
Abstract
Phylogenomics of eukaryote supergroups suggest a highly complex last common ancestor of eukaryotes and a key role of mitochondrial endosymbiosis in the origin of eukaryotes.
Similar content being viewed by others
Eukaryotes
The origin of eukaryotes is a huge enigma and a major challenge for evolutionary biology [1–3]. There is a sharp divide in the organizational complexity of the cell between eukaryotes, which have complex intracellular compartmentalization, and even the most sophisticated prokaryotes (archaea and bacteria), which do not [4–6]. A typical eukaryotic cell is about 1,000-fold bigger by volume than a typical bacterium or archaeon, and functions under different physical principles: free diffusion has little role in eukaryotic cells, but is crucial in prokaryotes [7, 8]. The compartmentalization of eukaryotic cells is supported by an elaborate endomembrane system and by the actin-tubulin-based cytoskeleton [9, 10]. There are no direct counterparts of these organelles in archaea or bacteria. The other hallmark of the eukaryotic cell is the presence of mitochondria, which have a central role in energy transformation and perform many additional roles in eukaryotic cells, such as in signaling and cell death.
The conservation of the major features of cellular organization and the existence of a large set of genes that are conserved across eukaryotes leave no doubt that all extant eukaryotic forms evolved from a last eukaryote common ancestor (LECA; see below). All eukaryotes that have been studied in sufficient detail possess either mitochondria or organelles derived from mitochondria [11–13], so it is thought that LECA already possessed mitochondria (see below). Plants and many unicellular eukaryotes also have another type of organelle, plastids.
The organizational complexity of the eukaryotic cells is complemented by extremely sophisticated, cross-talking signaling networks [14]. The main signaling systems in eukaryotes are the kinase-phosphatase machinery that regulates protein function through phosphorylation and dephosphorylation [15–18]; the ubiquitin network that governs protein turnover and localization through reversible protein ubiquitylation [19–21]; regulation of translation by microRNAs [22–24]; and regulation of transcription at the levels of individual genes and chromatin remodeling [24–27]. Eukaryotes all share the main features of cellular architecture and the regulatory circuitry that clearly differentiate them from prokaryotes, although the ancestral forms of some signature eukaryotic systems are increasingly detected in prokaryotes, as discussed below. Phylogenomic reconstructions show that the characteristic eukaryotic complexity arose almost 'ready made', without any intermediate grades seen between the prokaryotic and eukaryotic levels of organization [9, 28–30]. Explaining this apparent leap in complexity at the origin of eukaryotes is one of the principal challenges of evolutionary biology.
The key to the origin of eukaryotes will undoubtedly be found using comparative genomics of eukaryotes, archaea and bacteria. Complete genome sequences from all three domains of cellular life are accumulating exponentially, albeit at markedly different paces. As of March 2010, the NCBI genome database contained over 1,000 bacterial genomes, about 100 archaeal genomes, and about 100 genomes of eukaryotes [31]. Here, I discuss some of the main insights that have come from comparative analysis of these genomes, which may help to shed light on the origin and the early stages of evolution of eukaryotes. So far, the comparative genomics era has brought fascinating clues but no decisive breakthrough.
The supergroups of eukaryotes and the root of the eukaryotic evolutionary tree
Although several eukaryotic kingdoms, such as animals, fungi, plants and ciliates, are well defined and seem to be monophyletic beyond reasonable doubt, deciphering the evolutionary relationships between these kingdoms and numerous other groups of unicellular eukaryotes (also called protists) turned out to be daunting. For many years, evolutionary biologists tended to favor the so called crown group phylogeny [2, 32]. The 'crown' of this evolutionary tree included animals (Metazoa) and plants (Viridiplantae), fungi and various assortments of protists, depending on the methods used for tree construction [33, 34]. The rest of the protists, such as microsporidia, diplomonads and parabasalia, were considered 'early branching eukaryotes'; for some of them, this conclusion was reached because they appeared to lack mitochondria and were therefore thought to have evolved before the mitochondrial symbiosis. The scenario resulting from the crown group phylogeny was called the archezoan scenario: the archaezoan was defined as a hypothetical ancestral form that lacked mitochondria but possessed the other signature features of the eukaryotic cell. However, during the past decade, the early branching groups have lost their positions at the root of the eukaryotic tree, one after another [35–37]. The improved taxon sampling as a result of genome sequencing together with new, more robust methods for phylogenetic analysis indicate that the deep placing of these groups seen in early trees was a long-branch artifact caused by the fast evolution of the respective organisms [37–39]. At the same time, comparative-genomic and ultrastructural studies destroyed the biological underpinning of the near-root positions of the (former) early branching groups of protists by showing that none of them ancestrally lack mitochondria, as they all have genes of apparent mitochondrial origin and mitochondria-related organelles, such as hydrogenosomes and mitosomes [11, 12, 13, 40].
There are therefore no grounds to consider any group of eukaryotes primitive, a presymbiotic archezoan. Rather, taking into account the small genomes and high rate of evolution characteristic of most of the protist groups thought to be early branching, and their parasitic lifestyle, it is becoming increasingly clear that most or perhaps all of them evolved from more complex ancestral forms by reductive evolution [37, 39]. Reductive evolution refers to the evolutionary modality typical of parasites: they tend to lose genes, organelles and functions when the respective functionalities are taken over by the host. So the archezoan (crown group) phylogeny seems to have been disproved, and deep phylogeny and the theories of the origin of eukaryotes effectively had to start from scratch.
This time phylogenomic approaches were mainly used, that is, phylogenetic analysis of genome-wide sets of conserved genes; this was made possible by the much larger number of genomes that had been sequenced [41, 42]. The key accomplishment at this new stage was the proposal of 'supergroups' of eukaryotes that are suggested to combine highly diverse groups of organisms in a monophyletic group [36, 43–45]. Most of the phylogenomic analyses published so far converge on five supergroups (or six if the Amoebozoa and Opisthokonts do not form a single supergroup, the Unikonts; Figure 1). Although proving monophyly is non-trivial for these groups [46–48], the general structure of the tree, with a few supergroups forming a star-like phylogeny (Figure 1), is reproduced consistently, and the latest results [49–52] seem to support the monophyly of the five supergroups.

Evolution of the eukaryotes. The relationship between the five eukaryotic supergroups - Excavates, Rhizaria, Unikonts, Chromalveolates and Plantae - are shown as a star phylogeny with LECA placed in the center. The 4,134 genes assigned to LECA are those shared by the free-living excavate amoeboflagellate Naegleria gruberi with representatives of at least one other supergroup [67]. The numbers of these putative ancestral genes retained in selected lineages from different supergroups are also indicated. Branch lengths are arbitrary. Two putative root positions are shown: I, the Unikont-Bikont rooting [56, 57]; II, rooting at the base of Plantae [60].
The relationship between the supergroups is a formidable problem as the internal branches are extremely short, suggesting that the radiation of the supergroups occurred rapidly (on the evolutionary scale), perhaps resembling an evolutionary 'big bang' [53–55]. Two recent, independent phylogenetic studies [51, 52] each analyzed over 130 conserved proteins from several dozen eukaryotic species and, after exploring the effects of removing fast-evolving taxa, arrived at a three-megagroup structure of the eukaryotic tree. The megagroups consist of Unikonts, Excavates, and the assemblage of Plantae, Chromalveolata and Rhizaria [51, 52].
