
Abstract
A subset of X-linked genes escapes silencing by X inactivation and is expressed from both X chromosomes in mammalian females. Species-specific differences in the identity of these genes have recently been discovered, suggesting a role in the evolution of sex differences. Chromatin analyses have aimed to discover how genes remain expressed within a repressive environment.
Similar content being viewed by others

Review
The difference in sex-chromosome make-up between mammalian males (XY) and females (XX) has led to the evolution of two main dosage-compensation mechanisms: upregulation of the active X chromosome (Xa) in both sexes to balance X expression with the autosomes; and inactivation of one X chromosome in females to avoid X hyperexpression and correct for the difference in gene dosage between the sexes [1–3] (see Table 1). These mechanisms evolved to compensate for the presence of only one copy (haploinsufficiency) of X-linked genes in males due to degeneration of the Y chromosome from its origin as an X homolog [4]. Suppression of recombination between the sex chromosomes was apparently mediated by large Y inversions, as deduced by remnant X/Y homology. This led to Y degeneration due to accumulation of mutations and inability to restore the correct DNA sequence [5, 6]. Only small regions of homology and pairing between the sex chromosomes remain, called pseudoautosomal regions (PARs) because genes within these regions behave like autosomal genes.
Initiation of X inactivation in female embryos depends on the transcription of the long noncoding RNA XIST/Xist (X-inactive specific transcript) from one chromosome (which will become the inactive X (Xi)) and recruitment of a protein complex important for X-chromosome silencing and heterochromatin formation [7, 8]. In humans, XIST (17 kb in size) is located in the long arm of the X chromosome, whereas in mice where there is only one arm, Xist (15 kb in size) is in the middle of the chromosome. Xist RNA spreads along the X chromosome in cis and recruits a protein complex responsible for deposition of repressive histone modifications onto the Xi [9–11]. As a result the Xi becomes heterochromatic, silent and condensed. Before implantation, X inactivation is imprinted, with the paternal X chromosome always being silenced. At the blastocyst stage, the paternal X reactivates and random X inactivation takes place (see Table 1).
Although most genes on the Xi are silenced, some genes remain expressed from both the Xa and the Xi. Not surprisingly, genes that retain a Y-linked copy - for example, Kdm5c and Kdm5d (which encode histone demethylases) - escape X inactivation and thus have two expressed alleles in both male and female somatic tissues. However, not all 'escaping' genes have a Y copy, for example Car5b (carbonic anhydrase). Recent reports have shown striking differences between human and mouse regarding the identity and number of these 'escape' genes in somatic tissues [12, 13]. Why are there such species differences? Structural differences between the X chromosomes may play a role as well as selective pressure to maintain sex differences.
Escape from X inactivation is not limited to female somatic cells. Indeed, another type of silencing of the X takes place in male germ cells and is known as meiotic sex chromosome inactivation (MSCI; see Table 1). MSCI results in silencing of protein-coding messenger RNAs from the X chromosome, but a majority of the X-linked microRNAs (miRNAs) escape MSCI, suggesting that they play a role in male meiosis [14]. How do genes escape silencing on the heterochromatic X chromosome, whether in somatic or germ cells? Many studies have shown that epigenetics plays a crucial role in X inactivation and escape [7, 15]. In this review, we will summarize recent progress made in the field of escape from X inactivation, compare the number and distribution of human and mouse escape genes, and discuss possible molecular mechanisms involved in genes escaping X inactivation.
Differences in escape genes between humans and mice
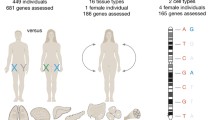
We shall first deal with the main type of X inactivation - that is, random X-chromosome inactivation in female somatic cells (see Table 1). In humans, about 15% of X-linked genes consistently escape this type of X inactivation, as determined from their expression in rodent x human hybrid cells that retain the human Xi, and on measurements of relative expression of allelic polymorphisms in primary fibroblasts [12]. Many human genes escaping X inactivation have already lost their corresponding Y copy. This suggests either that establishment of X inactivation may lag behind Y degeneration, or that specific mechanisms may exist to maintain expression of a subset of genes from the Xi as the result of selective advantages. In the mouse, we have recently shown that only 3% of genes escape X inactivation using next-generation RNA sequencing to survey allele-specific expression of X-linked genes. We derived a cell line from a mouse resulting from a cross between two species of mice, Mus spretus and Mus musculus, which are separated by as much as 7 million years of evolution and thus differ by numerous DNA sequence variants (about one variant for every 100 base pairs). These variant sequences were exploited to determine expression from each allele of X-linked genes after RNA sequencing. Because X inactivation is random, we selected for cells with the M. musculus X chromosome inactive to achieve 100% skewing of X inactivation [13]. Following this approach, any gene with RNA sequence reads from both species of mice was classified as an escape gene. From this study we conclude that compared to humans, X inactivation in the mouse is more complete (Figure 1).

