
Abstract
Background
Accurate catalogs of structural variants (SVs) in mammalian genomes are necessary to elucidate the potential mechanisms that drive SV formation and to assess their functional impact. Next generation sequencing methods for SV detection are an advance on array-based methods, but are almost exclusively limited to four basic types: deletions, insertions, inversions and copy number gains.
Results
By visual inspection of 100 Mbp of genome to which next generation sequence data from 17 inbred mouse strains had been aligned, we identify and interpret 21 paired-end mapping patterns, which we validate by PCR. These paired-end mapping patterns reveal a greater diversity and complexity in SVs than previously recognized. In addition, Sanger-based sequence analysis of 4,176 breakpoints at 261 SV sites reveal additional complexity at approximately a quarter of structural variants analyzed. We find micro-deletions and micro-insertions at SV breakpoints, ranging from 1 to 107 bp, and SNPs that extend breakpoint micro-homology and may catalyze SV formation.
Conclusions
An integrative approach using experimental analyses to train computational SV calling is essential for the accurate resolution of the architecture of SVs. We find considerable complexity in SV formation; about a quarter of SVs in the mouse are composed of a complex mixture of deletion, insertion, inversion and copy number gain. Computational methods can be adapted to identify most paired-end mapping patterns.
Similar content being viewed by others


Background
The identification of structural variants (SVs) in mammalian genomes [1–4] has important implications for our understanding of genetic diversity, has elucidated the concept of genomic disorders [5, 6] and has improved the analysis of genetic association in common and rare diseases [7–12], cancer development [13] and genomic evolution [14, 15]. However, the accurate identification of SVs in mammalian genomes remains challenging.
Next generation sequencing provides a novel approach for identifying structural variants [16] and exploits read-pair information [17, 18], split reads [19, 20], read depth [21] and sequence assembly [22] to localize SVs. Typically, variation in the expected number of reads mapping to the reference sequence is used to identify copy number variants while deviations from the expected distance between reads, and the orientation of reads, is used to infer the presence and type of structural variant at a locus. These methods presuppose that sequencing reads form characteristic patterns for different types of structural variants [23]. For example, when the two sequenced ends of a fragment map back to the reference genome in the correct orientation, but at a distance that is significantly larger than the size of the fragment itself (as inferred from the library insert size distribution), this indicates a deletion.
Algorithms that use whole-genome sequence reads make assumptions about the paired-end mapping (PEM) patterns they will encounter, even though we know that the molecular architecture of structural variants can be remarkably complex [24, 25]. For example, deletion and inversion events that appear simple may contain additional sequence at breakpoints and different types of structural variants sometimes occur together, so that, for example, an inversion immediately abuts a deletion [26]. However, current automated methods to identify SVs are unable to differentiate basic patterns (for example, a simple inversion) from more complex ones (for example, an inversion right next to a deletion), resulting in some SVs being incorrectly classified while others are missed altogether.
Past studies have described several forms of complex structural variants, ranging from multiple rearrangements at large genomic loci [27, 28], to deletions, inversions, insertions and duplications that fall in close proximity [29]. More recently, a subtle form of complex SVs has been characterized by micro-insertions or micro-deletions at the breakpoints of larger SVs [30]. In our present study, we describe complex SVs as two or more structural variants co-occurring at the same locus, without intervening DNA of normal structure between the variants (SVs are directly adjacent to each other) and without distinction by SV size (complex SVs can be two or more large SVs right next to each other or a small SV right next to a larger SV).
Here we combine visual inspection of PEM data from 17 mouse genomes [31] with experimental validation to resolve the molecular architecture of SVs and to guide genome-wide computational analysis [32]. We provide a comprehensive catalog of 21 PEM patterns derived from simple and complex SVs, and show how these patterns may provide insights into the fine-scale molecular architecture of SV formation.
Results
Catalog of paired-end mapping patterns
We started by generating a set of validated PEM patterns that we could use to guide genome-wide computational analysis. To do this, we visually examined short-read PEM patterns and manually called SVs from 100 Mbp in 17 inbred strains of mice [31, 32] (A/J, AKR/J, BALB/cJ, C3H/HeJ, C57BL/6NJ, CBA/J, DBA/2J, LP/J, 129S5SvEvBrd, 129P2/OlaHsd, 129S1/SvImJ, NOD/ShiLtJ, NZO/HlLtJ, CAST/EiJ, PWK/PhJ, WSB/EiJ and SPRET/EiJ) that included the whole of mouse chromosome 19 (61 Mbp in size), and a random set of other chromosomal regions. We provide an overview of the procedure to catalog PEM patterns in Figure 1a, b.