Furthermore, there have been several attempts to infer the position of the root of the eukaryotic tree (Figure 1). The first alternative to the crown group tree was proposed by Cavalier-Smith and coworkers [56–58], who used rare genomic changes (RGCs) [59], such as the fusion of two enzyme genes [56, 57] and the domain structure of myosins [58], to place the root between the Unikonts and the rest of eukaryotes (I (red arrow) in Figure 1). This separation seems biologically plausible because Unikont cells have a single cilium, whereas all other eukaryotic cells have two. However, this conclusion could be suspect because the use of only a few RGCs makes it difficult to rule out homoplasy (parallel emergence of the same RGC, such as gene fusion or fission, in different lineages). Rogozin and coworkers [60] used a different RGC approach based on rare replacements of highly conserved amino acid residues requiring two nucleotide substitutions and inferred the most likely position of the root to be between Plantae and the rest of eukaryotes (II (green arrow) in Figure 1). Again, this seems biologically plausible because the cyanobacterial endosymbiosis that gave rise to plastids occurred on the Plantae lineage.
The controversy about the root position and the lack of consensus regarding the monophyly of at least some of the supergroups, let alone the megagroups, indicate that, despite the emerging clues, the deep phylogeny of eukaryotes currently should be considered unresolved. In a sense, given the likely 'big bang' of early eukaryote radiation, the branching order of the supergroups, in itself, might be viewed as relatively unimportant [61]. However, the biological events that triggered these early radiations are of major interest, so earnest attempts to resolve the deepest branches of the eukaryotic tree will undoubtedly continue with larger and further improved datasets and methods.
The last common ancestor of eukaryotes
Comparative analysis of representative genomes from different eukaryotic supergroups enables the reconstruction of the gene complement of LECA using maximum parsimony (MP) or more sophisticated maximum likelihood (ML) methods [62–64]. Essentially, genes that are represented in diverse extant representatives of different supergroups, even though lost in some lineages, can be mapped back to LECA. The results of all these reconstructions consistently point to a complex LECA, in terms of both the sheer number of ancestral genes and, perhaps even more importantly, the ancestral presence of the signature functional systems of the eukaryotic cell (see below). A MP reconstruction based on phyletic patterns in clusters of orthologous genes of eukaryotes mapped 4,137 genes to LECA (Figure 1) [63, 65, 66]. Remarkably, an even simpler estimation, based on the recent analysis of the genome of Naegleria gruberi, the first sequenced genome of a free-living excavate [67], revealed about a nearly identical number of genes, 4,134, that are shared by Naegleria and at least one other supergroup of eukaryotes, suggesting that these genes are part of the LECA heritage (Figure 1). Such estimates are highly conservative as they do not account for lineage-specific loss of ancestral genes, a major aspect in the evolution of eukaryotes. Indeed, even animals and plants, the eukaryotic kingdoms that seem to be the least prone to gene loss, have still lost about 20% of the putative ancestral genes identified in the unicellular Naegleria (Figure 1). Given that the current estimate for the gene complement of LECA must be conservative, the genome of LECA is likely to have been as complex as those of typical extant free-living unicellular eukaryotes [68].
This conclusion is supported by reconstructions from comparative genomics of the ancestral composition of the key functional systems of the LECA, such as the nuclear pore [28, 69], the spliceosome [29], the RNA interference machinery [70], the proteasome and the ubiquitin signaling system [71], and the endomembrane apparatus [10]. The outcomes of these reconstructions are all straightforward and consistent, even when different topologies of the phylogenetic tree of eukaryotes were used as the scaffold for the reconstruction: LECA already possessed all these structures in its fully functional state, possibly as complex as the counterparts in modern eukaryotes.
Reconstruction of other aspects of the genomic composition and architecture of LECA similarly points to a highly complex ancestral genome. Comparative-genomic analysis of intron positions in orthologous genes within and between supergroups suggests high intron densities in the ancestors of the supergroups and in LECA, at least as dense as in modern free-living unicellular eukaryotes [72–75]. A systematic analysis of widespread gene duplications in eukaryotes indicates that hundreds of duplications predate LECA, especially duplications of genes involved in protein turnover [63, 65, 66]. Taken together, these results clearly indicate that LECA was a typical, fully developed eukaryotic cell. The subsequent evolution of eukaryotes has seemingly shown no consistent trend toward increased complexity, except for lineage-specific embellishments, such as those seen in animals and plants. There was obviously an important stage of evolution on the 'stem' of eukaryotes, after they first evolved but before LECA, which included extensive duplication of numerous essential genes, so that the set of ancestral genes approximately doubled [63, 65, 66].
The archaeal and bacterial roots of eukaryotes
Eukaryotes are hybrid organisms in terms of both their cellular organization and their gene complement. All eukaryotes seem to possess mitochondria or related organelles derived from α-proteobacteria, whereas Plantae and many groups of Chromalveolata additionally have cyanobacteria-derived plastids [76, 77]. The gene complement of eukaryotes is an uneven mix of genes of apparent archaeal origin, genes of probable bacterial origin, and genes that so far seem eukaryote-specific, without convincing evidence of ancestry in either of the two prokaryote domains (Figure 2). Paradoxical as this might appear, although trees based on rRNA genes and concatenated alignments of information-processing proteins, such as polymerases or splicing proteins, both put archaea and eukaryotes together, genome-wide analyses consistently and independently show that there are three or more times more genes with bacterial homologs than with archaeal homologs [62, 63, 78, 79] (Figure 2). The archaeal subset is strongly enriched in information processing functions (translation, transcription, replication, splicing), whereas the bacterial subset consists largely of metabolic enzymes [62, 78] (see below for more details).

Breakdown of the genes from two eukaryotes by the putative evolutionary affinities. (a) Yeast and (b) red algae. The putative origin of genes was tentatively inferred from the best hits obtained by searching the NCBI non-redundant protein sequence database using the BLASTP program [125], with all protein sequences from the respective organisms used as queries. Although sequence similarity searches are often regarded as a very rough approximation of the phylogenetic position [126], the previous analysis of the yeast genome showed a high level of congruence between the best hits and phylogenomic results [78]. Major archaeal and bacterial groups are color-coded and denoted 1 to 18; the number of proteins with the best hit to the given groups is indicated. The groups are: 1, Euryarchaeota; 2, Crenarchaeota-Thaumarchaeota-Nanoarchaeota; 3, Firmicutes; 4, γ-Proteobacteria; 5, α-Proteobacteria; 6, δ- and ε-Proteobacteria; 7, β-Proteobacteria; 8, unclassified Proteobacteria; 9, Cyanobacteria; 10, Actinobacteria; 11, Bacteroides-Chlorobi group; 12, Chloroflexi; 13, Planctomycetes; 14, Verrucomicrobia-Chlamydiae-Spirochetes; 15, Deinococcus-Thermus group; 16, Aquificacae and Thermotogae; 17, other bacteria; 18, no archaeal or bacterial homologs.
