski NG: The evolution of human skin colouration and its relevance to health in the modern world. J R Coll Phys Edin. 2012, 42: 58-63. 10.4997/JRCPE.2012.114.
Jablonski NG, Chaplin G: Human skin pigmentation, migration and disease susceptibility. Philos Trans R Soc Lond Biol Sci. 2012, 367: 785-792. 10.1098/rstb.2011.0308.
Yuen AW, Jablonski NG: Vitamin D: in the evolution of human skin colour. Mel Hypotheses. 2010, 74: 39-44. 10.1016/j.mehy.2009.08.007.
Robins AH: The evolution of light skin color: role of vitamin D disputed. Am J Phys Anthropol. 2009, 139: 447-450. 10.1002/ajpa.21077.
Nessvi S, Johansson L, Jopson J, Stewart A, Reeder A, McKenzie R, Scragg RK: Association of 25-hydroxyvitamin D3 levels in adult New Zealanders with ethnicity, skin color and self-reported skin sensitivity to sun exposure. Photochem Photobiol. 2011, 87: 1173-1178. 10.1111/j.1751-1097.2011.00956.x.
Ahn J, Yu K, Stolzenberg-Solomon R, Simon KC, McCullough ML, Gallicchio L, Jacobs EJ, Ascherio A, Helzlsouer K, Jacobs KB, Li Q, Weinstein SJ, Purdue M, Virtamo J, Horst R, Wheeler W, Chanock S, Hunter DJ, Hayes RB, Kraft P, Albanes D: Genome-wide association study of circulating vitamin D levels. Hum Mol Gene. 2010, 19: 2739-2745. 10.1093/hmg/ddq155.
Wang TJ, Zhang F, Richards JB, Kestenbaum B, van Meurs JB, Berry D, Kiel DP, Streeten EA, Ohlsson C, Koller DL, Peltonen L, Cooper JD, O'Reilly PF, Houston DK, Glazer NL, Vandenput L, Peacock M, Shi J, Rivadeneira F, McCarthy MI, Anneli P, de Boer IH, Mangino M, Kato B, Smyth DJ, Booth SL, Jacques PF, Burke GL, Goodarzi M, Cheung CL, et al: Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet. 2010, 376: 180-188. 10.1016/S0140-6736(10)60588-0.
Berry DJ, Vimaleswaran KS, Whittaker JC, Hingorani AD, Hypponen E: Evaluation of genetic markers as instruments for mendelian randomization studies on vitamin D. PloS One. 2012, 7: e37465-10.1371/journal.pone.0037465.
Tanaka T, Scheet P, Giusti B, Bandinelli S, Piras MG, Usala G, Lai S, Mulas A, Corsi AM, Vestrini A, Sofi F, Gori AM, Abbate R, Guralnik J, Singleton A, Abecasis GR, Schlessinger D, Uda M, Ferrucci L: Genome-wide association study of vitamin B6, vitamin B12, folate, and homocysteine blood concentrations. Am J Hum Genet. 2009, 84: 477-482. 10.1016/j.ajhg.2009.02.011.
Gambichler T, Bader A, Sauermann K, Altmeyer P, Hoffmann K: Serum folate levels after UVA exposure: a two-group parallel randomised controlled trial. BMC Dermatol. 2001, 1: 8-10.1186/1471-5945-1-8.
Moan J, Nielsen KP, Juzeniene A: Immediate pigment darkening: its evolutionary roles may include protection against folate photosensitization. FASEB J. 2012, 26: 971-975. 10.1096/fj.11-195859.
Elias PM, Menon G, Wetzel BK, Williams JJ: Evidence that stress to the epidermal barrier influenced the development of pigmentation in humans. Pigment Cell Melanoma Res. 2009, 22: 420-434. 10.1111/j.1755-148X.2009.00588.x.
Gunathilake R, Schurer NY, Shoo BA, Celli A, Hachem JP, Crumrine D, Sirimanna G, Feingold KR, Mauro TM, Elias PM: pH-regulated mechanisms account for pigment-type differences in epidermal barrier function. J Invest Dermatol. 2009, 129: 1719-1729. 10.1038/jid.2008.442.
Cheli Y, Luciani F, Khaled M, Beuret L, Bille K, Gounon P, Ortonne JP, Bertolotto C, Ballotti R: {alpha}MSH and cyclic AMP elevating agents control melanosome pH through a protein kinase A-independent mechanism. J Biol Chem. 2009, 284: 18699-18706. 10.1074/jbc.M109.005819.
Fuller BB, Spaulding DT, Smith DR: Regulation of the catalytic activity of preexisting tyrosinase in black and Caucasian human melanocyte cell cultures. Exp Cell Res. 2001, 262: 197-208. 10.1006/excr.2000.5092.
Smith DR, Spaulding DT, Glenn HM, Fuller BB: The relationship between Na(+)/H(+) exchanger expression and tyrosinase activity in human melanocytes. Exp Cell Res. 2004, 298: 521-534. 10.1016/j.yexcr.2004.04.033.
Fumagalli M, Sironi M, Pozzoli U, Ferrer-Admetlla A, Pattini L, Nielsen R: Signatures of environmental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution. PLoS Genet. 2011, 7: e1002355-10.1371/journal.pgen.1002355.
Frost P: European hair and eye color: A case of frequency-dependent sexual selection?. Evol Hum Behav. 2006, 27: 85-103. 10.1016/j.evolhumbehav.2005.07.002.
Gardiner E, Jackson CJ: Eye color predicts disagreeableness in North Europeans: Support in favor of Frost (2006). Curr Psychol. 2010, 2 (9): 1-9.
Provencio I: The hidden organ in your eyes. Sci Am. 2011, 304: 54-59.
Roecklein KA, Rohan KJ, Duncan WC, Rollag MD, Rosenthal NE, Lipsky RH, Provencio I: A missense variant (P10L) of the melanopsin (OPN4) gene in seasonal affective disorder. J Affect Disord. 2009, 114: 279-285. 10.1016/j.jad.2008.08.005.
Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen TS, Altshuler D, Lander ES: Positive natural selection in the human lineage. Science. 2006, 312: 1614-1620. 10.1126/science.1124309.
Pritchard JK, Pickrell JK, Coop G: The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol. 2010, 20: R208-215. 10.1016/j.cub.2009.11.055.
O'Connell MJ: Selection and the cell cycle: positive Darwinian selection in a well-known DNA damage response pathway. J Mol Evol. 2010, 71: 444-457. 10.1007/s00239-010-9399-y.
Beleza S, Dos Santos AM, McEvoy B, Alves I, Martinho C, Cameron E, Shriver MD, Parra EJ, Rocha J: The timing of pigmentation lightening in Europeans. Mol Biol Evol. 2012, doi 10.1093/molbev/mss207
Schallreuter KU, Wazir U, Kothari S, Gibbons NC, Moore J, Wood JM: Human phenylalanine hydroxylase is activated by H2O2: a novel mechanism for increasing the l-tyrosine supply for melanogenesis in melanocytes. Biochem Biophys Res Comm. 2004, 322: 88-92. 10.1016/j.bbrc.2004.07.082.
Acknowledgements
Richard A Sturm and David L Duffy are NHMRC Senior Research Fellows.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Sturm, R.A., Duffy, D.L. Human pigmentation genes under environmental selection. Genome Biol 13, 248 (2012). https://doi.org/10.1186/gb-2012-13-9-248
Accepted:
Published:
DOI: https://doi.org/10.1186/gb-2012-13-9-248