
Abstract
Recent studies have identified both heritable DNA methylation effects and differential methylation in disease-discordant identical twins. Larger sample sizes, replication, genetic-epigenetic analyses and longitudinal assays are now needed to establish the role of epigenetic variants in disease.
Similar content being viewed by others
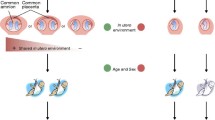


Twins provide a unique opportunity to study DNA methylation, because they are matched controls for nearly all genetic variants and many environmental factors. The study of twins in epigenetics is valuable from two perspectives: first, it can provide information about the underlying biological mechanisms that drive and maintain variation in DNA methylation; and second, in the context of epigenome-wide association studies (EWAS), it can give insights into epigenetic effects in complex disease. Over the past year there has been a surge of studies reporting genome-wide DNA methylation profiles in twins. Here, we briefly discuss recent findings and their implications, which raise important new questions in the field.
DNA methylation twin heritability estimates
Early studies of epigenetic profiles in twins examined DNA methylation at particular genomic regions and showed that monozygotic twin concordance in methylation was greater than the concordance observed between dizygotic pairs or pairs of unrelated individuals, but that rates varied across genes and in general decreased with age [1]. Several recent studies have estimated DNA methylation twin heritability (Box 1), and the contribution of environmental effects to variation in DNA methylation at individual CpG sites across the genome. The studies were conducted across different cells and tissues, and across a wide age range, from newborns to middle-aged twins. They all apply recently developed genome-wide DNA methylation assays, which target more regions at finer-scale resolution and measure DNA methylation at each CpG site as a quantitative trait aiming to reflect the proportion of methylated cells in the sample.
Gervin et al. [2] examined local DNA methylation variability and heritability in the major histocompatibility complex (MHC) region in middle-aged twins. They reported a low overall rate of DNA methylation heritability in CD4+ lymphocytes in 49 monozygotic and 40 dizygotic twin pairs using extensive bisulfite sequencing of the MHC region. Their estimates show evidence for modest genetic effects on DNA methylation at specific CpG sites, but the majority of DNA methylation patterns across the MHC were attributed to non-genetic factors and showed extensive variability.
Box 1. DNA methylation heritability estimates from twin studies
DNA methylation heritability refers to the proportion of locus-specific DNA methylation variance in the population that is due to genetic variation. Twin studies estimate the narrow sense heritability (h2), which is the proportion of the total locus-specific DNA methylation variance in the population that is attributed to additive genetic effects. Twin-based heritability estimates compare correlations within monozygotic and dizygotic twins and can be calculated as h2=(rMZ-rDZ), where r is the correlation in DNA methylation levels at a particular locus in each pair type (monozygotic (MZ) or dizygotic (DZ)). The classical twin study allows not only for the estimation of genetic effects on locus-specific DNA methylation variability, but also for the differentiation of shared and unique environmental components, which are of interest because monozygotic and dizygotic twins share the same uterus and birth date and are exposed to similar environmental factors in early life. Heritability estimates are population and environment specific, but in the context of DNA methylation they are also specific to the type of cell, tissue, locus and developmental stage. Interpreting twin-based DNA methylation heritabilities should avoid common misconceptions (see [46, 47]) and assumptions of generalizability to trans-generational inheritance at all genomic regions (see [48]).
Shifting towards genome-wide assays, Gordon et al. [3] examined the methylome of neonatal twins in three tissues - cord blood mononuclear cells (CBMCs), human umbilical vascular endothelial cells (HUVECs) and placenta - using a promoter-specific genome-wide methylation array (Illumina HumanMethylation27 DNA Analysis BeadChip assay (Illumina27K), Illumina, San Diego, CA, USA). They also found that DNA methylation heritability estimates were relatively low across the genome, and that the majority of DNA methylation variation could be attributed to non-shared intra-uterine environment and stochastic effects. However, individual CpG sites showed a wide range of heritability estimates, and the top 5% most heritable probes within tissues had high heritability (h2 > 0.49). Surprisingly, relatively few of the highly heritable probes were shared across tissues, and only three probes were highly heritable in all three tissues.
