
Abstract
Analysis of the genomes of multiple Candida species reveals shared traits associated with pathogenesis, but also striking differences in sexual reproduction.
Similar content being viewed by others

Yeasts belonging to the Candida group of species are opportunistic pathogens responsible for both debilitating mucosal infections (such as oral thrush and vaginitis) as well as potentially life-threatening systemic infections in humans. These fungi represent a diverse collection of hemiascomycetes, although the majority of Candida species are grouped together in a single clade, termed the Candida clade. In a recent study, Butler et al. [1] have sequenced the genomes of six species from the Candida clade and compared them with genomes from non-pathogenic yeasts. They show that gene families associated with pathogenesis have expanded in species from the Candida clade, suggesting that this expansion coevolved with the ability to cause infection of the host. In addition, the authors demonstrate a surprising divergence in the mechanisms regulating mating and meiosis in this clade. These studies provide an exciting entry point for comparative analysis of Candida species; future studies will compare and contrast how these species acquired pathogenicity as well as potentially novel modes of sexual reproduction in these yeasts.
Candidagenomes
The six species sequenced from the Candida clade included an isolate of Candida albicans, the most frequently isolated species from humans, as well as the closely related pathogens C. parapsilosis and C. tropicalis (Figure 1). A fourth species, Lodderomyces elongisporus, was originally thought to be non-pathogenic but has recently been isolated from multiple bloodstream infections [2]. Each of these four species exists primarily in the diploid state. In addition, the genomes of two emerging haploid pathogens, C. guilliermondii and C. lusitaniae, were analyzed. These sequences were compared to the genome of the non-pathogen Debaryomyces hansenii, a halotolerant yeast found in the environment, as well as multiple species from the Saccharomyces clade.

Phylogenetic tree of the Candida clade of species, together with the related hemiascomycete S. cerevisiae. The six species sequenced in the Butler et al. study [1] were C. albicans, C. tropicalis, C. parapsilosis, L. elongisporus, C. guilliermondii, and C. lusitaniae. These sequences were compared to those of non-pathogenic yeasts, including D. hansenii and species from the S. cerevisiae clade. Note that L. elongisporus has now been isolated from bloodstream infections (a rare pathogen), and that C. albicans undergoes efficient mating but a conventional meiosis has not been identified (parasexual reproduction). Figure adapted from the web site of the Broad Institute. Dip, diploid genome; Hap, haploid genome.
The Candida genomes showed a broad distribution of sizes, ranging from 10.6 to 15.5 megabases (Mb), although the predicted number of protein-coding genes was very similar in each (5,733 to 6,318). This apparent discrepancy was attributed to variability in intergenic spacing among species. General chromosomal configurations were similar (each species contained between six and nine chromosomes), although synteny has been disrupted by intra-chromosomal rearrangements since these species diverged. Interestingly, only C. albicans and C. tropicalis contained the major repeat sequence (MRS) elements. These are important sites for genetic recombination in C. albicans as they promote chromosome translocations and increase genomic diversity [3]. The discovery of MRS elements in C. tropicalis suggests that these repeats play a similar role in karyotypic variation in these strains [4], although the contribution of these changes to pathogenesis is not known.
A characteristic feature of the diploid C. albicans genome is significant sequence divergence between chromosome homologs. Thus, one single nucleotide polymorphism (SNP) was found every 330 bp in the most commonly studied strain, SC5314, and similarly, one SNP occurred every 390 bp in the clinical WO-1 isolate. The other diploid Candida clade species also exhibit SNPs, ranging from one SNP every 222 bases in L. elongisporus to one SNP per 15,553 bases in C. parapsilosis. Thus, heterozygosity is common in the diploid Candida species, but the extent of polymorphism varies greatly between them. Three species also show long stretches of homozygosity, and this was most dramatic in the C. albicans WO-1 strain, with 30% of the genome lacking SNPs. It is unclear if these homogenous regions are due to break-induced replication or are the result of progress through a parasexual or sexual cycle.