At a coarse level, these observations are best compatible with genome fusion scenarios [79, 80] whereby the eukaryotic genome emerged through a fusion between two ancestral genomes, an archaeal or archaea-related one, and a bacterial, most likely α-proteobacterial, one, given the well-established ancestry of the mitochondrial endosymbiont [81]. However, attempts to pinpoint the specific archaeal and bacterial 'parents' of eukaryotes reveal complicated evolutionary relationships. Although many of the bacterial-like genes in eukaryotes have α-proteobacterial homologues, these are far from dominant amongst the bacterial-like genes which show apparent evolutionary affinities with a variety of bacterial groups (Figure 2). An important cause of this complicated breakdown of the bacterial-like component of the eukaryotic gene complement is the large size of the α-proteobacterial pangenome, that is, of the combined genes found in all α-proteobacteria, and the associated diversity of the gene sets in individual members of this group [82]. Thus, without knowing the exact identity within the α-proteobacteria of the bacterial endosymbiont that gave rise to the eukaryotic mitochondria, it is hard to delineate its genetic contribution. Apart from this uncertainty about the gene complement of the endosymbiont, it is impossible to rule out multiple sources of the bacterial-like genes in eukaryotes [83], which may have origins other than the genome of the bacterial endosymbiont. In particular, whatever the actual nature of the archaeal-like ancestor, it probably lived at moderate temperatures and non-extreme conditions and was consequently in contact with a diverse bacterial community. Modern archaea with such lifestyles have numerous genes of diverse bacterial origins, indicating extensive horizontal acquisition of genes from bacteria [84, 85]. Thus, the archaeal-like host of the endosymbiont could have already had many bacterial genes, partly explaining the observed pattern.
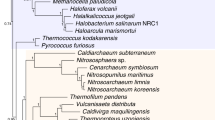
The case of the archaeal(-like) parent is far more difficult than that of the bacterial ancestor(s) as there are no data on the ancestral lineage that would parallel the unambiguous origin of mitochondria from α-proteobacteria. Phylogenomic studies using different methods point to different archaeal lineages - Crenarchaeota [86, 87], Euryarchaeota [88], or an unidentified deep branch [89, 90] - as the candidates for the eukaryote ancestor (Figure 3). Unequivocal resolution of such deep evolutionary relationships is extremely difficult. Moreover, at least one of these analyses [89] explicitly suggests the possibility that the archaeal heritage of eukaryotes is genuinely mixed, with the largest contribution coming from a deep lineage, followed by the contributions from Crenarchaeota (Thaumoarchaeota) and the Euryarchaeota (Figure 3). In the next section I examine the possibility of multiple archaeal and bacterial ancestors of the eukaryotes with respect to distinct functional systems of eukaryotic cells.

Possible archaeal origins of eukaryotic genes. The archaeal tree is shown as a bifurcation of Euryarchaeota and the putative second major branch combining Crenarchaeota, Thaumarchaeota, and Korarchaeota [127]; deep, possibly extinct lineages are shown as a single stem.
Mixed origins of the key functional systems of eukaryotes
Some of the most compelling indications on the course of evolution and the nature of ancestral forms come from signature genes that are uniquely shared by two or more major lineages and from detailed evolutionary analysis of well characterized functional systems, in particular the signature systems of the eukaryotic cell. Comparative genome sequence analysis has revealed that some of the key molecular machines of the eukaryotes, and not only those directly involved in information processing, can be confidently derived from archaeal ancestors (Table 1 and Figure 4). Strikingly, this archaeal heritage seems to be patchy with respect to the specific origins, with apparent evolutionary affinities to different groups of archaea (Table 1 and Figure 4). For instance, comparative analysis of the translation system components tends to suggest an affinity between eukaryotes and Crenarchaeota [91]. Similarly, the core transcription machinery of eukaryotes shares some important proteins with Crenarchaeota, Thaumarchaeota and Korarchaeota, to the exclusion of Euryarchaeota [92–94]. By contrast, the histones, the primary components of nucleosomes, are missing in most of the Crenarchaeota but invariably conserved in Euryarchaeota (and also present in Korarchaeum and some Thaumarchaeota) [95].

Apparent complex origins of some key functional systems of eukaryotes. The likely origins of proteins and domains are shown by color code for three key functional systems of the eukaryotic cell: (a) B-family DNA polymerases comprising the core of the replication apparatus (triangles show Zn-finger modules; crosses indicate inactivated enzymatic domains; pol, polymerase; exo, exonuclease) [100]; (b) RNA interference (RNAi) machinery (RdRp, RNA-dependent RNA polymerase) [70]; and (c) cell division apparatus (the Vps4 ATPase and Snf7-like proteins comprise the ESCRT-III machinery) and cytoskeleton [97, 98, 105, 113]. The domains are not drawn to scale. The light blue color of the three amino-terminal domains of Polε indicates the substantial sequence divergence from the homologous domains of other eukaryotic polymerases.
Eukaryotic cell division components are also conserved in several but not all of the major archaeal lineages. For example, homologs of the ESCRT-III complex, which performs key roles in vesicle biogenesis and cytokinesis in eukaryotes, are responsible for cell division in the Crenarchaeota but are missing in most of the Euryarchaeota, which possess a bacterial-like division mechanism using the GTPase FtsZ, a distant homolog of tubulin [96, 97]. However, a few members of the Euryarchaeota have both systems, with FtsZ probably responsible for division and ESCRT-III for vesicle biogenesis [98].
Eukaryote B-family DNA polymerases, a group of four paralogs that are collectively responsible for genome replication, show a complex pattern of ancestry (Figure 4): one branch of the eukaryotic polymerases seems to have evolved from archaeal PolBI, which is conserved in all archaea, whereas the other branch appears to derive from the Crenarchaea-specific PolBII [99, 100]. Surprisingly, the eukaryotic polymerases additionally contain a Zn-finger domain homologous to that of PolD, which is restricted to Euryarchaeota [100]; furthermore, the small subunits of eukaryotic Polα and Polδ are inactivated derivatives of the exonuclease subunit of PolD [101].
Another major theme emerging from these studies is the bacterial contribution and the formation of archaeao-bacterial chimeras (Table 1 and Figure 4). A clear-cut case of a chimeric eukaryotic system is the RNA interference machinery, in which one of the key proteins, the endonuclease Dicer, consists of two bacterial RNAse III domains and a helicase domain of apparent euryarchaeal origin, and the other essential protein, Argonaute, also shows a euryarchaeal affinity (Figure 4) [70, 102]. The nuclear pore complex, a quintessential eukaryotic molecular machine, does not show any indications of archaeal ancestry but rather consists of proteins of apparent bacterial origin combined with proteins consisting of simple repeats whose provenance is difficult to ascertain [28].
These observations suggest that the archaeal ancestor of eukaryotes combined a variety of features found separately in diverse extant archaea. This inference is consistent with the results of phylogenomic analysis and evolutionary reconstruction discussed above. Thus, the currently existing archaeal lineages probably evolved by differential streamlining, or reductive evolution of the complex ancestral forms, whereas eukaryotes largely retained the ancestral complexity. The diverse origins of eukaryotic functional systems has major implications for how eukaryotes originated, as explained below.
Eukaryogenesis: where did the eukaryotes come from?
The results of comparative genomics and ultrastructural studies do not yet definitively show where the eukaryotic cell came from, but they do offer important insights. Box 1 lists the key observations that must be included in any evolutionary scenario for the evolution of eukaryotes (called eukaryogenesis) and summarizes the two alternative scenarios, which are depicted in Figure 5. The main issue revolves around the role of endosymbiosis [2, 3, 103, 104]: was it the cause of the entire chain of events that led to the emergence of LECA (the stem phase of evolution), as proposed by the symbiogenesis scenario, or was it a step in the evolution of the already formed eukaryotic cell, as proposed by the archaezoan scenario? In other words, was the host of the α-proteobacterial symbiont (the future mitochondrion) a prokaryote (as in the symbiogenesis scenario) or an amitochondrial eukaryote, an archaezoan?