Another recent study of middle-aged twins and unrelated individuals examined whole-blood DNA methylation on the promoter-specific genome-wide DNA methylation array (Illumina27K) [4]. The authors estimated DNA methylation heritabilities and identified DNA methylation quantitative trait loci (meQTLs), which are genetic variants that associate DNA methylation levels at CpG sites, typically in cis. The mean CpG-site heritability across the genome was relatively low, but individual sites had high heritability estimates, and 1,537 CpG sites were found to associate with meQTL SNPs in cis.
Recent findings are in line with previous reports of greater similarity in DNA methylation levels between monozygotic compared to dizygotic twins at specific regions in the genome [5, 6]. Recent estimates of mean genome-wide CpG-site-specific DNA methylation heritability are 12 to 18% in blood [3, 4], 5% in placenta [3], and 7% in HUVECs [3]. Overall, the mean DNA methylation heritability across the genome is consistently estimated as relatively low, but locus-specific levels are quite variable, and specific CpG sites show strong evidence for heritability. Previous reports of high monozygotic twin concordance in DNA methylation levels in early life were based on assays that examined fewer genomic regions at relatively low resolution in a small sample of young twins [1], whereas recent technologies include more loci at single-CpG-level resolution and have been performed in larger samples across a wide age range. High DNA methylation heritability at a subset of regions is also concordant with results from allele-specific methylation (ASM) studies across multiple tissues and samples [7–11]. So far, consistent evidence of strong heritability at a subset of CpG sites exists, but heritable sites constitute only a small proportion of all CpG sites assayed. However, detecting low to modest heritability is dependent on sample size, and estimates of the proportion of heritable CpG sites are strongly influenced by the selection of regions included in the methylation arrays used. The majority of results so far are based on promoter-specific assays (Illumina 27k) or custom arrays [5, 6]. Future work needs to estimate methylation heritabilities in unselected genome-wide data in larger samples.
Despite the relatively small proportion of CpG sites that show evidence for DNA methylation heritability, the heritable effects are convincing because they are concordant with results from multiple meQTL studies in unrelated individuals in brain tissue [12, 13], whole blood [4], and lymphoblastoid cell lines (LCLs) [14, 15]. To assess whether twin heritability findings are consistent with meQTLs in unrelated individuals of similar genetic backgrounds, we assessed whether CpG sites with meQTLs were also heritable in twins. As expected, we estimated greater heritability at the 1,537 CpG sites with meQTLs identified in whole blood in middle-aged individuals, and some of these CpG sites also showed evidence for meQTLs in independent samples [4]. For example, approximately 30% of CpG sites with meQTLs identified in whole blood in twins [4] overlap with CpG sites with meQTLs from unrelated subjects in different tissues, including brain [12] and transformed cells (LCLs) [14]. This suggests that specific CpG sites are under strict genetic control, and are stable and shared across tissues within individuals.
To understand the mechanisms that likely underlie heritable DNA methylation effects, it is worth looking at the characteristics of CpG sites with meQTLs, and of SNPs that are meQTLs. The genome-wide meQTL studies published to date report that the majority of promoter-specific CpG sites with meQTLs have associations with SNPs in cis [12, 14, 15]. A recent report has identified the presence of small methylation-determining regions in promoters, which are necessary and sufficient to regulate DNA methylation depending on developmental state, the presence of specific DNA-binding motifs, and a critical CpG density [16]. Further work is needed to assess whether the enrichment of cis meQTL associations also occurs at non-promoter CpG sites with meQTLs. CpG sites with meQTLs also appear to be population specific, stressing the importance of genetic background on epigenetic effects [15].
DNA methylation heritability and meQTL findings also relate to reports identifying similar genetic effects in different epigenetic mechanisms, such as histone modifications [17], transcription-factor binding [18], and chromatin structure [19, 20]. These results provide insights into the complex interplay across different levels of epigenetic mechanisms and the mechanisms that control chromatin conformation [19]. More studies are needed to help distinguish between the epigenetic processes that are drivers of chromatin structure changes, and those that are markers of these changes.
Disease-discordant twin EWAS
The second general advantage of studying epigenetic patterns in twins is in identifying epigenetic variants that are linked to disease, using EWAS of disease-discordant identical twins. The disease-discordant twin approach holds great promise and has proven to be successful in identifying a number of epidemiological and environmental risk factors in complex phenotypes [21, 22]. Disease-discordant identical twins can be seen as an ideal model, because twins are matched for most genetic variants, as well as many non-genetic effects such as early environment, maternal effects, and age and cohort effects. Furthermore, rates of twin discordances are higher than commonly believed, and are generally >50% for even the most common complex traits studied (Figure 1).