Evolution of pathogenicity
Butler et al. [1] identified a number of expanded gene families in the Candida clade that are associated with pathogenesis. Out of 9,209 gene families, 21 were enriched in the pathogenic species compared to the non-pathogens. These included three families of cell-wall proteins, most of which contain putative carboxy-terminal glycosyl phosphatidylinositol (GPI) anchors that mediate covalent attachment to the fungal cell wall. The ALS gene family encodes eight cell-wall proteins that have been studied in detail in C. albicans. These proteins (particularly Als1 and Als3) pro mote adhesion to epithelial cells, although other functions for this family have emerged, including a role in biofilm formation [5–7]. Similarly, the family of Hyr/Iff proteins was enriched in the Candida clade. At least one member of this family, IFF11, was recently shown to lack a GPI anchor and is secreted in C. albicans. Loss of IFF11 results in defective cell-wall structure and reduced virulence [8]. The third family of cell-wall proteins enriched in Candida includes members related to Pga30. Less is known about this family of proteins, although several members were identified in the C. albicans cell-wall proteome [9]. All three families of cell-wall proteins show high mutation rates, suggesting that these are rapidly evolving factors that provide Candida species with a selective advantage for invasion and infection of the mammalian host.
Interestingly, the family of Hwp1-related cell-wall proteins is limited to only a subset of Candida species. In C. albicans, Hwp1 plays an important role in biofilm formation and the covalent attachment of hyphae to host epithelial cells via mammalian transglutaminases [7, 10]. However, genes encoding Hwp1-like proteins were found only in the diploid Candida species. In addition, only the C. albicans Hwp1 protein contained the amino-terminal transglutaminase-substrate domain [1]. Comparative genome analyses can therefore provide revealing insights into why Candida species differ in their ability to colonize and infect the human body. Another striking example of a species-specific gene set was that encoding Fgr38-like family members, with 33 genes present in C. albicans and only one in all the other Candida species combined. Fgr38 was originally identified in a screen for factors that affect hyphal growth [11]. Such large-scale amplification of a gene family in C. albicans suggests that it might play an important, but as yet largely undefined, role in promoting the fitness of this species.
Expansion of multiple gene families associated with extra-cellular enzymes and transmembrane transporters was also noted in Candida [1]. These include oligopeptide transporters, amino-acid permeases, lipases, superoxide dismutases, ferric reductases and GPI-anchored yapsin proteases. Extracellular hydrolytic enzymes (for example, secreted aspartyl proteases, lipases and phospholipases) are known to be necessary for virulence in Candida, and are involved in nutrient acquisition, tissue invasion and evasion of the immune response [12, 13]. The selective expansion of these gene families in Candida again attests to the importance of these genes in colonization and pathogenesis.
It is also revealing that C. glabrata, a prevalent pathogen that is closely related to Saccharomyces cerevisiae, did not show the same expansion of genes as the other Candida species. Thus, while C. albicans had 161 representatives in the 21 gene families enriched in pathogenic Candida, C. glabrata contained only three genes from these families [1]. These observations demonstrate that C. glabrata has evolved alternative mechanisms from most of the other Candida species for infection of the host. This view is also consistent with studies on C. glabrata that have established that traits associated with virulence in C. albicans (for example, hyphal growth and secreted proteinase activity) are not utilized by C. glabrata [12]. Despite these fundamental differences, C. glabrata and C. albicans do share general features necessary for virulence, including phenotypic plasticity and expanded numbers of host adhesins [12].
Sexual identity, mating and meiosis
Mating in the hemiascomycete yeast is regulated by transcription factors encoded at the mating-type (MAT) loci. These are master regulators of cell identity, defining the programs of sexual differentiation in these species. The two idiomorphs of the locus are MAT a and MATα, which direct cells to mate as a or α cell types, respectively. Interestingly, although these loci have been shown to regulate sexual identity in both S. cerevisiae and C. albicans [14, 15], the transcriptional circuitry by which they do so has diverged between these species [16].
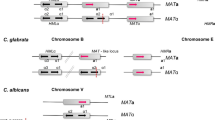
Analysis of the MTL (mating-type-like) locus in different Candida species revealed a surprising diversity in MTL organization [1]. Although synteny surrounding the MTL locus has been largely conserved, the presence of the master transcriptional regulators themselves is highly variable. In C. albicans, which appears to have retained the ancestral MTL configuration, MTL a encodes a1 and a2 transcription factors, and expression of a2 is required for the cell to mate as an a-type cell. Conversely, MTLα includes α1 and α2, and expression of α1 is necessary for α mating-type characteristics [16]. In both S. cerevisiae and C. albicans, a1 forms a heterodimer with α2 that represses expression of mating genes, while in S. cerevisiae a1/α2 also facilitates entry of diploid cells into meiosis [14, 16]. Curiously, the MTLα2 gene was found to be missing from all three haploid Candida species [1]. This was not associated with an inability to undergo sexual reproduction; Reedy et al. [17] recently confirmed that C. lusitaniae undergoes efficient mating and meiosis even in the absence of α2, and C. guilliermondii and D. hansenii also have complete sexual cycles ([18] and references therein). Rather, these experiments indicate that the transcriptional regulation of meiosis has been rewired in these species, with another α-specific factor (perhaps α1) taking the place of α2 [17].