More genes escape X inactivation in humans than in the mouse. Distribution of genes subject to X inactivation (blue) and of 'escape' genes (orange) in human and mouse. The position of the pseudoautosomal regions (PAR1 and 2 in human, PAR in mouse), of the centromeres (cen, purple bar), and of the X-inactivation center encoding the long noncoding RNA XIST/Xist (black bar) are indicated. Note that as the centromere is located at one end of the mouse X chromosome, there is no short arm or long arm. Data from Carrel and Willard [12] and Yang et al. [13].
Escape from X inactivation in other mammalian species has not been extensively characterized. Nonetheless, escape genes have been identified in marsupials, which differ from eutherian mammals in terms of key features of X inactivation - Xist is absent and the paternal X always silenced. At least four X-linked genes encoding glucose-6-phosphate dehydrogenase (G6PD), hypoxanthine guanine phosphoribosyl transferase (HPRT), phosphoglycerate kinase (PGK1), and a monocarboxylic acid transporter (SLC16A2) show incomplete silencing in a tissue- and species-dependent manner in marsupial females [16, 17].
Significant differences exist in terms of the distribution of escape genes in human and mouse. In humans, most escape genes are located on the X short arm. One reason for this could be because the short arm has most recently diverged from the Y, and so these genes have only recently (in evolutionary terms) lost their Y paralogs [5, 6, 12]. Alternatively, the centromeric heterochromatin might exert a barrier effect that would prevent sufficient spreading of XIST RNA, which is generated from the X-inactivation center located in the long arm [18]. In contrast, escape genes are randomly distributed along the mouse X chromosome, which has its centromere located at one end [13]. In humans, escape genes are clustered (as many as 13 adjacent genes in large domains ranging in size between approximately 100 kb and 7 Mb), whereas in mouse, single genes are embedded in regions of silenced chromatin (Figure 2a). This suggests that escape from X inactivation in mouse is controlled at the level of individual genes rather than chromatin domains [12, 13, 19].

Silenced and escape regions have distinct chromatin marks. (a) Chromatin containing escape genes is excluded from the condensed heterochromatic body of the Xi. In mouse, individual escape genes are surrounded by inactivated chromatin. In contrast, human escape genes exist in domains comprising clusters of genes. Orange bars represent escape genes and blue bars inactivated genes. (b) Silenced chromatin in the Xi is coated by Xist RNA potentially via specific DNA motifs (green). Repressive histone modifications and histone variants (for example, H3K27me3, H3K9me3, H4K20me3, and macroH2A1) are recruited and DNA methylation modifies the CpG islands. This type of chromatin structure prevents transcription (blue bar below). In contrast, escape gene regions are enriched for permissive histone marks (for example, H3K4me3, and H3 and H4 acetylation) and RNA polymerase II (RNA pol II) and are hypomethylated at their CpG islands. Insulator sites bound by the insulator protein CTCF, together with unknown factors (as denoted by the '?'), may separate inactivated genes (blue bar) from active genes (orange bar). CTCF binding may block CpG methylation and the spread of repressive chromatin and/or may organize the chromatin into loops.
In both human and mouse, many of the genes that escape X inactivation are expressed more strongly in females. In fact, one study has identified escape genes on the basis of expression levels in women with different numbers of X chromosomes [20]. However, in both humans and mice, differences in levels of expression of the escape genes between males and females are small, indicating partial repression of the escape genes on the Xi [21, 22]. This was confirmed by measuring allele-specific expression of escape genes in humans and in mice [12, 13]. We hypothesize that the Xi allele is either partially silenced by adjacent repressive modifications or might lack modifications associated with X upregulation of the Xa. As we do not know yet what these modifications are, this hypothesis remains to be tested. It is expected that, compared with mice, men and women would demonstrate greater sex differences in X-linked gene expression as a result of the large number of escape genes. Whether such sex differences provide an evolutionary advantage remains to be explored. Possible evolutionary advantages would be, for example, higher expression in female reproductive organs or in neurological tissues, which could influence behavior. It should be noted that most studies about escape from X inactivation have been done using cell lines; thus, tissue-specific effects have not been fully addressed.