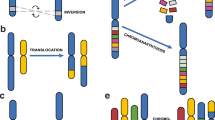
An overview of the procedure to characterize structural variants. A flow diagram of the different steps we used to characterize structural variants. (a) We first inspected 100 Mbp of next generation sequencing (NGS) data in 17 genomes. We examined chromosome 19 in its entirety and a random set of other chromosomal regions. (b) We identified 21 PEM patterns across 1,494 SV sites. We show two examples of PEM patterns, H1 and H2, as visualized using the LookSeq tool [38]. The H1 deletion is on chromosome 1 and has strain distribution pattern 11110101 (1 means presence and 0 absence of the deletion) in the following strain order: A/J, AKR/J, BALB/cJ, C3H/HeJ, C57BL/6J, CBA/J, DBA/2J and LP/J. The H2 deletion is on chromosome 8 and is present in seven strains. (c) We randomly selected 662 SV sites for PCR-validation to investigate all PEM patterns and show the results for the two deletion SVs. (d) We randomly selected 261 SV sites for analysis of breakpoint sequence features using Sanger-based sequencing technology. We show sequencing data of a simple deletion of type H1 on chromosome 10 (20,467,348-20,467,894). Sequence analysis confirmed the deletion of 547 bp but also revealed an insertion of 34 bp.
Based on read depth and anomalous PEM, we identified 21 patterns, as described in Table 1 and Additional file 1. We unambiguously classified 11 PEM patterns, referred to as 'H' patterns, for high confidence. While some of the H patterns are typical and have already been described [23], others (H3, H5, H9 and H11) are novel. Figure 2a shows the novel PEM pattern H5, an inversion directly flanked by two deletions. Note that depending on the size of the inversion, the H5 pattern of paired-end reads will differ: for instance, suppose the length of the inversion is small, H5 reads will span both deletions and inversions, giving a PEM pattern of a typical deletion; suppose now the inversion is larger, H5 reads will individually span each deletion, giving a PEM pattern of an inversion.

Novel PEM patterns. (a) PEM pattern of a Del(s)+Inv (H5). We draw paired-end reads (black arrows) and how they map to the reference (Ref.) genome C57BL/6J (dashed grey lines). Blue boxes represent deletions and orange boxes inversions. Green arrows represent primers used for PCR amplification and sequencing reactions. (b) PEM pattern of an Inv+Ins (Q8), with PCR data across eight classical strains (A/J, AKR/J, BALB/cJ, C3H/HeJ, C57BL/6J, CBA/J, DBA/2J and LP/J). The pink box represents de novo sequence insertion. The amplicon size for BALB/cJ, C3H/HeJ, CBA/J and DBA/2J is about 500 bp larger than the other strains, indicative of the insertion.
Interpretation of the remaining ten PEM patterns was ambiguous. We refer to these as type Q ('Questionable') patterns (Q1 to Q10; Table 1; Additional file 1). With the exception of Q4 (large deletion), the remaining Q patterns have not been described before. Two patterns appeared false positives (Q6 and Q10). Q1 and Q7 were due to variable number tandem repeats. Q5 and Q9 were difficult to interpret: read-pair information suggested a structural variant while read depth did not. Q2, Q3 and Q8 had partial patterns compared to typical SV patterns. For example, Q2 has a read depth of zero flanked by regions of normal read depth but it does not have paired-end reads spanning the length of the variant as does a typical deletion.
We found that partial PEM patterns were caused by the presence of a de novo insertion right next to a deletion or inversion. Figure 2b shows an example of an inversion flanked by a de novo insertion. A paired-end read spanning the first breakpoint of the inversion is mapped as expected to the reference genome. However, one end of the fragment spanning the second breakpoint of the inversion does not map to the reference genome (because it lies within the inserted sequence), creating a signature of an 'orphaned' read mate.
From the 100 Mbp we visually inspected, we identified a total of 1,494 SVs that matched the 21 PEM patterns (Additional file 1). Because visual identification of H6 and H7 patterns was more difficult than for the other variants, we excluded them from our analysis of chromosome 19 and identified 872 deletions (631 type H and 241 type Q) bigger than 100 bp, 15 inversions (2 type H and 13 type Q) and 3 copy number gains (all type H) (Additional file 2). In addition to the 890 SVs identified on chromosome 19, we found 604 on the other chromosomes. We refer to these lists on chromosome 19 and other chromosomal regions as our gold-standard list of PEM patterns.
Since we examined the whole of chromosome 19 in eight strains, we looked at the distribution of SVs along the chromosome in the context of regional features. To do this, we counted the number of SVs overlapping protein-coding genes, coding exons and repeat regions on chromosome 19 using Ensembl build 65, and we compared this to a null distribution of the expected number of overlaps, obtained by performing a permutation analysis. Across all strains, we found a non-random distribution of SVs along the chromosome (Additional file 3) with enrichment (P < 0.01, fold change 2.2) in repeat regions and depletion (P < 0.01, fold change 0.25) in coding regions. We found only two SVs on chromosome 19 that affect one, or more, coding exons of genes involved either in immunity or olfaction (Additional file 4).