Other differences at the MTL locus were noted, with the most striking occurring in L. elongisporus, where all four mating-type regulators (a1, a2, α1 and α2) have been lost. This fungus is thought to be homothallic (self-mating) [2] and may be the first ascomycete identified to undergo sexual reproduction in the absence of either MTL a or MTLα [1]. D. hansenii is also homothallic, and in this case both MTL loci have become fused into a single locus. Similar fusion of MTL loci has been described in other homothallic ascomycetes, suggesting that this is a common mechanism by which homothallism can evolve from a heterothallic (cross-mating) ancestor ([18] and references therein).
The meiotic program has undergone considerable changes in the Candida lineage. The transcription factor gene IME1, the major regulator of meiosis in S. cerevisiae, is absent, yet the three established sexual species (C. lusitaniae, C. guilliermondii and D. hansenii) can undergo meiosis without it [1, 17]. C. albicans strains undergo efficient mating between a and α forms [14, 15], yet completion of the sexual cycle occurs by a parasexual mechanism of random chromosome loss rather than conventional meiosis ([19] and references therein). On the basis of genomic evidence, C. albicans either has a cryptic meiotic program that has yet to be identified, or conserved 'meiosis-specific' genes have been reprogrammed to function in the parasexual cycle [1]. C. guilliermondii and C. lusitaniae have also lost multiple components of the meiotic machinery. These include parts of the synaptonemal complex (Hop1, Red1, Mek1, Zip1 and Zip2), together with the Dmc1-dependent pathway of meiotic recombination, and the crossover-formation pathway mediated by the MutS homologs Msh4/Msh5. These findings indicate that meiosis in Candida is fundamentally distinct from that in the model hemiascomycete, S. cerevisiae, in terms of both its regulation and its molecular apparatus. At least the role of Spo11 in directing DNA double-strand breaks appears to be conserved in Candida, as this protein mediates meiotic recombination in C. lusitaniae [17] as well as parasexual recombination in C. albicans [19].
Finally, two Candida species, C. parapsilosis and C. tropicalis, were found to contain intact pheromone signaling transduction pathways by genomic analysis but have never been observed to mate ([18] and references therein). Either these species recently lost the ability to undergo sexual reproduction (as evidenced by mutation of MTL a1 in C. parapsilosis [20]) or cryptic mating pathways have yet to be uncovered in these species.
The wealth of information provided by Butler et al. [1] has opened the door for comparative studies to further illuminate Candida biology. Already, multiple traits associated with Candida pathogenesis have been inferred, including expansion of gene families associated with colonization and infection, as well as species-specific features that make each Candida unique. In the latter case, it is particularly intriguing that mechanisms of mating and sexual reproduction have rapidly diverged between Candida species. Future studies will no doubt address how these differences evolved and what role they play in the lifestyles of these opportunistic yeast.
References
Butler G, Rasmussen MD, Lin MF, Santos MA, Sakthikumar S, Munro CA, Rheinbay E, Grabherr M, Forche A, Reedy JL, Agrafioti I, Arnaud MB, Bates S, Brown AJ, Brunke S, Costanzo MC, Fitzpatrick DA, de Groot PW, Harris D, Hoyer LL, Hube B, Klis FM, Kodira C, Lennard N, Logue ME, Martin R, Neiman AM, Nikolaou E, Quail MA, Quinn J, et al: Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009, 459: 657-662. 10.1038/nature08064.
Lockhart SR, Messer SA, Pfaller MA, Diekema DJ: Lodderomyces elongisporus masquerading as Candida parapsilosis as a cause of bloodstream infections. J Clin Microbiol. 2008, 46: 374-376. 10.1128/JCM.01790-07.
Chibana H, Beckerman JL, Magee PT: Fine-resolution physical mapping of genomic diversity in Candida albicans. Genome Res. 2000, 10: 1865-1877. 10.1101/gr.148600.
Zhang J, Hollis RJ, Pfaller MA: Variations in DNA subtype and antifungal susceptibility among clinical isolates of Candida tropicalis. Diagn Microbiol Infect Dis. 1997, 27: 63-67. 10.1016/S0732-8893(97)00002-3.