Role of escape genes in disease
Escape genes play important roles in human diseases as women with a single X chromosome (X-chromosome monosomy; 45,X) have Turner syndrome, with severe phenotypes including ovarian dysgenesis, short stature, webbed neck, and other physical abnormalities [23]. In addition, as many as 99% of 45,X embryos die in utero [24]. Deficiency in escape genes is thought to play a major role in phenotypes observed in Turner patients [25]. Because the Y chromosome protects men from these deficiencies, the most likely candidate genes would have a Y copy, except for genes that control female-specific phenotypes such as ovarian failure and thus, by definition, would not affect men. So far, the pseudoautosomal gene SHOX (SHORT STATURE HOMEBOX), which encodes a homeodomain transcription factor, is the only gene directly implicated in the short-stature phenotype [26]. Interestingly, early lethality of 45,X embryos may be due to a defect in placenta differentiation, which is supported by the finding that many placental genes have much higher expression in 46,XX versus 45,X cells in differentiated human embryonic stem (ES) cells [27]. Notably, the pseudoautosomal gene CSF2RA (colony-stimulating factor 2 receptor, alpha), which encodes a receptor for a hematopoietic differentiation factor, has more than ninefold higher expression in 46,XX versus 45,X cells, suggesting that this gene may be involved in placenta differentiation defects [27]. In contrast, X0 mice have a near-normal phenotype and are fertile, although the number of oocytes is reduced, potentially as a result of the lack of sex-chromosome pairing [28]. Meiotic arrest due to lack of pairing could be attenuated in mouse compared with human single-X oocytes because of self-pairing of the X in mouse [29].
The fact that few escape genes exist in the mouse is consistent with the significant differences in the impact of X-chromosome monosomy in female mice and in women [13]. Genes that escape from X inactivation in humans but are subject to X inactivation in the mouse may be good candidates for genes responsible for Turner syndrome severe phenotypes. Pseudoautosomal genes may play a prominent role in these phenotypes, as already demonstrated for SHOX, and possibly for CSF2RA. Indeed, the mouse pseudoautosomal region contains only one gene, Sts (steroid sulfatase) [30], whereas all genes located in the pseudoautosomal region in humans are autosomal in the mouse and thus are not affected in X0 mice [31].
Another potential role for escape from X inactivation is in aging. Inappropriate reactivation of an X-linked gene, Otc, which encodes a urea cycle enzyme called ornithine transcarbamoylase, has been reported in mouse tissues [32]. Furthermore, a recent study has found epigenetic alterations including X reactivation in a mouse model of accelerated aging due to telomere shortening [33]. So far, no such reactivation of X-linked genes has been observed in humans. It will be important to determine whether environmental factors could cause inappropriate escape from X inactivation due to changes in epigenetic marks.
Chromatin modifications and escape from X inactivation
The Xi is distinguishable from its active counterpart by its epigenetic marks, including coating with Xist RNA. This is the earliest event in X inactivation during embryogenesis, and gene silencing follows within one or two cell cycles [7]. Interestingly, Xist-induced silencing can only be achieved in early differentiating ES cells, and reaches a point of irreversibility. Just how Xist RNA is spread along the Xi is still not fully understood. One hypothesis suggests that long interspersed repetitive elements (L1) repeats are overrepresented on the X and may serve as 'booster' elements by anchoring Xist RNA to the chromosome, thus aiding spreading [34]. Consistent with this hypothesis, human genes that escape X inactivation have fewer L1 repeats [6, 35, 36]. These genes are also enriched in specific sequence motifs such as Alu repeats and short motifs containing ACG/CGT at their 5' ends [37]. In the mouse, another type of repeat - long terminal repeats (LTRs) - appears to be depleted on escape genes [19]. These observations imply that Xist RNA coating could be deficient at genes escaping X inactivation. This was recently demonstrated in mouse myoblasts using RNA tagging and recovery of associated DNA (modified TRAP) method for identification of targets [38]. In this study, escapees Kdm5c and Kdm6a, which encode chromatin-modifying histone lysine demethylases, were shown to be devoid of Xist RNA coating over their promoters and transcribed regions. Conversely, genes subjected to X inactivation, and L1 repeat elements themselves, recruited Xist RNA [38] (Figure 2b). Taken together, these studies support the idea that specific DNA sequence motifs are involved in recruitment of Xist RNA to the Xi.