The two alternative scenarios of eukaryogenesis. (a) The archaezoan scenario; (b) the symbiogenesis scenario. The putative archaeal or archaezoan hosts of the α-proteobacterial endosymbiont are shown with elements of their cytoskeleton and cell division apparatus colored as in Figure 4.

General concepts in the evolution of the eukaryotes
Given that eukaryogenesis may have been a unique event and that intermediate stages in the process cannot be seen, these questions are enormously difficult, and final answers might not be attainable. But the symbiogenesis scenario seems to be more plausible than the archaezoan scenario [105], for three main reasons. First, under the archaezoan scenario, there is no plausible selective force behind the evolution of the nucleus, and in particular the elaborate nuclear pore complex. The nucleus disrupts the transcription-translation coupling that is typical of bacteria and archaea [106–108] and necessitates the evolution of the time- and energy-consuming mechanism of nucleocytosolic transport of mRNA. By contrast, the symbiogenesis hypothesis offers a plausible selective factor: defense against the invasion of the host genome by Group II self-splicing introns, which are abundant in α-proteobacteria and could have been unleashed as a result of exposure of the archaeal host genome to the bacterial endosymbiont DNA; these would disrupt gene expression unless transcription and translation were decoupled and compartmentalized [106]. At least some additional innovations of eukaryogenesis, such as the evolution of the nonsense-mediated decay of transcripts containing premature stop codons and expansion of the ubiquitin system, can be envisaged as part of the same chain of adaptations to the intron bombardment as the origin of the nucleus [109] (Figure 5).
Second, functional studies in prokaryotes, particularly archaea, show that not only the molecular components of the several signature eukaryotic systems but also their actual structures and functions have evolved in archaea and thus predate eukaryogenesis. These include the archaeal proteasome [110], exosome [111] and Sm-protein complex, the progenitor of the spliceosome [112], the ESCRT-III membrane remodeling system [113, 114], actin-like proteins [105] and a prototype of the ubiquitin system of protein modification [115]. Each of these molecular machines found in different groups of archaea has been shown or predicted to be mechanistically similar to the eukaryotic counterpart, but they all function within the prokaryotic cell. The endomembrane system and the nucleus are dramatic exceptions, and so are the mitochondria themselves. It is tempting to connect these dots by proposing that eukaryogenesis was triggered by endosymbiosis, and that the endomembrane systems including the nucleus evolved as defense against invasion of Group II introns and perhaps foreign DNA in general [106, 109]. It does not seem accidental that many key components of these endomembrane systems seem to be of bacterial origin whereas others are repetitive proteins that might have evolved de novo [28]. Under the symbiogenesis scenario, diverse pre-existing systems of the archaeal host were co-opted and expanded within the emerging eukaryotic cellular organization [66].
Several arguments can be and have been put forward against the symbiogenesis scenario. First, prokaryotic endosymbionts in prokaryotic hosts are not widespread, prompting the view that phagocytosis, which is apparently unique to eukaryotic cells, was critical for the acquisition of the mitochondrion [3]. This argument is not compelling because: (1) eukaryogenesis is extremely rare, probably unique, in the history of life; (2) endosymbiotic bacteria within other bacteria are rare but known [116–118], and intracellular bacterial predation has been suggested as a potential route to endosymbiosis [119]; and (3) recent observations on membrane remodeling systems and actin-like proteins in archaea suggest the possibility of still unexplored mechanisms for engulfment of other prokaryotes, perhaps resembling primitive phagocytosis [105].
Second, a potentially strong argument against the symbiogenesis scenario could be the existence of a substantial number of eukaryote signature proteins (ESPs), so far found only in eukaryotes [120]. The provenance of ESPs is an intriguing question. However, on many occasions, careful sequence and structure searches have revealed archaeal and/or bacterial homologs of proteins originally considered ESPs, or else the existence of such homologs became obvious with the appearance of new genomes [66]. The discovery of prokaryotic homologs of tubulin, actin and ubiquitin are well known examples [71, 97], and more recent cases include the GINS proteins, which are involved in DNA replication [121], the ESCRT-III systems and the subunits of the TRAPP3 complex, which have a key role in eukaryotic vesicle trafficking [122]. Under the symbiogenesis scenario, the former and remaining ESPs result primarily from acceleration of evolution of genes whose functions have substantially changed during eukaryogenesis.
A third, potentially serious difficulty with the symbiogenesis scenario is that neither archaeal-like nor bacterial-like genes can be traced to a single prokaryotic lineage (although the origin of the mitochondria from α-proteobacteria is well established). However, the pangenomes of prokaryotes are large whereas the gene composition of individual organisms is highly flexible [123, 124], so reconstruction of the actual partners of the endosymbiosis that led to eukaryogenesis might not be feasible from a limited set of extant genomes. Moreover, many if not most archaea and bacteria might have evolved by streamlining, so eukaryogenesis could have been triggered by symbiosis between two prokaryotes with complex genomes.
In short, it is currently impossible to strictly rule out the possibility that the key eukaryotic innovations evolved independently from and prior to the mitochondrial endosymbiosis. In other words, the host of the endosymbiont might have been an archaezoan. However, the archaezoan scenario does not provide a plausible staging of events during the evolution of the complex internal organization of the eukaryotic cell, does not offer a raison d'être for the nucleus, and does not account for the presence of signature functional systems of eukaryotes in different archaeal lineages. In contrast, the symbiogenesis scenario can tie all these diverse lines of evidence into a coherent, even if still woefully incomplete, narrative.
Comparative genomics has so far neither solved the enigma of eukaryogenesis nor offered a definitive picture of the primary radiation of the major eukaryote lineages. However, although falling short of decisive answers, phylogenomic analysis has yielded many insights into the origin and earliest stages of evolution of eukaryotes. Recent findings indicate that several key cellular systems of eukaryotes exist in archaea. The scattering of these systems among different archaeal lineages, along with the phylogenies of conserved proteins, suggests that the archaeal ancestor of eukaryotes belonged to a deep, possibly extinct archaeal branch with a highly complex genome and diverse cellular functionalities. In contrast, the endomembrane systems of eukaryotes, and in particular the nucleus with its elaborate nuclear pore complex, are not found in archaea, and seem to be derived, at least in part, from bacterial ancestral components. These findings seem to be best compatible with a symbiogenesis scenario for the origin of eukaryotes under which eukaryogenesis was triggered by the endosymbiosis of an α-proteobacterium with an ancestral archaeon, with the nucleus evolving as a defense against intron invasion.
Phylogenomic analysis has clarified the evolutionary links between major groups of eukaryotes and led to the delineation of five or six supergroups. The relationships between the supergroups and the root position in the tree of eukaryotes remain extremely difficult to decipher, probably owing to a compressed cladogenesis or 'big bang' phase of evolution that followed eukaryogenesis. The expanding sampling of genomes from diverse branches of life is far from being a trivial pursuit, but has rather delivered unexpected biological insights.
References
Dacks JB, Doolittle WF: Reconstructing/deconstructing the earliest eukaryotes: how comparative genomics can help. Cell. 2001, 107: 419-425. 10.1016/S0092-8674(01)00584-0.