Monozygotic twin discordance rates for common disease. Estimates of mean monozygotic twin discordance rates from the literature and the TwinsUK cohort for a series of common diseases, such as colon cancer and breast cancer [32], rheumatoid arthritis (RA) [33, 34], osteoarthritis (OA) [35], psoriasis [36], cardiovascular disease (CVD) and myocardial infarction (MI) [37], type 1 diabetes [38, 39], type 2 diabetes [38, 40], autism and autism spectrum disorders (ASD) [41–44], and schizophrenia [45].
Several EWAS in disease-discordant twins have been published within the past year and the results show a trend - each study reported modest, but consistent, differential methylation in moderate to large numbers of genes relevant to the phenotype. We briefly describe results from three recent studies of common diseases in discordant twins, which were performed on the same promoter-specific DNA methylation platform (Illumina27K).
Dempster et al. [23] examined whole-blood DNA methylation patterns in 22 monozygotic twin pairs discordant for schizophrenia or bipolar disorder. They identified many differentially methylated regions (DMRs), and pathway analysis of the top loci showed a significant enrichment for gene networks directly relevant to psychiatric disorders and neurodevelopment. The mean methylation difference between affected and unaffected co-twins was 6% at the top DMR, but varied considerably across the sample. Assuming a conservative Bonferroni-adjusted threshold (α = 1.9 × 10-6), standard paired-analysis results did not surpass the multiple testing correction, but - taking into account heterogeneity across families - resulted in genome-wide significant associations at the top DMRs.
Rakyan et al. [24] examined DNA methylation in CD14+ monocytes from 15 type 1 diabetes (T1D) discordant monozygotic twin pairs. Assuming a conservative Bonferroni-adjusted threshold (α = 2.2 × 10-6), standard paired-analysis results did not surpass the multiple testing correction. However, the authors followed up the top 132 DMRs in four additional T1D-discordant monozygotic pairs and observed a similar direction of association effects. Pathway analysis indicated that several of the genes associated with the 132 DMRs were linked to T1D or the immune response. The authors also obtained longitudinal DNA methylation profiles in two additional datasets, which showed that the DMR variants were enriched in individuals both before and after disease onset, suggesting that the DMR effects arise early on in the etiological process that leads to T1D.
Gervin et al. [25] assessed DNA methylation and gene expression differences in psoriasis-discordant monozygotic twin pairs, using samples from CD4+ (17 monozygotic pairs) and CD8+ (13 monozygotic pairs) cells. The authors observed many DMRs and differentially expressed regions with small effects, which were not significant genome-wide. However, combined analysis of DNA methylation and gene expression identified genes where differences in DNA methylation were correlated with differences in gene expression, and several of the top-ranked genes were known to be associated with psoriasis. Gene ontology analysis revealed an enrichment of genes involved in biological processes associated with the immune response and in pathways comprising cytokines and chemokines, which have a clear role in psoriasis.
In each of the three studies there were many DMRs with modest effects, but these were often located in genes that are either known candidates for, or have apparent biological relevance to, the trait. These findings are especially exciting because of the overlap with molecular studies and genome-wide association study (GWAS) results, which imply that epigenetic studies of disease may prove to reveal not just markers of the disease process, but a novel approach to studying risk factors and mechanisms of complex phenotype susceptibility and progression. EWAS could therefore provide another route for the discovery of novel disease-associated SNPs. The EWAS performed to date have identified epigenetic variants with effect sizes larger than typical GWAS effects. For example, a recent DNA methylation study of smoking identified a DMR in a CpG site in the F2RL3 gene, coding for protease-activated receptor-4 (PAR4), at which median DNA methylation levels were 83% in heavy smokers and 95% in non-smokers, giving a difference of 12% methylation between the two groups [26]. This corresponds to an odds ratio of 3.9 of the epigenetic variant [27], which is approximately 3.5-fold greater than reported GWAS effects. However, EWAS findings also raise two important questions: first, why have genome-wide significant EWAS signals not yet been identified in known candidate genes; and second, are the identified changes causal or secondary to the trait?