While Xist RNA coating is important in the initiation of X inactivation, many other epigenetic modifications follow to silence the X and maintain silencing. An early repressive chromatin mark, tri-methylation of lysine 27 on histone H3 (H3K27me3), is recruited by the Polycomb complex of chromatin-modifying proteins, resulting in compaction of the silenced portion of the Xi (Figure 2a). Other repressive marks include H3K9me3 and the histone variant macroH2A1, which are also enriched on the Xi (Figure 2b) [7, 39]. Concomitantly, 'active' marks such as acetylation of histone H3 and H4 are lost from the silenced chromatin [7, 40]. Modifications characteristic of silenced genes contrast with those within escape genes, which remain euchromatic and harbor histone H3 and H4 acetylation [7, 41]. H3K4me3, another mark associated with transcriptional activity, is absent from most of the Xi except at discrete regions corresponding to areas of escape, as shown in female lymphoblasts [42] (Figure 2b). We recently demonstrated a lack of H3K27me3 at escape genes in mouse, which shows complete concordance in the cell line used to assay allelic expression [13].
The existence of discrete areas of 'escape chromatin' adjacent to silenced chromatin suggests the need for boundary elements, such as insulator sequences, that may block the spreading of heterochromatin into escape regions or prevent repressive marks from being added to escape domains (Figure 2). Supporting this idea are our findings that the insulator protein CTCF (CCCTC-binding factor), which binds known insulator sequences, binds to the transition region between the escape gene Kdm5c and the inactivated gene Iqsec2 (IQ motif and SEC7 domain-containing protein 2) in mouse, whereas in humans, the corresponding region between the same genes, which both escape X inactivation, does not bind CTCF [43]. Furthermore, we have found that the CpG island at the 5' end of Kdm5c remains hypomethylated throughout mouse development, possibly because it is rendered inaccessible to DNA methyltransferases by CTCF binding (Figure 2b). CTCF-binding sites were also identified in other transition areas between escape and inactivated genes, suggesting that CTCF may play a role in the insulation of escape domains [43]. However, a subsequent study showed that insertion of CTCF-binding sites from the HS4 insulator site (from the chicken β-globin gene cluster) at each end of a short reporter gene was not sufficient to protect it from silencing when inserted within an inactivated gene on the Xi in mouse cells [44]. A more recent study reported that a bacterial artificial chromosome clone containing Kdm5c and its flanking regions retains its properties of escape even when inserted at other sites that are normally inactivated on the Xi in mouse cells [45]. CTCF-binding sites may turn out not to be sufficient for insulation, and other elements within or around escape genes may be important.
In particular, the structure of chromatin may have an important role in insulation by looping specific regions out of the condensed Xi (Figure 2a) [46]. Our recent X-chromatin profiles show a discontinuous distribution of the repressive chromatin mark H3K27me3 along the Xi, consistent with the presence of insulator elements and/or specific attachment sites for looped chromatin [13]. However, in human × mouse hybrid cell lines, where the human X can be distinguished from the rodent background, repressive chromatin marks were found to be progressively diminished in the intergenic region between the inactivated RBM10 (RNA-binding motif protein 10) and the escape gene UBA1/UBE1 (ubiquitin-like modifier activating enzyme). Specifically, H3K9me3 and another histone modification associated with gene silencing, H4K20me3, were enriched in the last RBM10 exon but were already depleted approximately 2 kb upstream of UBA1/UBE1 [41].