Architecture of SVs using PEM inspection
Next we tested the molecular architecture of SVs as inferred by our visual inspection of PEM patterns (Figure 1c). To do this, we applied a primer design strategy depending on type and length of the SV (Additional file 5) and confirmed the underlying molecular structure of all 21 PEM patterns using PCR- and Sanger-based sequencing across 8 (A/J, AKR/J, BALB/cJ, C3H/HeJ, C57BL/6J, CBA/J, DBA/2J and LP/J) of the 17 Mouse Genomes Project strains [31]. These eight strains are the progenitors of the HS (heterogeneous stock) [33], an outbred population we have used to achieve genome-wide high-resolution mapping of multiple phenotypes [34].
We designed 742 pairs of primers (Additional file 6) and successfully amplified 662 SV sites (Additional file 7). It should be noted that we excluded failed designs (due to the presence of SNP(s) in the primer sequences) and designed 80 additional primer pairs to amplify sites when one primer pair yielded no information or only part of the answer - for instance, for a deletion adjacent to an insertion predicted by visual inspection of the PEM. With the exception of insertions (excluded from our chromosome 19 data set), SV sites we analyzed were representative of the overall distribution of PEM categories (Additional file 8).
We defined simple SVs as those whose structural interpretation is straightforward and consists of one SV type: insertions, deletions and inversions (Figure 3a). We also identify another type of insertion, a copy number gain, as consisting of non-repetitive DNA that is present in multiple copies relative to the reference genome. When this sequence occurs immediately adjacent to its original, it is annotated as tandem duplication; when it is small and close to another copy, it is annotated as a linked gain.

Architecture of structural variants. (a) Simple SVs: deletion (Del), insertion (Ins), inversion (Inv), tandem duplication (tandem Dup) and other types of copy number gains. Linked gain is a small copy number gain at close proximity to its copy. Inverted linked gain (not drawn) is similar to a linked gain but the copy is inverted. Del+Nml+Del is two deletions separated by a normal copy of small size. (b) Complex SVs: deletion co-occurring with insertion (Del+Ins), inversion with flanking deletions (Del(s)+Inv), inversion with insertion (Inv+Ins), deletion within a copy number gain (Del in gain) and inversion within a copy number gain (Inv in gain).
In contrast to previous SV studies that use the number of breakpoints that fall in close proximity, our definition of complex SVs is based on the mixture of SV types (of small or large size) that directly abut each other, with no intervening DNA, since these might be the progeny of a single process (marked as Del+Ins, Del(s)+Inv and Inv+Ins in Figure 3b). We also separately identify an SV within a copy number gain (termed 'Del in gain' and 'Inv in gain' in Figure 3b) since the probability of coincidence is less than one event per genome.
Our categorization of predicted SV structures, based on manual inspection of PEM patterns, resulted in the highly confident identification of a structural variant for 18 of the 21 patterns that we examined by PCR: 12 were indicative of a simple SV and 6 of a complex SV (Table 1). Two patterns did not represent structural variants (Q6 and Q10), but were due to the presence of a retrotransposed pseudogene, which caused false SV patterns. SVs of type Q7 (55 cases) were due to variable number tandem repeats, for which we could not predict the number of repeats or molecular structure (Additional file 8).
We estimated the relative proportions of simple and complex SVs by manual inspection of PEM patterns on chromosome 19. Assuming an equal number of deletions and insertions on chromosome 19, then about 88% of SVs are composed of one SV, 2.5% of two adjacent SVs at the same locus and 9.5% are variable number tandem repeats (Additional file 8). Note that we have not recorded SVs on chromosome 19 where three (or possibly more) different types of SVs co-occurred (for example, a deletion right next to an inversion and insertion), although about a dozen rearrangements had three different types of SVs based on their PEM patterns. Consequently, our estimate of the number of complex SVs on chromosome 19 based on PEM inspection is likely to be conservative.
Fine-scale architecture of simple SVs
To gain insights into the fine scale architecture of simple SVs as inferred by our visual inspection of PEM patterns, we randomly selected 261 simple SV sites and analyzed their breakpoints (Figure 1d; Additional file 8). Using the rat as an outgroup species, we inferred SV ancestry (as described in [32]), and classified SVs into two groups (ancestral insertion or deletion). We found 105 ancestral insertions and 142 ancestral deletions; the remaining 14 were either inversions (n = 6) or tandem duplications (n = 8). We sequenced a total of 4,176 breakpoints at 261 selected SV regions in 8 strains using PCR-based Sanger sequencing (Additional file 9).
We found additional complexities at breakpoints of 62 SVs (24%): small additional (micro-insertions) and small missing (micro-deletions) sequences (Table 2). Figure 1d shows an example of a simple H1 deletion; sequence analysis at the nucleotide level confirmed the deletion of 547 bp but also revealed an insertion of 34 bp.