We believe that the first issue is a question of power. None of the studies so far have used large samples or high-resolution methylation (or other epigenetic) assays. Typically, studies have either used very small samples (n < 5) with high-resolution approaches such as bisulfite sequencing [28], or lower-resolution assays, such as Illumina27K, with modest sample sizes (n = 13 to 25) [4, 23–25]. The power of these studies to detect disease-related DNA differential methylation effects will depend on many factors. These include variables describing the biology of DNA methylation, such as the initial trigger of the epigenetic variant and its stability through cell division, its effect size on the disease (or of the disease on the methylation variant), the coverage of the methylation assay, and sample size and study design. Kaminsky et al. [29] estimated the power of the discordant twin study design, using a particular CpG-island microarray methylation variant in a candidate gene, and found reasonable power to detect DMRs with 15 twin pairs. However, formal power calculations for more extensive genome-wide coverage have not yet been reported in twins. Preliminary estimates from published DMRs report low (35%) to reasonable (>80%) power to detect DMRs at specific CpG sites, at methylation differences of 5 to 6% between affected and unaffected twins [4, 23]. The observed variability of the reported methylation differences at the CpG site of interest (and distribution of DNA methylation levels in the sample) will also impact power, as has been observed in traditional case-control DNA methylation power analysis [27, 30].
The second disease-related differential methylation question is whether it is possible to distinguish epigenetic changes that are causal from those that arise secondary to disease. The identification of potential causal effects is exciting, but secondary effects can also help us to understand complex phenotype progression, and may lead to the determination of early diagnostic or prognostic markers. In both cases the therapeutic value of the results has great potential.
We propose two approaches to disentangle potential epigenetic cause from consequence in disease: first, integrating genetic-epigenetic data in phenotype analysis; and second, obtaining longitudinal epigenetic data before and after disease onset. Genetic-epigenetic studies would identify cases where genetic effects on the trait are potentially mediated by DNA methylation, and DNA methylation is therefore likely to be causal to the trait. In these cases genetic variants that are associated with the trait would also tend to be meQTLs for the CpG site, at which DNA methylation is also associated with the phenotype. However, the proportion of CpG sites in the genome where DNA methylation is under the influence of genetic effects seems to be relatively small (albeit based on low-resolution scans so far). In addition, the majority of genetic-epigenetic effects on the phenotype may already be identified in gene mapping studies of disease, and EWAS findings would in some cases only clarify potential mechanisms of action of already-identified GWAS signals. It is also possible that the genetic variant interacts with the epigenetic variant in disease susceptibility; for example, DMR effects may affect only disease-discordant monozygotic twins of a particular genotype. However, although genetic-epigenetic disease results imply causality, this is not necessarily always the case. It is possible that genetic associations lead to the phenotype of interest, which in turn drives changes in methylation and alters gene expression as a consequence.
The most conclusive approach to disentangle potential cause versus consequence of DNA methylation changes associated with disease is to perform longitudinal studies. In this case, the underlying cause of the DNA methylation effect can be genetic or non-genetic, and should be examined before, during, and after disease onset to help understand its role in disease onset and progression. Longitudinal studies are crucial to understanding epigenetic effects in disease and should be a priority when samples are available, which sadly is often not the case.
The main goal of longitudinal DNA methylation studies is to identify whether the DNA methylation change arose prior to disease onset and is therefore likely to be causal. If that is the case, it is important to note the timing of the change both before the appearance of the phenotype and potentially during intermediate pre-clinical phenotype states prior to final disease (for example, normoglycemic, pre-diabetic, diabetic). Obtaining such data will inform the biological model of epigenetic effects on disease. For instance, is there a threshold model similar to the second hit in retinoblastoma [31], which can be applied to DNA methylation effects during phenotype onset? If a threshold model is correct, then identifying the threshold of deleterious DNA methylation changes for each phenotype will be of clinical value. If longitudinal methylation studies identify effects that are likely to be causal to disease, then another immediate question is whether reversing these methylation effects during or after disease onset can help prevent, delay, or ameliorate the disease.
On the other hand, if longitudinal studies predominantly find that observed methylation changes are probably consequences of disease, then these findings can give insights into the mechanisms involved in disease progression. A related question is whether reversal of such changes can also reverse disease or prevent exacerbation of disease symptoms. This becomes further complicated in the case of relapsing diseases such as bipolar disorder, multiple sclerosis, or psoriasis, where there is a known or unknown trigger of the condition.