Dranginis AM, Rauceo JM, Coronado JE, Lipke PN: A biochemical guide to yeast adhesins: glycoproteins for social and antisocial occasions. Microbiol Mol Biol Rev. 2007, 71: 282-294. 10.1128/MMBR.00037-06.
Hoyer LL, Green CB, Oh SH, Zhao X: Discovering the secrets of the Candida albicans agglutinin-like sequence (ALS) gene family - a sticky pursuit. Med Mycol. 2008, 46: 1-15. 10.1080/13693780701435317.
Nobile CJ, Nett JE, Andes DR, Mitchell AP: Function of Candida albicans adhesin Hwp1 in biofilm formation. Eukaryot Cell. 2006, 5: 1604-1610. 10.1128/EC.00194-06.
Bates S, de la Rosa JM, MacCallum DM, Brown AJ, Gow NA, Odds FC: Candida albicans Iff11, a secreted protein required for cell wall structure and virulence. Infect Immun. 2007, 75: 2922-2928. 10.1128/IAI.00102-07.
Castillo L, Calvo E, Martinez AI, Ruiz-Herrera J, Valentin E, Lopez JA, Sentandreu R: A study of the Candida albicans cell wall proteome. Proteomics. 2008, 8: 3871-3881. 10.1002/pmic.200800110.
Staab JF, Bradway SD, Fidel PL, Sundstrom P: Adhesive and mammalian transglutaminase substrate properties of Candida albicans Hwp1. Science. 1999, 283: 1535-1538. 10.1126/science.283.5407.1535.
Uhl MA, Biery M, Craig N, Johnson AD: Haploinsufficiency-based large-scale forward genetic analysis of filamentous growth in the diploid human fungal pathogen C. albicans. EMBO J. 2003, 22: 2668-2678. 10.1093/emboj/cdg256.
Kaur R, Domergue R, Zupancic ML, Cormack BP: A yeast by any other name: Candida glabrata and its interaction with the host. Curr Opin Microbiol. 2005, 8: 378-384. 10.1016/j.mib.2005.06.012.
Naglik J, Albrecht A, Bader O, Hube B: Candida albicans proteinases and host/pathogen interactions. Cell Microbiol. 2004, 6: 915-926. 10.1111/j.1462-5822.2004.00439.x.
Hull CM, Raisner RM, Johnson AD: Evidence for mating of the "asexual" yeast Candida albicans in a mammalian host. Science. 2000, 289: 307-310. 10.1126/science.289.5477.307.
Magee BB, Magee PT: Induction of mating in Candida albican s by construction of MTLa and MTLα strains. Science. 2000, 289: 310-313. 10.1126/science.289.5477.310.
Tsong AE, Miller MG, Raisner RM, Johnson AD: Evolution of a combinatorial transcriptional circuit: a case study in yeasts. Cell. 2003, 115: 389-399. 10.1016/S0092-8674(03)00885-7.
Reedy JL, Floyd AM, Heitman J: Mechanistic plasticity of sexual reproduction and meiosis in the Candida pathogenic species complex. Curr Biol. 2009, 19: 891-899. 10.1016/j.cub.2009.04.058.
Heitman J, Kronstad JW, Taylor JW, Casselton LA: Sex in Fungi. 2007, Washington DC: ASM Press
Forche A, Alby K, Schaefer D, Johnson AD, Berman J, Bennett RJ: The parasexual cycle in Candida albicans provides an alternative pathway to meiosis for the formation of recombinant strains. PLoS Biol. 2008, 6: e110-10.1371/journal.pbio.0060110.
Logue ME, Wong S, Wolfe KH, Butler G: A genome sequence survey shows that the pathogenic yeast Candida parapsilosis has a defective MTLa1 allele at its mating type locus. Eukaryot Cell. 2005, 4: 1009-1017. 10.1128/EC.4.6.1009-1017.2005.
Acknowledgements
I thank Kevin Alby, Joseph Bliss, Marlowe Tessmer, and Racquel Sherwood for comments on the manuscript. RJB is supported by a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease award.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Bennett, R.J. A Candida-based view of fungal sex and pathogenesis. Genome Biol 10, 230 (2009). https://doi.org/10.1186/gb-2009-10-7-230
Published:
DOI: https://doi.org/10.1186/gb-2009-10-7-230