We determined the extent and content of micro-insertions and micro-deletions and identified three patterns of SV breakpoints (classified in Table 2). The first pattern is characterized by micro-deletions at SV breakpoints. At 15% of ancestral insertions there were missing nucleotides at the breakpoints, ranging from 1 to 289 bp.
The second pattern includes SVs that have sequence inserted at their breakpoints; 27% of ancestral deletions showed a micro-insertion, with size ranging from 1 to 107 bp. We report the origin of the sequence involved in micro-insertions in Additional file 9. There were three cases: (i) intra- or (ii) inter-chromosomal copy number gain of small size, or (iii) insertion of retrotransposons.
The third pattern of SV breakpoints is characterized by simple SVs with both occurrence of micro-deletion and micro-insertion. One third of inversions had this pattern at their breakpoint. Size of SVs was not correlated to one particular pattern, nor to micro-deletion/micro-insertion length and type.
Our analysis of breakpoint sequence features in multiple strains also allowed us to look for a relationship between sequence variants and SV formation. In particular, we addressed the question as to whether sequence variants at breakpoints were associated with SV formation.
In all cases, the presence of SNPs in the micro-homology region (short length of identical sequence at an ancestral deletion's start and end points) was correlated with the presence of the SV (Figure 4). The SNP elongates the micro-homology, or, alternatively, the micro-homology reflects a hyper-mutable state associated with break-induced replication around the SV [35]. However, this phenomenon is rare: we only observed five (4.5%) cases amongst our manually curated ancestral deletions (Additional file 9) where a SNP and SV formation co-segregate. We found a similar relationship between a SNP formed at the target site duplication and the presence of an ancestral insertion. Fifteen ancestral insertions (16%) had SNPs or short indels within their target site duplication, coincident with the insertion (Additional file 9).

Relationship between SNP and SV formation. Two SNPs lying on the 6 bp micro-homology of an ancestral deletion of 64 bp (chr12:27,040,459-27,040,522) correlated with the presence of the SV. Sequencing traces are shown for a test strain (A/J) and the reference strain (C57BL/6J). Note that all other test strain traces are identical to the one shown here. The black asterisks indicate the micro-homology of 6 bp (GAACTA). The presence of two SNPs (C->G and T->A, shown using red asterisks) in all test strains (here only shown in A/J) is associated with the presence of the structural variant.
Discussion
Our findings are important in two ways. First, we show that an integrative approach using experimental analyses to train computational SV calling is essential for the accurate characterization of SV architecture. Second, we find a considerable complexity in SV formation; about a quarter of SVs in the mouse are composed of a complex mixture of deletion, insertion, inversion and copy number gain.
In contrast to studies that start by identifying SVs using automated genome-wide methods, followed by experimental validation, we started by experimentally determining a set of SVs and then applied this information to interpret whole-genome automated SV detection [32]. Laboratory-based efforts proved essential for two main reasons. First, they allowed the correct interpretation of the PEM patterns. Without knowing how to interpret the underlying molecular structure of each PEM, some patterns would be missed or classified incorrectly by computational methods alone. Second, our laboratory efforts allowed the recognition of a diversity of PEM patterns. Otherwise we would not be able to distinguish between simple and complex SVs.
Finer-scale breakpoint sequence analysis reveals that 24% of simple SVs have smaller rearrangements at the nucleotide level (micro-insertions or micro-deletions at the breakpoint of a larger SV). This raises questions about the likely mechanisms of SV formation.
We know that retrotransposition is the commonest mechanism of SV formation in the mouse [32]. We also know that retrotransposons (LINEs, SINEs and long terminal repeats) are typically characterized by flanking target site duplications and a poly(A) tail or poly(T) head. However, we observed that 15% of retrotransposon SVs do not have target site duplications and truncated or absent poly(A) tails or poly(T) heads (Additional file 9). Moran and colleagues [36] observed a similar phenomenon in the human genome and suggested that retrotransposons, such as LINE-1 elements, integrate into DNA lesions, resulting in retrotransposon-mediated DNA repair. We suggest that about 15% of retrotransposon SVs in the mouse genome formed through a similar mechanism involving DNA repair.
It is reasonable to assume that the complexities (micro-insertions and micro-deletions) we see at the breakpoints of ancestral deletions, inversions and gains (we call these 'complex' non-retrotransposon SVs) (Table 2) will correlate with a complex mechanism of formation. A DNA replication fork stalling and template switching/microhomology-mediated break-induced replication (FoSTeS/MMBIR) mechanism has been proposed to generate such complex SVs in the human genome [37]. In addition, about half of our complex non-retrotransposon SVs have microhomology (short sequence of identical bases) ranging from 3 to 25 bp (Additional file 9), compatible with a microhomology-mediated break-induced replication process. It could be that the complex non-retrotransposon SVs are also the progeny of mutational processes during DNA replication.