In conclusion, the early twin EWAS have provided us with fascinating insights into the potential power of the identical disease-discordant twin model to find novel susceptibility genes as well as novel disease mechanisms and potential drug targets. These results call for larger samples, replication, and more in-depth analyses, including genetic-epigenetic analyses and longitudinal assays, to establish the role of epigenetic variants in disease. Epigenetic effects may also play an important role in relapsing diseases such as bipolar disorder, multiple sclerosis and psoriasis, where there is a known or unknown trigger of the condition.
Abbreviations
- AMS:
-
allele-specific methylation
- CBMC:
-
cord blood mononuclear cell
- DMR:
-
differentially methylated region
- EWAS:
-
epigenome-wide association studies
- GWAS:
-
genome-wide association studies
- HUVEC:
-
human umbilical vascular endothelial cell
- Illumina27K:
-
Illumina HumanMethylation27 DNA Analysis BeadChip assay
- LCL:
-
lymphoblastoid cell line
- meQTL:
-
methylation quantitative trait locus
- MHC:
-
major histocompatibility complex
- QTL:
-
quantitative trait locus
- SNP:
-
single nucleotide polymorphism
- T1D:
-
type 1 diabetes.
References
Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M: Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005, 102: 10604-10609. 10.1073/pnas.0500398102.
Gervin K, Hammero M, Akselsen HE, Moe R, Nygard H, Brandt I, Gjessing HK, Harris JR, Undlien DE, Lyle R: Extensive variation and low heritability of DNA methylation identified in a twin study. Genome Res. 2011, 21: 1813-1821. 10.1101/gr.119685.110.
Gordon L, Joo JE, Powell JE, Ollikainen M, Novakovic B, Li X, Andronikos R, Cruickshank MN, Conneely KN, Smith AK, Alisch RS, Morley R, Visscher PM, Craig JM, Saffery R: Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence. Genome Res. 2012, 22: 1395-1340. 10.1101/gr.136598.111.
Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, Shin SY, Dempster EL, Murray RM, Grundberg E, Hedman AK, Nica A, Small KS, MuTHER Consortium, Dermitzakis ET, McCarthy MI, Mill J, Spector TD, Deloukas P: Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012, 8: e1002629-10.1371/journal.pgen.1002629.
Boks MP, Derks EM, Weisenberger DJ, Strengman E, Janson E, Sommer IE, Kahn RS, Ophoff RA: The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS One. 2009, 4: e6767-10.1371/journal.pone.0006767.
Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, Wong AH, Feldcamp LA, Virtanen C, Halfvarson J, Tysk C, McRae AF, Visscher PM, Montgomery GW, Gottesman II, Martin NG, Petronis A: DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009, 41: 240-245. 10.1038/ng.286.
Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, Parker SL, Kucera KS, Willard HF, Myers RM: Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011, 7: e1002228-10.1371/journal.pgen.1002228.
Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, Li K, Murty VV, Schupf N, Vilain E, Morris M, Haghighi F, Tycko B: Genomic surveys by methylationsensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008, 40: 904-908. 10.1038/ng.174.
Meaburn EL, Schalkwyk LC, Mill J: Allele-specific methylation in the human genome: implications for genetic studies of complex disease. Epigenetics. 2010, 5: 578-582. 10.4161/epi.5.7.12960.
Schalkwyk LC, Meaburn EL, Smith R, Dempster EL, Jeffries AR, Davies MN, Plomin R, Mill J: Allelic skewing of DNA methylation is widespread across the genome. Am J Hum Genet. 2010, 86: 196-212. 10.1016/j.ajhg.2010.01.014.
Shoemaker R, Deng J, Wang W, Zhang K: Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 2010, 20: 883-889. 10.1101/gr.104695.109.
Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, Johnson R, Zielke HR, Ferrucci L, Longo DL, Cookson MR, Singleton AB: Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010, 6: e1000952-10.1371/journal.pgen.1000952.
Zhang D, Cheng L, Badner JA, Chen C, Chen Q, Luo W, Craig DW, Redman M, Gershon ES, Liu C: Genetic control of individual differences in gene-specific methylation in human brain. Am J Hum Genet. 2010, 86: 411-419. 10.1016/j.ajhg.2010.02.005.
Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, Gilad Y, Pritchard JK: DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011, 12: R10-10.1186/gb-2011-12-1-r10.
Fraser HB, Lam LL, Neumann SM, Kobor MS: Population-specificity of human DNA methylation. Genome Biol. 2012, 13: R8-10.1186/gb-2012-13-2-r8.
Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, Schubeler D: Identification of genetic elements that autonomously determine DNA methylation states. Nat Genet. 2011, 43: 1091-1097. 10.1038/ng.946.
Kadota M, Yang HH, Hu N, Wang C, Hu Y, Taylor PR, Buetow KH, Lee MP: Allelespecific chromatin immunoprecipitation studies show genetic influence on chromatin state in human genome. PLoS Genet. 2007, 3: e81-10.1371/journal.pgen.0030081.
Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, Habegger L, Rozowsky J, Shi M, Urban AE, Hong MY, Karczewski KJ, Huber W, Weissman SM, Gerstein MB, Korbel JO, Snyder M: Variation in transcription factor binding among humans. Science. 2010, 328: 232-235. 10.1126/science.1183621.
Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, Stephens M, Gilad Y, Pritchard JK: DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012, 482: 390-394. 10.1038/nature10808.
McDaniell R, Lee BK, Song L, Liu Z, Boyle AP, Erdos MR, Scott LJ, Morken MA, Kucera KS, Battenhouse A, Keefe D, Collins FS, Willard HF, Lieb JD, Furey TS, Crawford GE, Iyer VR, Birney E: Heritable individual-specific and allelespecific chromatin signatures in humans. Science. 2010, 328: 235-239. 10.1126/science.1184655.
Hopper JL, Seeman E: The bone density of female twins discordant for tobacco use. N Engl J Med. 1994, 330: 387-392. 10.1056/NEJM199402103300603.
Greenfield JR, Samaras K, Jenkins AB, Kelly PJ, Spector TD, Gallimore JR, Pepys MB, Campbell LV: Obesity is an important determinant of baseline serum C-reactive protein concentration in monozygotic twins, independent of genetic influences. Circulation. 2004, 109: 3022-3028. 10.1161/01.CIR.0000130640.77501.79.
Dempster E, Pidsley R, Schalkwyk L, Toulopoulou T, Picchioni M, Kravariti E, Murray R, Mill J: Methylomic profiling in twins discordant for major psychosis. Twin Res Hum Genet. 2010, 13: 253-
Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, Daunay A, Busato F, Mein CA, Manfras B, Dias KR, Bell CG, Tost J, Boehm BO, Beck S, Leslie RD: Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet. 2011, 7: e1002300-10.1371/journal.pgen.1002300.
Gervin K, Vigeland MD, Mattingsdal M, Hammero M, Nygard H, Olsen AO, Brandt I, Harris JR, Undlien DE, Lyle R: DNA methylation and gene expression changes in monozygotic twins discordant for psoriasis: identification of epigenetically dysregulated genes. PLoS Genet. 2012, 8: e1002454-10.1371/journal.pgen.1002454.
Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H: Tobacco-smokingrelated differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011, 88: 450-457. 10.1016/j.ajhg.2011.03.003.
Rakyan VK, Down TA, Balding DJ, Beck S: Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011, 12: 529-541. 10.1038/nrg3000.
Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA, Zhang L, Farmer AD, Bell CJ, Kim RW, May GD, Woodward JE, Caillier SJ, McElroy JP, Gomez R, Pando MJ, Clendenen LE, Ganusova EE, Schilkey FD, Ramaraj T, Khan OA, Huntley JJ, Luo S, Kwok PY, Wu TD, Schroth GP, Oksenberg JR, Hauser SL, Kingsmore SF: Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010, 464: 1351-1356. 10.1038/nature08990.
Kaminsky Z, Petronis A, Wang SC, Levine B, Ghaffar O, Floden D, Feinstein A: Epigenetics of personality traits: an illustrative study of identical twins discordant for risk-taking behavior. Twin Res Hum Genet. 2008, 11: 1-11. 10.1375/twin.11.1.1.
Wang S: Method to detect differentially methylated loci with case-control designs using Illumina arrays. Genet Epidemiol. 2011, 35: 686-694. 10.1002/gepi.20619.