Embley TM, Martin W: Eukaryotic evolution, changes and challenges. Nature. 2006, 440: 623-630. 10.1038/nature04546.
Kurland CG, Collins LJ, Penny D: Genomics and the irreducible nature of eukaryote cells. Science. 2006, 312: 1011-1014. 10.1126/science.1121674.
Ovadi J, Saks V: On the origin of intracellular compartmentation and organized metabolic systems. Mol Cell Biochem. 2004, 256-257: 5-12. 10.1023/B:MCBI.0000009855.14648.2c.
Martin W: Evolutionary origins of metabolic compartmentalization in eukaryotes. Philos Trans R Soc Lond B Biol Sci. 365: 847-855.
Jekely G: Origin of eukaryotic endomembranes: a critical evaluation of different model scenarios. Adv Exp Med Biol. 2007, 607: 38-51. full_text.
Hudder A, Nathanson L, Deutscher MP: Organization of mammalian cytoplasm. Mol Cell Biol. 2003, 23: 9318-9326. 10.1128/MCB.23.24.9318-9326.2003.
Guigas G, Kalla C, Weiss M: The degree of macromolecular crowding in the cytoplasm and nucleoplasm of mammalian cells is conserved. FEBS Lett. 2007, 581: 5094-5098. 10.1016/j.febslet.2007.09.054.
Dacks JB, Peden AA, Field MC: Evolution of specificity in the eukaryotic endomembrane system. Int J Biochem Cell Biol. 2009, 41: 330-340. 10.1016/j.biocel.2008.08.041.
Field MC, Dacks JB: First and last ancestors: reconstructing evolution of the endomembrane system with ESCRTs, vesicle coat proteins, and nuclear pore complexes. Curr Opin Cell Biol. 2009, 21: 4-13. 10.1016/j.ceb.2008.12.004.
Giezen van der M, Tovar J: Degenerate mitochondria. EMBO Rep. 2005, 6: 525-530. 10.1038/sj.embor.7400440.
Hjort K, Goldberg AV, Tsaousis AD, Hirt RP, Embley TM: Diversity and reductive evolution of mitochondria among microbial eukaryotes. Philos Trans R Soc Lond B Biol Sci. 365: 713-727.
Giezen van der M: Hydrogenosomes and mitosomes: conservation and evolution of functions. J Eukaryot Microbiol. 2009, 56: 221-231. 10.1111/j.1550-7408.2009.00407.x.
Hunter T: The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 2007, 28: 730-738. 10.1016/j.molcel.2007.11.019.
Hubbard SR, Till JH: Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000, 69: 373-398. 10.1146/annurev.biochem.69.1.373.
Pawson T, Kofler M: Kinome signaling through regulated protein-protein interactions in normal and cancer cells. Curr Opin Cell Biol. 2009, 21: 147-153. 10.1016/j.ceb.2009.02.005.
Pawson T: Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004, 116: 191-203. 10.1016/S0092-8674(03)01077-8.
Johnson SA, Hunter T: Kinomics: methods for deciphering the kinome. Nat Methods. 2005, 2: 17-25. 10.1038/nmeth731.
Dikic I, Wakatsuki S, Walters KJ: Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol. 2009, 10: 659-671. 10.1038/nrm2767.
Hershko A, Ciechanover A: The ubiquitin system. Annu Rev Biochem. 1998, 67: 425-479. 10.1146/annurev.biochem.67.1.425.
Ciechanover A, Orian A, Schwartz AL: Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays. 2000, 22: 442-451. 10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q.
Liu Q, Paroo Z: Biochemical principles of small RNA pathways. Annu Rev Biochem. 2010,
Chapman EJ, Carrington JC: Specialization and evolution of endogenous small RNA pathways. Nat Rev Genet. 2007, 8: 884-896. 10.1038/nrg2179.
Amaral PP, Dinger ME, Mercer TR, Mattick JS: The eukaryotic genome as an RNA machine. Science. 2008, 319: 1787-1789. 10.1126/science.1155472.
Jacquier A: The complex eukaryotic transcriptome: unexpected pervasive transcription and novel small RNAs. Nat Rev Genet. 2009, 10: 833-844. 10.1038/nrg2683.
D'Alessio JA, Wright KJ, Tjian R: Shifting players and paradigms in cell-specific transcription. Mol Cell. 2009, 36: 924-931. 10.1016/j.molcel.2009.12.011.
Wilson MD, Odom DT: Evolution of transcriptional control in mammals. Curr Opin Genet Dev. 2009, 19: 579-585. 10.1016/j.gde.2009.10.003.
Mans BJ, Anantharaman V, Aravind L, Koonin EV: Comparative genomics, evolution and origins of the nuclear envelope and nuclear pore complex. Cell Cycle. 2004, 3: 1612-1637.
Collins L, Penny D: Complex spliceosomal organization ancestral to extant eukaryotes. Mol Biol Evol. 2005, 22: 1053-1066. 10.1093/molbev/msi091.
Dagan T, Artzy-Randrup Y, Martin W: Modular networks and cumulative impact of lateral transfer in prokaryote genome evolution. Proc Natl Acad Sci USA. 2008, 105: 10039-10044. 10.1073/pnas.0800679105.
Entrez Genome. [http://www.ncbi.nlm.nih.gov/sites/genome]
Roger AJ: Reconstructing early events in eukaryotic evolution. Am Nat. 1999, 154 Suppl 4: S146-S163. 10.1086/303290.
Sogin ML, Morrison HG, Hinkle G, Silberman JD: Ancestral relationships of the major eukaryotic lineages. Microbiologia. 1996, 12: 17-28.
Baldauf SL, Roger AJ, Wenk-Siefert I, Doolittle WF: A kingdom-level phylogeny of eukaryotes based on combined protein data. Science. 2000, 290: 972-977. 10.1126/science.290.5493.972.
Baldauf SL: The deep roots of eukaryotes. Science. 2003, 300: 1703-1706. 10.1126/science.1085544.
Roger AJ, Hug LA: The origin and diversification of eukaryotes: problems with molecular phylogenetics and molecular clock estimation. Philos Trans R Soc Lond B Biol Sci. 2006, 361: 1039-1054. 10.1098/rstb.2006.1845.
Brinkmann H, Philippe H: The diversity of eukaryotes and the root of the eukaryotic tree. Adv Exp Med Biol. 2007, 607: 20-37. full_text.
Keeling PJ, McFadden GI: Origins of microsporidia. Trends Microbiol. 1998, 6: 19-23. 10.1016/S0966-842X(97)01185-2.
Philippe H, Germot A, Moreira D: The new phylogeny of eukaryotes. Curr Opin Genet Dev. 2000, 10: 596-601. 10.1016/S0959-437X(00)00137-4.
Minge MA, Silberman JD, Orr RJ, Cavalier-Smith T, Shalchian-Tabrizi K, Burki F, Skjaeveland A, Jakobsen KS: Evolutionary position of breviate amoebae and the primary eukaryote divergence. Proc Biol Sci. 2009, 276: 597-604. 10.1098/rspb.2008.1358.
Boussau B, Daubin V: Genomes as documents of evolutionary history. Trends Ecol Evol. 2009, 25: 224-232. 10.1016/j.tree.2009.09.007.
Delsuc F, Brinkmann H, Philippe H: Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet. 2005, 6: 361-375. 10.1038/nrg1603.