Interestingly, our estimate that 24% of SVs have micro-structures at their breakpoint is the same as that reported by Eichler and colleagues [30] in a study of human structural variation. Another sequencing-based study of SVs in two mouse strains (DBA/2J and C57BL/6J) examined 3,316 breakpoints and reported that 16% of non-transposon structural variants are complex, as defined by multiple breakpoints mapped to within 1 kbp of each other [29]. However, we were not able to directly compare these results to ours since we have not used the same classification criteria (we used a classification based on SVs being right next to each other, whereas Hall and colleagues' [29] was based on SVs being at close proximity).
Ideally, sequencing longer reads would typically be required to resolve the complex architecture of structural variants we report in this study, something that goes beyond the current generation sequencing platforms. Our findings offer an intermediate solution between next generation sequencing analysis and complete de novo assembly of genomes.
Materials and methods
Visual identification of PEM patterns
We visually inspected short-read sequencing data using LookSeq [38] and manually detected PEM patterns across mouse chromosome 19 in its entirety and a random set of other chromosomal regions, accounting for 100 Mbp of total genomic regions. We analyzed molecular architecture of these PEM patterns at nucleotide-level resolution using PCR and Sanger-based sequencing.
Experimental validation of SV architecture
Primers were designed using Primer3 [39] and purchased from MWG (Ebersberg, Germany). Primer design strategy was dependent on the type and size of the structural variant (Additional file 5). Three independent PCR reactions were carried out with Hotstar Taq obtained from Qiagen (Hilden, Germany). Reactions were performed as previously described [40]. LongRange PCR kit (Qiagen) was used for genomic regions > 2 kbp. PCR gel images were then assessed for quality of primer design and performance of PCR reaction. Representative PCR examples are given in Additional file 1. We provide interested readers with PCR results for each SV site (Additional file 7). Complex SV sites were subject to several rounds of primer design and PCR. PCR products were then purified in a 96-well Millipore purification plate, resuspended in 30 μl of H2O and sequenced as previously described [40]. All sequencing reactions were run out on an ABI3700 sequencer and assembled by using PHRED/PHRAP [41].
Assembly of Sanger-based sequencing data
Consed was used for visualization and editing of the assembly [42]. Strains with and without the SV were aligned into one contig. Breakpoint analysis was mostly based on visual inspection of the alignment and BLAT search. The first breakpoint was identified when the strains with and without the SV stopped aligning and the second breakpoint when they started aligning again. Micro-insertion at the breakpoint was defined as any additional sequence relative to the reference genome (MGSCv37) in the strains with a deletion variant. Micro-deletion at the breakpoint was defined as any missing sequence relative to the reference genome (MGSCv37) in the strains with insertion variant. We also recorded the longest stretch of sequence identity at both breakpoints, which we defined as sequence micro-homology. When micro-homology occurred, we recorded the SV so that start and end coordinates were the smallest. For blunt ended SV, one set of start and end coordinates was recorded. BLAT was used to get the exact start and end coordinates of the SV.
Genome-wide detection of structural variants
Genome-wide structural variants were detected using four methods: split-read mapping (Pindel) [20], mate-pair analysis (BreakDancer) [18], single-end cluster analysis (SECluster and RetroSeq, unpublished), and read-depth (CND) [21]. Details of the complete pipeline, SVMerge, is described elsewhere [43]. We used in-house Perl scripts to detect genome-wide complex structural variants [32].
Data
Data sets described in this study are available under study accession number 'estd185' from the Database of Genomic Variants Archive (DGVa) [44]. Our previous genome-wide data of structural variants [32] are also available from DGVa under accession number 'estd118'.
Abbreviations
- bp:
-
base pair
- Del:
-
deletion
- Dup:
-
duplication
- kbp:
-
kilobase pair
- Ins:
-
insertion
- Inv:
-
inversion
- LINE:
-
long interspersed nuclear element
- Mbp:
-
megabase pair
- Nml:
-
normal
- PCR:
-
polymerase chain reaction
- SINE:
-
short interspersed nuclear element
- PEM:
-
paired-end mapping
- SNP:
-
single nucleotide polymorphism
- SV:
-
structural variant.
References
Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AW, Robson S, Stirrups K, Valsesia A, Walter K, Wei J, Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME: Origins and functional impact of copy number variation in the human genome. Nature. 2010, 464: 704-712. 10.1038/nature08516.
Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, Haugen E, Zerr T, Yamada NA, Tsang P, Newman TL, Tuzun E, Cheng Z, Ebling HM, Tusneem N, David R, Gillett W, Phelps KA, Weaver M, Saranga D, Brand A, Tao W, Gustafson E, McKernan K, Chen L, Malig M, et al: Mapping and sequencing of structural variation from eight human genomes. Nature. 2008, 453: 56-64. 10.1038/nature06862.
Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, Chinwalla A, Conrad DF, Fu Y, Grubert F, Hajirasouliha I, Hormozdiari F, Iakoucheva LM, Iqbal Z, Kang S, Kidd JM, Konkel MK, Korn J, Khurana E, Kural D, Lam HY, Leng J, Li R, Li Y, Lin CY, Luo R, et al: Mapping copy number variation by population-scale genome sequencing. Nature. 2011, 470: 59-65. 10.1038/nature09708.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, et al: Global variation in copy number in the human genome. Nature. 2006, 444: 444-454. 10.1038/nature05329.
Lupski JR: Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998, 14: 417-422. 10.1016/S0168-9525(98)01555-8.
Lupski JR: Genomic disorders ten years on. Genome Med. 2009, 1: 42-10.1186/gm42.
McCarroll SA: Extending genome-wide association studies to copy-number variation. Hum Mol Genet. 2008, 17: R135-142. 10.1093/hmg/ddn282.
McCarroll SA, Altshuler DM: Copy-number variation and association studies of human disease. Nat Genet. 2007, 39: S37-42. 10.1038/ng2080.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BH, Cochrane L, Corsello C, et al: Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010, 466: 368-372. 10.1038/nature09146.
Bochukova EG, Huang N, Keogh J, Henning E, Purmann C, Blaszczyk K, Saeed S, Hamilton-Shield J, Clayton-Smith J, O'Rahilly S, Hurles ME, Farooqi IS: Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010, 463: 666-670. 10.1038/nature08689.
Jarick I, Vogel CI, Scherag S, Schafer H, Hebebrand J, Hinney A, Scherag A: Novel common copy number variation for early onset extreme obesity on chromosome 11q11 identified by a genome-wide analysis. Hum Mol Genet. 2011, 20: 840-852. 10.1093/hmg/ddq518.
Xu B, Woodroffe A, Rodriguez-Murillo L, Roos JL, van Rensburg EJ, Abecasis GR, Gogos JA, Karayiorgou M: Elucidating the genetic architecture of familial schizophrenia using rare copy number variant and linkage scans. Proc Natl Acad Sci USA. 2009, 106: 16746-16751. 10.1073/pnas.0908584106.
Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, Cole K, Mosse YP, Wood A, Lynch JE, Pecor K, Diamond M, Winter C, Wang K, Kim C, Geiger EA, McGrady PW, Blakemore AI, London WB, Shaikh TH, Bradfield J, Grant SF, Li H, Devoto M, Rappaport ER, Hakonarson H, Maris JM: Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009, 459: 987-991. 10.1038/nature08035.
Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE: Diversity of human copy number variation and multicopy genes. Science. 2010, 330: 641-646. 10.1126/science.1197005.
Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, London SJ, Eichler EE: De novo rates and selection of large copy number variation. Genome Res. 2010, 20: 1469-1481. 10.1101/gr.107680.110.
Alkan C, Coe BP, Eichler EE: Genome structural variation discovery and genotyping. Nat Rev Genet. 2011, 12: 363-376. 10.1038/nrg2958.
Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, Taillon BE, Chen Z, Tanzer A, Saunders AC, Chi J, Yang F, Carter NP, Hurles ME, Weissman SM, Harkins TT, Gerstein MB, Egholm M, Snyder M: Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007, 318: 420-426. 10.1126/science.1149504.
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, Shi X, Fulton RS, Ley TJ, Wilson RK, Ding L, Mardis ER: BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009, 6: 677-681. 10.1038/nmeth.1363.
Albers CA, Lunter G, MacArthur DG, McVean G, Ouwehand WH, Durbin R: Dindel: accurate indel calls from short-read data. Genome Res. 2011, 21: 961-973. 10.1101/gr.112326.110.
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z: Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009, 25: 2865-2871. 10.1093/bioinformatics/btp394.
Simpson JT, McIntyre RE, Adams DJ, Durbin R: Copy number variant detection in inbred strains from short read sequence data. Bioinformatics. 2010, 26: 565-567. 10.1093/bioinformatics/btp693.
She X, Jiang Z, Clark RA, Liu G, Cheng Z, Tuzun E, Church DM, Sutton G, Halpern AL, Eichler EE: Shotgun sequence assembly and recent segmental duplications within the human genome. Nature. 2004, 431: 927-930. 10.1038/nature03062.
Medvedev P, Stanciu M, Brudno M: Computational methods for discovering structural variation with next-generation sequencing. Nat Methods. 2009, 6: S13-20. 10.1038/nmeth.1374.
Liu P, Erez A, Nagamani SC, Dhar SU, Kolodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, Bacino CA, Campos-Acevedo LD, Delgado MR, Freedenberg D, Garnica A, Grebe TA, Hernandez-Almaguer D, Immken L, Lalani SR, McLean SD, Northrup H, Scaglia F, Strathearn L, Trapane P, Kang SH, Patel A, Cheung SW, Hastings PJ, Stankiewicz P, Lupski JR, et al: Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011, 146: 889-903. 10.1016/j.cell.2011.07.042.