Knudson AG: Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971, 68: 820-823. 10.1073/pnas.68.4.820.
Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K: Environmental and heritable factors in the causation of cancer - analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000, 343: 78-85. 10.1056/NEJM200007133430201.
MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, Silman AJ: Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000, 43: 30-37. 10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B.
Svendsen AJ, Holm NV, Kyvik K, Petersen PH, Junker P: Relative importance of genetic effects in rheumatoid arthritis: historical cohort study of Danish nationwide twin population. BMJ. 2002, 324: 264-266. 10.1136/bmj.324.7332.264.
Spector TD, Cicuttini F, Baker J, Loughlin J, Hart D: Genetic influences on osteoarthritis in women: a twin study. BMJ. 1996, 312: 940-943. 10.1136/bmj.312.7036.940.
Grjibovski AM, Olsen AO, Magnus P, Harris JR: Psoriasis in Norwegian twins: contribution of genetic and environmental effects. J Eur Acad Dermatol Venereol. 2007, 21: 1337-1343. 10.1111/j.1468-3083.2007.02268.x.
Zdravkovic S, Wienke A, Pedersen NL, de Faire U: Genetic susceptibility of myocardial infarction. Twin Res Hum Genet. 2007, 10: 848-852. 10.1375/twin.10.6.848.
Condon J, Shaw JE, Luciano M, Kyvik KO, Martin NG, Duffy DL: A study of diabetes mellitus within a large sample of Australian twins. Twin Res Hum Genet. 2008, 11: 28-40. 10.1375/twin.11.1.28.
Hyttinen V, Kaprio J, Kinnunen L, Koskenvuo M, Tuomilehto J: Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs: a nationwide follow-up study. Diabetes. 2003, 52: 1052-1055. 10.2337/diabetes.52.4.1052.
Lehtovirta M, Pietilainen KH, Levalahti E, Heikkila K, Groop L, Silventoinen K, Koskenvuo M, Kaprio J: Evidence that BMI and type 2 diabetes share only a minor fraction of genetic variance: a follow-up study of 23,585 monozygotic and dizygotic twins from the Finnish Twin Cohort Study. Diabetologia. 2010, 53: 1314-1321. 10.1007/s00125-010-1746-4.
Rosenberg RE, Law JK, Yenokyan G, McGready J, Kaufmann WE, Law PA: Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med. 2009, 163: 907-914. 10.1001/archpediatrics.2009.98.
Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, Miller J, Fedele A, Collins J, Smith K, Lotspeich L, Croen LA, Ozonoff S, Lajonchere C, Grether JK, Risch N: Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011, 68: 1095-1102. 10.1001/archgenpsychiatry.2011.76.
Kates WR, Burnette CP, Eliez S, Strunge LA, Kaplan D, Landa R, Reiss AL, Pearlson GD: Neuroanatomic variation in monozygotic twin pairs discordant for the narrow phenotype for autism. Am J Psychiatry. 2004, 161: 539-546. 10.1176/appi.ajp.161.3.539.
Lichtenstein P, Carlstrom E, Rastam M, Gillberg C, Anckarsater H: The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am J Psychiatry. 2010, 167: 1357-1363. 10.1176/appi.ajp.2010.10020223.
Beckmann H, Franzek E: The genetic heterogeneity of "schizophrenia". World J Biol Psychiatry. 2000, 1: 35-41. 10.3109/15622970009150564.
Boomsma D, Busjahn A, Peltonen L: Classical twin studies and beyond. Nat Rev Genet. 2002, 3: 872-882. 10.1038/nrg932.
Visscher PM, Hill WG, Wray NR: Heritability in the genomics era - concepts and misconceptions. Nat Rev Genet. 2008, 9: 255-266.
Petronis A: Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010, 465: 721-727. 10.1038/nature09230.
Acknowledgements
We thank PC Tsai for helping compile the estimates of effect size and twin discordance rates. This work was supported by the European Research Council senior researcher award to TDS (ERC 250157).
Author information
Authors and Affiliations
Corresponding authors
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Bell, J.T., Spector, T.D. DNA methylation studies using twins: what are they telling us?. Genome Biol 13, 172 (2012). https://doi.org/10.1186/gb-2012-13-10-172
Published:
DOI: https://doi.org/10.1186/gb-2012-13-10-172