Escape from X inactivation can vary between different tissues and/or individuals and the escape status can also be developmentally regulated. In humans, about 10% of X-linked genes show variation in escape in different tissues and/or individuals [12, 47]. Some escape genes may have a different chromatin structure throughout development, as suggested by the lack of promoter-restricted H3K4me2 in undifferentiated ES cells before X inactivation [48]. Other escape genes may be initially silenced, and only reactivate in some tissues or with aging [33]. Individual cells may also vary: in an analysis of single-cell allelic expression of Kdm5c in mouse, significant silencing in individual embryonic cells was observed in contrast to consistent expression from both alleles in adult cells [49]. Differences in H3K27me3 enrichment on some genes in a tissue and developmental-stage-specific manner also suggest variability in escape [13]. For example, enrichment in H3K27me3 along Mid1 (midline 1) in mouse embryos but not in adult liver suggests removal of the repressive mark in a tissue-specific manner. It is possible that the recently identified histone demethylases KDM6A and KDM6B may facilitate the removal of H3K27me3 at escape genes [50–52].
Escape from early imprinted paternal X inactivation
Imprinted X inactivation silences the paternal X during the preimplantation stage (see Table 1). This imprinting is reversed in the inner cell mass, and is followed by random X inactivation [7]. It is not known whether imprinted X inactivation occurs in humans and the mechanisms for imprinted X inactivation in mice are still unclear. Are there genes that escape the initial imprinted X inactivation? Several recent studies have addressed this question by profiling transcriptional activity from the paternal X during early development. A specific set of genes apparently does escape imprinted X inactivation at the two-cell stage [53, 54]. However, another subset of genes shows a variable escape status during development and in a lineage-specific manner. For example, Huwe1 (HECT, UBA and WWE domain containing 1) shows no evidence of silencing during pre-implantation stages but is efficiently silenced after implantation, whereas Kdm5c is partially inactivated during the preimplantation stage but escapes fully throughout the rest of development, and Atrx (alpha thalassemia/mental retardation syndrome X-linked) is expressed from both alleles in extraembryonic ectoderm but not in trophectoderm (the precursor of some extraembryonic tissues in the preimplantation embryo), or in later embryos [13, 49, 53].
Escape from male-specific meiotic sex-chromosome inactivation
In male spermatogenesis, yet another type of X-chromosome silencing takes place - MSCI [55] (see Table 1). Unlike X inactivation in female somatic cells, where extensive analyses have catalogued the proportion of genes that escape silencing, no such study has been done so far for MSCI. However, the permissive mark H3K4me3 is present in discrete regions of the X in mouse pachytene spermatocytes. Furthermore, immunofluorescence staining for RNA polymerase II in these cells revealed several regions of transcriptional activity, suggesting areas of escape from MSCI [42]. Another study revealed that up to 86% of the 72 known X-encoded miRNAs escape MSCI at different times during spermatogenesis. Some of the miRNAs were upregulated during MSCI and either downregulated or maintained in the context of postmeiotic sex chromatin [14]. Recent evidence suggests that repression of the X chromosome due to MSCI persists, at least in part, into the mature sperm [56], which could be important for suppression of oogenesis-specific genes and/or dosage compensation by potentially enabling transmission of a partially inactivated paternal X [57]. However, not all sex-linked genes remain inactivated following MSCI and evidence points to maintenance of post-meiotic X-chromosome repression being incomplete. In fact, about 18% of X-linked genes, especially multicopy genes, are expressed in postmeiotic cells [58].
X inactivation is an important process required to balance gene dosage in males and females. Equally important are those genes that escape X inactivation. Why is there a far greater number of X-linked genes that escape X inactivation in humans than in mice? Not only does the number of escape genes differ but also their location. Human escape genes exist in large domains of escape whereas mouse escape genes are scattered along the X chromosome. Their location in recent evolutionary strata in humans suggests a major role of sex chromosome evolution in the retention of escape genes. However, their retention may also be linked to their inherent ability to cause sex-specific differences in gene expression levels. We propose that the complexity of dosage compensation in mammals, which involves X upregulation, X inactivation, and escape from X inactivation, may have specific advantages in providing opportunities to modulate gene expression between the sexes in specific tissues. This may be especially advantageous in reproductive organs. Whether sex differences do lead to physiological effects remains to be determined. Specific epigenetic mechanisms may have evolved to ensure maintenance of escape from X inactivation. These may include the accumulation of repeats and DNA motifs to recruit or repel the silencing complex, as well as specific boundary elements. Future studies are needed to further characterize the chromatin structure of escape domains and to understand their role in evolution.
References
Nguyen DK, Disteche CM: Dosage compensation of the active X chromosome in mammals. Nat Genet. 2006, 38: 47-53. 10.1038/ng1705.