Quinlan AR, Hall IM: Characterizing complex structural variation in germline and somatic genomes. Trends Genet. 2011, 28: 43-53.
Conrad DF, Bird C, Blackburne B, Lindsay S, Mamanova L, Lee C, Turner DJ, Hurles ME: Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet. 2010, 42: 385-391. 10.1038/ng.564.
Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, et al: Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011, 144: 27-40. 10.1016/j.cell.2010.11.055.
Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, Onofrio R, Carter SL, Park K, Habegger L, Ambrogio L, Fennell T, Parkin M, Saksena G, Voet D, Ramos AH, Pugh TJ, Wilkinson J, Fisher S, Winckler W, Mahan S, Ardlie K, Baldwin J, Simons JW, Kitabayashi N, MacDonald TY, et al: The genomic complexity of primary human prostate cancer. Nature. 2011, 470: 214-220. 10.1038/nature09744.
Quinlan AR, Clark RA, Sokolova S, Leibowitz ML, Zhang Y, Hurles ME, Mell JC, Hall IM: Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res. 2010, 20: 623-635. 10.1101/gr.102970.109.
Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK, Eichler EE: A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell. 2010, 143: 837-847. 10.1016/j.cell.2010.10.027.
Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, Furlotte NA, Eskin E, Nellaker C, Whitley H, Cleak J, Janowitz D, Hernandez-Pliego P, Edwards A, Belgard TG, Oliver PL, McIntyre RE, Bhomra A, Nicod J, Gan X, Yuan W, van der Weyden L, Steward CA, Bala S, Stalker J, Mott R, et al: Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011, 477: 289-294. 10.1038/nature10413.
Yalcin B, Wong K, Agam A, Goodson M, Keane TM, Gan X, Nellaker C, Goodstadt L, Nicod J, Bhomra A, Hernandez-Pliego P, Whitley H, Cleak J, Dutton R, Janowitz D, Mott R, Adams DJ, Flint J: Sequence-based characterization of structural variation in the mouse genome. Nature. 2011, 477: 326-329. 10.1038/nature10432.
Talbot CJ, Nicod A, Cherny SS, Fulker DW, Collins AC, Flint J: High-resolution mapping of quantitative trait loci in outbred mice. Nat Genet. 1999, 21: 305-308. 10.1038/6825.
Valdar W, Solberg LC, Gauguier D, Burnett S, Klenerman P, Cookson WO, Taylor MS, Rawlins JN, Mott R, Flint J: Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat Genet. 2006, 38: 879-887. 10.1038/ng1840.
Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A: Break-induced replication is highly inaccurate. PLoS Biol. 2011, 9: e1000594-10.1371/journal.pbio.1000594.
Morrish TA, Gilbert N, Myers JS, Vincent BJ, Stamato TD, Taccioli GE, Batzer MA, Moran JV: DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat Genet. 2002, 31: 159-165. 10.1038/ng898.
Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR: The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009, 41: 849-853. 10.1038/ng.399.
Manske HM, Kwiatkowski DP: LookSeq: a browser-based viewer for deep sequencing data. Genome Res. 2009, 19: 2125-2132. 10.1101/gr.093443.109.
Rozen S, Skaletsky H: Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000, 132: 365-386.
Yalcin B, Willis-Owen SA, Fullerton J, Meesaq A, Deacon RM, Rawlins JN, Copley RR, Morris AP, Flint J, Mott R: Genetic dissection of a behavioral quantitative trait locus shows that Rgs2 modulates anxiety in mice. Nat Genet. 2004, 36: 1197-1202. 10.1038/ng1450.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8: 175-185.
Gordon D, Abajian C, Green P: Consed: a graphical tool for sequence finishing. Genome Res. 1998, 8: 195-202.
Wong K, Keane TM, Stalker J, Adams DJ: Enhanced structural variant and breakpoint detection using SVMerge by integration of multiple detection methods and local assembly. Genome Biol. 2010, 11: R128-10.1186/gb-2010-11-12-r128.
Database of Genomic Variants archive. [http://www.ebi.ac.uk/dgva/]
Smit AFAHR, Green P: RepeatMasker. [http://www.repeatmasker.org/]
Acknowledgements
We thank Polinka Hernandez-Pliego, Helen Whitley, James Cleak, Rebekah Dutton, Deborah Janowitz, Adam Whitley, Giles Durrant, Andrew Marc Hammond, Danica Joy Fabrigar, Lucia Chen, Martina Johannesson, Enzhao Cong and Glòria Blázquez for helping BY with various laboratory-based efforts. We also thank Xiangchao Gan for additional computational analysis. This project was supported by the Medical Research Council, UK, and the Wellcome Trust. DJA is supported by Cancer Research UK.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
DJA and JF directed the research. KW and TMK performed the genome-wide SV discovery. KW wrote computational methods to detect complex SVs. AB and BY analyzed Sanger-based sequencing data. BY cataloged PEM patterns, determined molecular architecture of SVs and led experimental analyses. MG carried out additional analyses. BY and JF wrote the paper. All authors read and approved the final manuscript.