Adl SM, Simpson AG, Farmer MA, Andersen RA, Anderson OR, Barta JR, Bowser SS, Brugerolle G, Fensome RA, Fredericq S, James TY, Karpov S, Kugrens P, Krug J, Lane CE, Lewis LA, Lodge J, Lynn DH, Mann DG, McCourt RM, Mendoza L, Moestrup O, Mozley-Standridge SE, Nerad TA, Shearer CA, Smirnov AV, Spiegel FW, Taylor MF: The new higher level classification of eukaryotes with emphasis on the taxonomy of protists. J Eukaryot Microbiol. 2005, 52: 399-451. 10.1111/j.1550-7408.2005.00053.x.
Keeling PJ: Genomics. Deep questions in the tree of life. Science. 2007, 317: 1875-1876. 10.1126/science.1149593.
Keeling PJ, Burger G, Durnford DG, Lang BF, Lee RW, Pearlman RE, Roger AJ, Gray MW: The tree of eukaryotes. Trends Ecol Evol. 2005, 20: 670-676. 10.1016/j.tree.2005.09.005.
Parfrey LW, Barbero E, Lasser E, Dunthorn M, Bhattacharya D, Patterson DJ, Katz LA: Evaluating support for the current classification of eukaryotic diversity. PLoS Genet. 2006, 2: e220-10.1371/journal.pgen.0020220.
Burki F, Shalchian-Tabrizi K, Minge M, Skjaeveland A, Nikolaev SI, Jakobsen KS, Pawlowski J: Phylogenomics reshuffles the eukaryotic supergroups. PLoS ONE. 2007, 2: e790-10.1371/journal.pone.0000790.
Yoon HS, Grant J, Tekle YI, Wu M, Chaon BC, Cole JC, Logsdon JM, Patterson DJ, Bhattacharya D, Katz LA: Broadly sampled multigene trees of eukaryotes. BMC Evol Biol. 2008, 8: 14-10.1186/1471-2148-8-14.
Moreira D, Heyden von der S, Bass D, Lopez-Garcia P, Chao E, Cavalier-Smith T: Global eukaryote phylogeny: combined small- and large-subunit ribosomal DNA trees support monophyly of Rhizaria, Retaria and Excavata. Mol Phylogenet Evol. 2007, 44: 255-266. 10.1016/j.ympev.2006.11.001.
Hackett JD, Yoon HS, Li S, Reyes-Prieto A, Rummele SE, Bhattacharya D: Phylogenomic analysis supports the monophyly of cryptophytes and haptophytes and the association of rhizaria with chromalveolates. Mol Biol Evol. 2007, 24: 1702-1713. 10.1093/molbev/msm089.
Hampl V, Hug L, Leigh JW, Dacks JB, Lang BF, Simpson AG, Roger AJ: Phylogenomic analyses support the monophyly of Excavata and resolve relationships among eukaryotic "supergroups". Proc Natl Acad Sci USA. 2009, 106: 3859-3864. 10.1073/pnas.0807880106.
Burki F, Shalchian-Tabrizi K, Pawlowski J: Phylogenomics reveals a new 'megagroup' including most photosynthetic eukaryotes. Biol Lett. 2008, 4: 366-369. 10.1098/rsbl.2008.0224.
Germot A, Philippe H: Critical analysis of eukaryotic phylogeny: a case study based on the HSP70 family. J Eukaryot Microbiol. 1999, 46: 116-124. 10.1111/j.1550-7408.1999.tb04594.x.
Rokas A, Kruger D, Carroll SB: Animal evolution and the molecular signature of radiations compressed in time. Science. 2005, 310: 1933-1938. 10.1126/science.1116759.
Koonin EV: The Biological Big Bang model for the major transitions in evolution. Biol Direct. 2007, 2: 21-10.1186/1745-6150-2-21.
Stechmann A, Cavalier-Smith T: Rooting the eukaryote tree by using a derived gene fusion. Science. 2002, 297: 89-91. 10.1126/science.1071196.
Stechmann A, Cavalier-Smith T: The root of the eukaryote tree pinpointed. Curr Biol. 2003, 13: R665-R666. 10.1016/S0960-9822(03)00602-X.
Richards TA, Cavalier-Smith T: Myosin domain evolution and the primary divergence of eukaryotes. Nature. 2005, 436: 1113-1118. 10.1038/nature03949.
Rokas A, Holland PW: Rare genomic changes as a tool for phylogenetics. Trends Ecol Evol. 2000, 15: 454-459. 10.1016/S0169-5347(00)01967-4.
Rogozin IB, Basu MK, Csuros M, Koonin EV: Analysis of rare genomic changes does not support the unikont-bikont phylogeny and suggests cyanobacterial symbiosis as the point of primary radiation of eukaryotes. Genome Biol Evol. 2009, 2009: 99-113.
Rokas A, Carroll SB: Bushes in the tree of life. PLoS Biol. 2006, 4: e352-10.1371/journal.pbio.0040352.
Koonin EV, Fedorova ND, Jackson JD, Jacobs AR, Krylov DM, Makarova KS, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Rogozin IB, Smirnov S, Sorokin AV, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA: A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5: R7-10.1186/gb-2004-5-2-r7.
Makarova KS, Wolf YI, Mekhedov SL, Mirkin BG, Koonin EV: Ancestral paralogs and pseudoparalogs and their role in the emergence of the eukaryotic cell. Nucleic Acids Res. 2005, 33: 4626-4638. 10.1093/nar/gki775.
Csuros M, Miklos I: Streamlining and large ancestral genomes in Archaea inferred with a phylogenetic birth-and-death model. Mol Biol Evol. 2009, 26: 2087-2095. 10.1093/molbev/msp123.
Ceulemans H, Beke L, Bollen M: Approaches to defining the ancestral eukaryotic protein complexome. Bioessays. 2006, 28: 316-324. 10.1002/bies.20373.
Aravind L, Iyer LM, Koonin EV: Comparative genomics and structural biology of the molecular innovations of eukaryotes. Curr Opin Struct Biol. 2006, 16: 409-419. 10.1016/j.sbi.2006.04.006.
Fritz-Laylin LK, Prochnik SE, Ginger ML, Dacks JB, Carpenter ML, Field MC, Kuo A, Paredez A, Chapman J, Pham J, Shu S, Neupane R, Cipriano M, Mancuso J, Tu H, Salamov A, Lindquist E, Shapiro H, Lucas S, Grigoriev IV, Cande WZ, Fulton C, Rokhsar DS, Dawson SC: The Genome of Naegleria gruberi illuminates early eukaryotic versatility. Cell. 2010, 140: 631-642. 10.1016/j.cell.2010.01.032.
Koonin EV: The incredible expanding ancestor of eukaryotes. Cell. 140: 606-608. 10.1016/j.cell.2010.02.022.
Bapteste E, Charlebois RL, MacLeod D, Brochier C: The two tempos of nuclear pore complex evolution: highly adapting proteins in an ancient frozen structure. Genome Biol. 2005, 6: R85-10.1186/gb-2005-6-10-r85.
Shabalina SA, Koonin EV: Origins and evolution of eukaryotic RNA interference. Trends Ecol Evol. 2008, 23: 578-587. 10.1016/j.tree.2008.06.005.
Hochstrasser M: Origin and function of ubiquitin-like proteins. Nature. 2009, 458: 422-429. 10.1038/nature07958.