Gupta V, Parisi M, Sturgill D, Nuttall R, Doctolero M, Dudko OK, Malley JD, Eastman PS, Oliver B: Global analysis of X-chromosome dosage compensation. J Biol. 2006, 5: 3-10.1186/jbiol30.
Lyon MF: Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961, 190: 372-373. 10.1038/190372a0.
Birchler JA, Fernandez HR, Kavi HH: Commonalities in compensation. BioEssays. 2006, 28: 565-568. 10.1002/bies.20408.
Lahn BT, Page DC: Four evolutionary strata on the human X chromosome. Science. 1999, 286: 964-967. 10.1126/science.286.5441.964.
Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, Platzer M, Howell GR, Burrows C, Bird CP, Frankish A, Lovell FL, Howe KL, Ashurst JL, Fulton RS, Sudbrak R, Wen G, Jones MC, Hurles ME, Andrews TD, Scott CE, Searle S, Ramser J, Whittaker A, Deadman R, Carter NP, Hunt SE, Chen R, Cree A, Gunaratne P, et al: The DNA sequence of the human X chromosome. Nature. 2005, 434: 325-337. 10.1038/nature03440.
Heard E, Disteche CM: Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev. 2006, 20: 1848-1867. 10.1101/gad.1422906.
Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R, Willard HF: A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature. 1991, 349: 38-44. 10.1038/349038a0.
Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT: Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008, 322: 750-756. 10.1126/science.1163045.
Brockdorff N, Ashworth A, Kay GF, McCabe VM, Norris DP, Cooper PJ, Swift S, Rastan S: The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell. 1992, 71: 515-526. 10.1016/0092-8674(92)90519-I.
Brown CJ, Hendrich BD, Rupert JL, Lafreniere RG, Xing Y, Lawrence J, Willard HF: The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992, 71: 527-542. 10.1016/0092-8674(92)90520-M.
Carrel L, Willard HF: X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005, 434: 400-404. 10.1038/nature03479.
Yang F, Babak T, Shendure J, Disteche CM: Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res. 2010, 20: 614-622. 10.1101/gr.103200.109.
Song R, Ro S, Michaels JD, Park C, McCarrey JR, Yan W: Many X-linked microRNAs escape meiotic sex chromosome inactivation. Nat Genet. 2009, 41: 488-493. 10.1038/ng.338.
Prothero KE, Stahl JM, Carrel L: Dosage compensation and gene expression on the mammalian X chromosome: one plus one does not always equal two. Chromosome Res. 2009, 17: 637-648. 10.1007/s10577-009-9063-9.
Deakin JE, Chaumeil J, Hore TA, Marshall Graves JA: Unravelling the evolutionary origins of X chromosome inactivation in mammals: insights from marsupials and monotremes. Chromosome Res. 2009, 17: 671-685. 10.1007/s10577-009-9058-6.
Duret L, Chureau C, Samain S, Weissenbach J, Avner P: The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science. 2006, 312: 1653-1655. 10.1126/science.1126316.
Disteche CM: Escapees on the X chromosome. Proc Natl Acad Sci USA. 1999, 96: 14180-14182. 10.1073/pnas.96.25.14180.
Tsuchiya KD, Greally JM, Yi Y, Noel KP, Truong JP, Disteche CM: Comparative sequence and x-inactivation analyses of a domain of escape in human Xp11.2 and the conserved segment in mouse. Genome Res. 2004, 14: 1275-1284. 10.1101/gr.2575904.
Sudbrak R, Wieczorek G, Nuber UA, Mann W, Kirchner R, Erdogan F, Brown CJ, Wöhrle D, Sterk P, Kalscheuer VM, Berger W, Lehrach H, Ropers HH: X chromosome-specific cDNA arrays: identification of genes that escape from X-inactivation and other applications. Hum Mol Genet. 2001, 10: 77-83. 10.1093/hmg/10.1.77.
Yang X, Schadt EE, Wang S, Wang H, Arnold AP, Ingram-Drake L, Drake TA, Lusis AJ: Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 2006, 16: 995-1004. 10.1101/gr.5217506.
Johnston CM, Lovell FL, Leongamornlert DA, Stranger BE, Dermitzakis ET, Ross MT: Large-scale population study of human cell lines indicates that dosage compensation is virtually complete. PLoS Genet. 2008, 4: e9-10.1371/journal.pgen.0040009.