Electronic supplementary material
13059_2011_2780_MOESM1_ESM.PDF
Additional file 1: 21 PEM patterns. We found 11 'high-confidence' patterns and 10 'questionable' patterns. For each PEM, we provide PEM details, illustration using LookSeq [38] and PCR results. We show paired-end reads (black arrows) and how they map to the reference genome (dashed grey lines). Green arrows represent primer pairs. PCR was carried out across the founder strains of the HS [33]. We used HyperladderII as size marker. (PDF 1 MB)
13059_2011_2780_MOESM2_ESM.XLS
Additional file 2: Chromosome 19 gold-standard data set. Columns 1 to 3: chromosome, approximate SV start and end coordinates (bp). Column 4: SV length (bp). Column 5: PEM pattern (Table 1; Additional file 1). Columns 6 to13: strain distribution pattern (SDP) across eight classical strains (1 = SV present; 0 = SV absent). Column 14: has SV been PCRed (1 = yes). (XLS 150 KB)
13059_2011_2780_MOESM3_ESM.PDF
Additional file 3: Distribution of manual SV calls along chromosome 19. The top horizontal tracks show the chromosomal distribution of manually identified structural variants (deletions, inversions and duplications) for specific mouse strains (A/J, AKR/J, BALB/cJ, C3H/HeJ, CBA/J, DBA/2J and LP/J). The bottom two tracks represent genes (Ensembl 65) and gaps on chromosome 19. (PDF 86 KB)
13059_2011_2780_MOESM4_ESM.XLS
Additional file 4: Chromosome 19 manual SV calls that affect coding regions. Column 1: chromosome. Columns 2 and 3: SV start and stop coordinates (bp). Column 4: SV event. Column 5: affected gene (a plus sign indicates that the gene is affected in its entirety). Column 6: description of the gene. (XLS 25 KB)
13059_2011_2780_MOESM5_ESM.PDF
Additional file 5: Primer design strategy. We applied a primer design strategy depending on type and length of the SV. Forward primer is in green and reverse primer in red. SV sites were repeat masked prior to primer design, using RepeatMasker [45]. Breakpoints were initially predicted using LookSeq [38]. Primer design is illustrated for: (a) tandem duplication, (b) insertion, (c) deletion and (d) inversion. (PDF 168 KB)
13059_2011_2780_MOESM6_ESM.XLS
Additional file 6: Primers. For each primer pair (PP), we provide a primer pair identification, name and sequence of forward and reverse primers. (XLS 204 KB)
13059_2011_2780_MOESM7_ESM.XLS
Additional file 7: PCR data in eight classical strains. Column 1: chromosome. Columns 2 and 3: SV start and end coordinates (bp). Column 4: SV length. Column 5: PEM pattern (Table 1; Additional file 1). Columns 6 to 13: 1 = presence or 0 = absence of the SV (2, 3 and 4 indicate multi-allelic SVs). Column 14: has the site been (= 1) or not (= 0) resolved at nucleotide level resolution (when column 14 = 1, columns 2 and 3 refer to the exact coordinates, otherwise they are estimates). Column 15: primer coverage (number of primer pairs designed per unique SV site). Column 16: primer pair used to amplify the SV region. (XLS 228 KB)
13059_2011_2780_MOESM8_ESM.XLS
Additional file 8: Summary data of PCR and Sanger-based sequencing for each of the 21 PEM patterns. Column 1: PEM pattern (Table 1; Additional file 1). Column 2: number of unique SV sites PCRed. Column 3: chromosome 19 data (some cells are marked NA (not applicable) because we have not systematically inspected H6 and H7 patterns). Column 4: predicted SV. Column 5: PCR validated SV. Column 6: number of SVs sequenced at nucleotide level. Column 7: type of SV as simple, complex, false or variable number tandem repeat. (XLS 34 KB)
13059_2011_2780_MOESM9_ESM.XLSX
Additional file 9: The 261 simple SV sites resolved at nucleotide level resolution using Sanger-based sequencing. Column 1: primer name. Columns 2 to 5: exact SV position. Column 6: PEM pattern. Column 7: length of any micro-deletion or micro-insertion at the SV breakpoint (bp). Columns 8 to 15: strain distribution pattern SDP. Columns 16 and 17: is there a SNP within the micro-homology (MH) or target site duplication (TSD) (0 = no, 1 = yes; NA, not applicable). Column 18: MH length (bp). Column 19: MH type. Column 20: TSD length. Column 21: origin of the inserted sequence. (XLSX 46 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Yalcin, B., Wong, K., Bhomra, A. et al. The fine-scale architecture of structural variants in 17 mouse genomes. Genome Biol 13, R18 (2012). https://doi.org/10.1186/gb-2012-13-3-r18
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/gb-2012-13-3-r18