Carmel L, Wolf YI, Rogozin IB, Koonin EV: Three distinct modes of intron dynamics in the evolution of eukaryotes. Genome Res. 2007, 17: 1034-1044. 10.1101/gr.6438607.
Csuros M, Rogozin IB, Koonin EV: Extremely intron-rich genes in the alveolate ancestors inferred with a flexible maximum-likelihood approach. Mol Biol Evol. 2008, 25: 903-911. 10.1093/molbev/msn039.
Roy SW: Intron-rich ancestors. Trends Genet. 2006, 22: 468-471. 10.1016/j.tig.2006.07.002.
Roy SW, Gilbert W: The evolution of spliceosomal introns: patterns, puzzles and progress. Nat Rev Genet. 2006, 7: 211-221.
Archibald JM: The puzzle of plastid evolution. Curr Biol. 2009, 19: R81-R88. 10.1016/j.cub.2008.11.067.
Elias M, Archibald JM: Sizing up the genomic footprint of endosymbiosis. Bioessays. 2009, 31: 1273-1279. 10.1002/bies.200900117.
Esser C, Ahmadinejad N, Wiegand C, Rotte C, Sebastiani F, Gelius-Dietrich G, Henze K, Kretschmann E, Richly E, Leister D, Bryant D, Steel MA, Lockhart PJ, Penny D, Martin W: A genome phylogeny for mitochondria among alpha-proteobacteria and a predominantly eubacterial ancestry of yeast nuclear genes. Mol Biol Evol. 2004, 21: 1643-1660. 10.1093/molbev/msh160.
Rivera MC, Lake JA: The ring of life provides evidence for a genome fusion origin of eukaryotes. Nature. 2004, 431: 152-155. 10.1038/nature02848.
Martin W, Embley TM: Evolutionary biology: early evolution comes full circle. Nature. 2004, 431: 134-137. 10.1038/431134a.
Gray MW, Burger G, Lang BF: Mitochondrial evolution. Science. 1999, 283: 1476-1481. 10.1126/science.283.5407.1476.
Esser C, Martin W, Dagan T: The origin of mitochondria in light of a fluid prokaryotic chromosome model. Biol Lett. 2007, 3: 180-184. 10.1098/rsbl.2006.0582.
Doolittle WF: You are what you eat: a gene transfer ratchet could account for bacterial genes in eukaryotic nuclear genomes. Trends Genet. 1998, 14: 307-311. 10.1016/S0168-9525(98)01494-2.
Galagan JE, Nusbaum C, Roy A, Endrizzi MG, Macdonald P, FitzHugh W, Calvo S, Engels R, Smirnov S, Atnoor D, Brown A, Allen N, Naylor J, Stange-Thomann N, DeArellano K, Johnson R, Linton L, McEwan P, McKernan K, Talamas J, Tirrell A, Ye W, Zimmer A, Barber RD, Cann I, Graham DE, Grahame DA, Guss AM, Hedderich R, Ingram-Smith C, et al: The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res. 2002, 12: 532-542. 10.1101/gr.223902.
Koonin EV: Horizontal gene transfer: the path to maturity. Mol Microbiol. 2003, 50: 725-727. 10.1046/j.1365-2958.2003.03808.x.
Cox CJ, Foster PG, Hirt RP, Harris SR, Embley TM: The archaebacterial origin of eukaryotes. Proc Natl Acad Sci USA. 2008, 105: 20356-20361. 10.1073/pnas.0810647105.
Foster PG, Cox CJ, Embley TM: The primary divisions of life: a phylogenomic approach employing composition-heterogeneous methods. Philos Trans R Soc Lond B Biol Sci. 2009, 364: 2197-2207. 10.1098/rstb.2009.0034.
Pisani D, Cotton JA, McInerney JO: Supertrees disentangle the chimerical origin of eukaryotic genomes. Mol Biol Evol. 2007, 24: 1752-1760. 10.1093/molbev/msm095.
Yutin N, Makarova KS, Mekhedov SL, Wolf YI, Koonin EV: The deep archaeal roots of eukaryotes. Mol Biol Evol. 2008, 25: 1619-1630. 10.1093/molbev/msn108.
Saruhashi S, Hamada K, Miyata D, Horiike T, Shinozawa T: Comprehensive analysis of the origin of eukaryotic genomes. Genes Genet Syst. 2008, 83: 285-291. 10.1266/ggs.83.285.
Vishwanath P, Favaretto P, Hartman H, Mohr SC, Smith TF: Ribosomal protein-sequence block structure suggests complex prokaryotic evolution with implications for the origin of eukaryotes. Mol Phylogenet Evol. 2004, 33: 615-625. 10.1016/j.ympev.2004.07.003.
Korkhin Y, Unligil UM, Littlefield O, Nelson PJ, Stuart DI, Sigler PB, Bell SD, Abrescia NG: Evolution of complex RNA polymerases: the complete archaeal RNA polymerase structure. PLoS Biol. 2009, 7: e102-10.1371/journal.pbio.1000102.
Blombach F, Makarova KS, Marrero J, Siebers B, Koonin EV, Oost van der J: Identification of an ortholog of the eukaryotic RNA polymerase III subunit RPC34 in Crenarchaeota and Thaumarchaeota suggests specialization of RNA polymerases for coding and non-coding RNAs in Archaea. Biol Direct. 2009, 4: 39-10.1186/1745-6150-4-39.
Daniels JP, Kelly S, Wickstead B, Gull K: Identification of a crenarchaeal orthologue of Elf1: implications for chromatin and transcription in Archaea. Biol Direct. 2009, 4: 24-10.1186/1745-6150-4-24.
Reeve JN, Bailey KA, Li WT, Marc F, Sandman K, Soares DJ: Archaeal histones: structures, stability and DNA binding. Biochem Soc Trans. 2004, 32: 227-230. 10.1042/BST0320227.
Vaughan S, Wickstead B, Gull K, Addinall SG: Molecular evolution of FtsZ protein sequences encoded within the genomes of archaea, bacteria, and eukaryota. J Mol Evol. 2004, 58: 19-29. 10.1007/s00239-003-2523-5.
Lowe J, Amos LA: Evolution of cytomotive filaments: the cytoskeleton from prokaryotes to eukaryotes. Int J Biochem Cell Biol. 2009, 41: 323-329. 10.1016/j.biocel.2008.08.010.
Lindas AC, Karlsson EA, Lindgren MT, Ettema TJ, Bernander R: A unique cell division machinery in the Archaea. Proc Natl Acad Sci USA. 2008, 105: 18942-18946. 10.1073/pnas.0809467105.
Filee J, Forterre P, Sen-Lin T, Laurent J: Evolution of DNA polymerase families: evidences for multiple gene exchange between cellular and viral proteins. J Mol Evol. 2002, 54: 763-773. 10.1007/s00239-001-0078-x.
Tahirov TH, Makarova KS, Rogozin IB, Pavlov YI, Koonin EV: Evolution of DNA polymerases: an inactivated polymerase-exonuclease module in Pol epsilon and a chimeric origin of eukaryotic polymerases from two classes of archaeal ancestors. Biol Direct. 2009, 4: 11-10.1186/1745-6150-4-11.
Aravind L, Koonin EV: Phosphoesterase domains associated with DNA polymerases of diverse origins. Nucleic Acids Res. 1998, 26: 3746-3752. 10.1093/nar/26.16.3746.