Bondy CA: Turner syndrome 2008. Horm Res. 2009, 71 (Suppl 1): 52-56. 10.1159/000178039.
Hook EB, Warburton D: The distribution of chromosomal genotypes associated with Turner's syndrome: livebirth prevalence rates and evidence for diminished fetal mortality and severity in genotypes associated with structural X abnormalities or mosaicism. Hum Genet. 1983, 64: 24-27. 10.1007/BF00289473.
Zinn AR, Ross JL: Molecular analysis of genes on Xp controlling Turner syndrome and premature ovarian failure (POF). Semin Reprod Med. 2001, 19: 141-146. 10.1055/s-2001-15394.
Clement-Jones M, Schiller S, Rao E, Blaschke RJ, Zuniga A, Zeller R, Robson SC, Binder G, Glass I, Strachan T, Lindsay S, Rappold GA: The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum Mol Genet. 2000, 9: 695-702. 10.1093/hmg/9.5.695.
Urbach A, Benvenisty N: Studying early lethality of 45,XO (Turner's syndrome) embryos using human embryonic stem cells. PLoS One. 2009, 4: e4175-10.1371/journal.pone.0004175.
Burgoyne PS, Baker TG: Perinatal oocyte loss in XO mice and its implications for the aetiology of gonadal dysgenesis in XO women. J Reprod Fertil. 1985, 75: 633-645.
Speed RM: Oocyte development in XO foetuses of man and mouse: the possible role of heterologous X-chromosome pairing in germ cell survival. Chromosoma. 1986, 94: 115-124. 10.1007/BF00286989.
Keitges E, Rivest M, Siniscalco M, Gartler SM: X-linkage of steroid sulphatase in the mouse is evidence for a functional Y-linked allele. Nature. 1985, 315: 226-227. 10.1038/315226a0.
Perry J, Palmer S, Gabriel A, Ashworth A: A short pseudoautosomal region in laboratory mice. Genome Res. 2001, 11: 1826-1832.
Wareham KA, Lyon MF, Glenister PH, Williams ED: Age related reactivation of an X-linked gene. Nature. 1987, 327: 725-727. 10.1038/327725a0.
Schoeftner S, Blanco R, de Silanes IL, Munoz P, Gomez-Lopez G, Flores JM, Blasco MA: Telomere shortening relaxes X chromosome inactivation and forces global transcriptome alterations. Proc Natl Acad Sci USA. 2009, 106: 19393-19398. 10.1073/pnas.0909265106.
Lyon MF: X-chromosome inactivation: a repeat hypothesis. Cytogenet Cell Genet. 1998, 80: 133-137. 10.1159/000014969.
Bailey JA, Carrel L, Chakravarti A, Eichler EE: Molecular evidence for a relationship between LINE-1 elements and X chromosome inactivation: the Lyon repeat hypothesis. Proc Natl Acad Sci USA. 2000, 97: 6634-6639. 10.1073/pnas.97.12.6634.
Carrel L, Park C, Tyekucheva S, Dunn J, Chiaromonte F, Makova KD: Genomic environment predicts expression patterns on the human inactive X chromosome. PLoS Genet. 2006, 2: e151-10.1371/journal.pgen.0020151.
Wang Z, Willard HF, Mukherjee S, Furey TS: Evidence of influence of genomic DNA sequence on human X chromosome inactivation. PLoS Comput Biol. 2006, 2: e113-10.1371/journal.pcbi.0020113.
Murakami K, Ohhira T, Oshiro E, Qi D, Oshimura M, Kugoh H: Identification of the chromatin regions coated by non-coding Xist RNA. Cytogenet Genome Res. 2009, 125: 19-25. 10.1159/000207514.
Changolkar LN, Pehrson JR: macroH2A1 histone variants are depleted on active genes but concentrated on the inactive X chromosome. Mol Cell Biol. 2006, 26: 4410-4420. 10.1128/MCB.02258-05.
Marks H, Chow JC, Denissov S, Francoijs KJ, Brockdorff N, Heard E, Stunnenberg HG: High-resolution analysis of epigenetic changes associated with X inactivation. Genome Res. 2009, 19: 1361-1373. 10.1101/gr.092643.109.