Makarova KS, Wolf YI, Oost van der J, Koonin EV: Prokaryotic homologs of Argonaute proteins are predicted to function as key components of a novel system of defense against mobile genetic elements. Biol Direct. 2009, 4: 29-10.1186/1745-6150-4-29.
Martin W, Dagan T, Koonin EV, Dipippo JL, Gogarten JP, Lake JA: The evolution of eukaryotes. Science. 2007, 316: 542-543. 10.1126/science.316.5824.542c. author reply 542-543
Poole A, Penny D: Eukaryote evolution: engulfed by speculation. Nature. 2007, 447: 913-10.1038/447913a.
Yutin N, Wolf MY, Wolf YI, Koonin EV: The origins of phagocytosis and eukaryogenesis. Biol Direct. 2009, 4: 9-10.1186/1745-6150-4-9.
Martin W, Koonin EV: Introns and the origin of nucleus-cytosol compartmentation. Nature. 2006, 440: 41-45. 10.1038/nature04531.
Martin W, Koonin EV: A positive definition of prokaryotes. Nature. 2006, 442: 868-10.1038/442868c.
French SL, Santangelo TJ, Beyer AL, Reeve JN: Transcription and translation are coupled in Archaea. Mol Biol Evol. 2007, 24: 893-895. 10.1093/molbev/msm007.
Koonin EV: The origin of introns and their role in eukaryogenesis: a compromise solution to the introns-early versus introns-late debate?. Biol Direct. 2006, 1: 22-10.1186/1745-6150-1-22.
Maupin-Furlow JA, Wilson HL, Kaczowka SJ, Ou MS: Proteasomes in the archaea: from structure to function. Front Biosci. 2000, 5: D837-D865. 10.2741/furlow.
Hartung S, Hopfner KP: Lessons from structural and biochemical studies on the archaeal exosome. Biochem Soc Trans. 2009, 37: 83-87. 10.1042/BST0370083.
Mura C, Kozhukhovsky A, Gingery M, Phillips M, Eisenberg D: The oligomerization and ligand-binding properties of Sm-like archaeal proteins (SmAPs). Protein Sci. 2003, 12: 832-847. 10.1110/ps.0224703.
Samson RY, Bell SD: Ancient ESCRTs and the evolution of binary fission. Trends Microbiol. 2009, 17: 507-513. 10.1016/j.tim.2009.08.003.
Ettema TJ, Bernander R: Cell division and the ESCRT complex: a surprise from the archaea. Commun Integr Biol. 2009, 2: 86-88.
Humbard MA, Miranda HV, Lim JM, Krause DJ, Pritz JR, Zhou G, Chen S, Wells L, Maupin-Furlow JA: Ubiquitin-like small archaeal modifier proteins (SAMPs) in Haloferax volcanii . Nature. 463: 54-60. 10.1038/nature08659.
von Dohlen CD, Kohler S, Alsop ST, McManus WR: Mealybug beta-proteobacterial endosymbionts contain gamma-proteobacterial symbionts. Nature. 2001, 412: 433-436. 10.1038/35086563.
Thao ML, Gullan PJ, Baumann P: Secondary (gamma-Proteobacteria) endosymbionts infect the primary (beta-Proteobacteria) endosymbionts of mealybugs multiple times and coevolve with their hosts. Appl Environ Microbiol. 2002, 68: 3190-3197. 10.1128/AEM.68.7.3190-3197.2002.
Hoffmeister M, Martin W: Interspecific evolution: microbial symbiosis, endosymbiosis and gene transfer. Environ Microbiol. 2003, 5: 641-649. 10.1046/j.1462-2920.2003.00454.x.
Davidov Y, Jurkevitch E: Predation between prokaryotes and the origin of eukaryotes. Bioessays. 2009, 31: 748-757. 10.1002/bies.200900018.
Hartman H, Fedorov A: The origin of the eukaryotic cell: a genomic investigation. Proc Natl Acad Sci USA. 2002, 99: 1420-1425. 10.1073/pnas.032658599.
MacNeill SA: Structure and function of the GINS complex, a key component of the eukaryotic replisome. Biochem J. 425: 489-500. 10.1042/BJ20091531.
Podar M, Wall MA, Makarova KS, Koonin EV: The prokaryotic V4R domain is the likely ancestor of a key component of the eukaryotic vesicle transport system. Biol Direct. 2008, 3: 2-10.1186/1745-6150-3-2.
Koonin EV, Wolf YI: Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008, 36: 6688-6719. 10.1093/nar/gkn668.
Lapierre P, Gogarten JP: Estimating the size of the bacterial pan-genome. Trends Genet. 2009, 25: 107-110. 10.1016/j.tig.2008.12.004.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Koski LB, Golding GB: The closest BLAST hit is often not the nearest neighbor. J Mol Evol. 2001, 52: 540-542.
Elkins JG, Podar M, Graham DE, Makarova KS, Wolf Y, Randau L, Hedlund BP, Brochier-Armanet C, Kunin V, Anderson I, Lapidus A, Goltsman E, Barry K, Koonin EV, Hugenholtz P, Kyrpides N, Wanner G, Richardson P, Keller M, Stetter KO: A korarchaeal genome reveals insights into the evolution of the Archaea. Proc Natl Acad Sci USA. 2008, 105: 8102-8107. 10.1073/pnas.0801980105.
Aravind L, Walker DR, Koonin EV: Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999, 27: 1223-1242. 10.1093/nar/27.5.1223.
Koonin EV, Makarova KS, Elkins JG: Orthologs of the small RPB8 subunit of the eukaryotic RNA polymerases are conserved in hyperthermophilic Crenarchaeota and "Korarchaeota". Biol Direct. 2007, 2: 38-10.1186/1745-6150-2-38.
Anantharaman V, Koonin EV, Aravind L: Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002, 30: 1427-1464. 10.1093/nar/30.7.1427.
Venancio TM, Balaji S, Iyer LM, Aravind L: Reconstructing the ubiquitin network: cross-talk with other systems and identification of novel functions. Genome Biol. 2009, 10: R33-10.1186/gb-2009-10-3-r33.
Koonin EV, Wolf YI, Aravind L: Prediction of the archaeal exosome and its connections with the proteasome and the translation and transcription machineries by a comparative-genomic approach. Genome Res. 2001, 11: 240-252. 10.1101/gr.162001.
Iyer LM, Anantharaman V, Wolf MY, Aravind L: Comparative genomics of transcription factors and chromatin proteins in parasitic protists and other eukaryotes. Int J Parasitol. 2008, 38: 1-31. 10.1016/j.ijpara.2007.07.018.
Aravind L, Iyer LM, Anantharaman V: The two faces of Alba: the evolutionary connection between proteins participating in chromatin structure and RNA metabolism. Genome Biol. 2003, 4: R64-10.1186/gb-2003-4-10-r64.
Gray MW, Burger G, Lang BF: The origin and early evolution of mitochondria. Genome Biol. 2001, 2: reviews1018-10.1186/gb-2001-2-6-reviews1018.
Acknowledgements
I thank Yuri Wolf for providing the data used in Figure 2, Bill Martin for helpful discussions and Tania Senkevich for critical reading of the manuscript. The author's research is supported by the DHHS (National Library of Medicine) intramural funds.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Koonin, E.V. The origin and early evolution of eukaryotes in the light of phylogenomics. Genome Biol 11, 209 (2010). https://doi.org/10.1186/gb-2010-11-5-209
Published:
DOI: https://doi.org/10.1186/gb-2010-11-5-209