Goto Y, Kimura H: Inactive X chromosome-specific histone H3 modifications and CpG hypomethylation flank a chromatin boundary between an X-inactivated and an escape gene. Nucleic Acids Res. 2009, 37: 7416-7428. 10.1093/nar/gkp860.
Khalil AM, Driscoll DJ: Trimethylation of histone H3 lysine 4 is an epigenetic mark at regions escaping mammalian X inactivation. Epigenetics. 2007, 2: 114-118. 10.4161/epi.2.2.4612.
Filippova GN, Cheng MK, Moore JM, Truong JP, Hu YJ, Nguyen DK, Tsuchiya KD, Disteche CM: Boundaries between chromosomal domains of X inactivation and escape bind CTCF and lack CpG methylation during early development. Dev Cell. 2005, 8: 31-42. 10.1016/j.devcel.2004.10.018.
Ciavatta D, Kalantry S, Magnuson T, Smithies O: A DNA insulator prevents repression of a targeted X-linked transgene but not its random or imprinted X inactivation. Proc Natl Acad Sci USA. 2006, 103: 9958-9963. 10.1073/pnas.0603754103.
Li N, Carrel L: Escape from X chromosome inactivation is an intrinsic property of the Jarid1c locus. Proc Natl Acad Sci USA. 2008, 105: 17055-17060. 10.1073/pnas.0807765105.
Heard E, Bickmore W: The ins and outs of gene regulation and chromosome territory organisation. Curr Opin Cell Biol. 2007, 19: 311-316. 10.1016/j.ceb.2007.04.016.
Brown CJ, Greally JM: A stain upon the silence: genes escaping X inactivation. Trends Genet. 2003, 19: 432-438. 10.1016/S0168-9525(03)00177-X.
Rougeulle C, Navarro P, Avner P: Promoter-restricted H3 Lys 4 di-methylation is an epigenetic mark for monoallelic expression. Hum Mol Genet. 2003, 12: 3343-3348. 10.1093/hmg/ddg351.
Lingenfelter PA, Adler DA, Poslinski D, Thomas S, Elliott RW, Chapman VM, Disteche CM: Escape from X inactivation of Smcx is preceded by silencing during mouse development. Nat Genet. 1998, 18: 212-213. 10.1038/ng0398-212.
Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K: Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci USA. 2007, 104: 18439-18444. 10.1073/pnas.0707292104.
Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K: UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007, 449: 731-734. 10.1038/nature06145.
Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, Iwase S, Alpatov R, Issaeva I, Canaani E, Roberts TM, Chang HY, Shi Y: A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007, 449: 689-694. 10.1038/nature06192.
Patrat C, Okamoto I, Diabangouaya P, Vialon V, Le Baccon P, Chow J, Heard E: Dynamic changes in paternal X-chromosome activity during imprinted X-chromosome inactivation in mice. Proc Natl Acad Sci USA. 2009, 106: 5198-5203. 10.1073/pnas.0810683106.
Kalantry S, Purushothaman S, Bowen RB, Starmer J, Magnuson T: Evidence of Xist RNA-independent initiation of mouse imprinted X-chromosome inactivation. Nature. 2009, 460: 647-651.
Turner JM: Meiotic sex chromosome inactivation. Development. 2007, 134: 1823-1831. 10.1242/dev.000018.
Namekawa SH, Park PJ, Zhang LF, Shima JE, McCarrey JR, Griswold MD, Lee JT: Postmeiotic sex chromatin in the male germline of mice. Curr Biol. 2006, 16: 660-667. 10.1016/j.cub.2006.01.066.
Huynh KD, Lee JT: Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature. 2003, 426: 857-862. 10.1038/nature02222.
Mueller JL, Mahadevaiah SK, Park PJ, Warburton PE, Page DC, Turner JM: The mouse X chromosome is enriched for multicopy testis genes showing postmeiotic expression. Nat Genet. 2008, 40: 794-799. 10.1038/ng.126.
Acknowledgements
This work was supported by grants from the National Institutes of Health to JBB (HD060402) and to CMD (GM046883 and GM079537).
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Berletch, J.B., Yang, F. & Disteche, C.M. Escape from X inactivation in mice and humans. Genome Biol 11, 213 (2010). https://doi.org/10.1186/gb-2010-11-6-213
Published:
DOI: https://doi.org/10.1186/gb-2010-11-6-213