
Abstract
A large-scale enhancer detection screen was performed in the zebrafish using a retroviral vector carrying a basal promoter and a fluorescent protein reporter cassette. Analysis of insertional hotspots uncovered areas around developmental regulatory genes in which an insertion results in the same global expression pattern, irrespective of exact position. These areas coincide with vertebrate chromosomal segments containing identical gene order; a phenomenon known as conserved synteny and thought to be a vestige of evolution. Genomic comparative studies have found large numbers of highly conserved noncoding elements (HCNEs) spanning these and other loci. HCNEs are thought to act as transcriptional enhancers based on the finding that many of those that have been tested direct tissue specific expression in transient or transgenic assays. Although gene order in hox and other gene clusters has long been known to be conserved because of shared regulatory sequences or overlapping transcriptional units, the chromosomal areas found through insertional hotspots contain only one or a few developmental regulatory genes as well as phylogenetically unrelated genes. We have termed these regions genomic regulatory blocks (GRBs), and show that they underlie the phenomenon of conserved synteny through all sequenced vertebrate genomes. After teleost whole genome duplication, a subset of GRBs were retained in two copies, underwent degenerative changes compared with tetrapod loci that exist as single copy, and that therefore can be viewed as representing the ancestral form. We discuss these findings in light of evolution of vertebrate chromosomal architecture and the identification of human disease mutations.
Similar content being viewed by others


Introduction
Insertional mutagenesis screening in zebrafish is a powerful technique because the inserted transgene allows immediate isolation of the target sequence and thus identification of the mutated gene [1] (for review [2, 3]). Several efficient insertional agents are now available for the zebrafish and make it the model organism of choice for transgenic manipulation at the genome level [2]. With the genome sequences of many species available, multiple insertions can be mapped to chromosomal neighborhoods, and fine resolution of genomic architecture has become possible. In addition to the ever increasing collection of protein coding and RNA genes, gene expression patterns, and mutant phenotypes, comparative genomics has recently shed light on noncoding elements and vertebrate genome evolution. The availability of both efficient insertional technologies in zebrafish and mouse and finished genome sequences has expedited genetic screens significantly; they have made possible assays of gene regulation in the context of defined chromosomal areas in the living embryo [4].
Comparative studies using cross-species genome alignments have identified a large number of noncoding elements, exceeding - in total length - the amount of sequence coding for protein [5, 6]. These sequences are candidate regulatory regions that direct gene expression, and a number of them have been tested by transgenic or transient assays [6–10] using fluorescent proteins, which allow live visualization of gene activity [11]. Recently, Ellingsen and coworkers [12] devised a retrovirus-based insertional system using yellow fluorescent protein (YFP) under the control of the zebrafish gata2 promoter. More than 15,000 insertions were screened, and more than 1,000 transgenic lines have been established; thus far, 340 of these integrations have been mapped to the zebrafish genome [12] (Rinkwitz S, et al., unpublished data). In essence, this approach allows visualization of cis regulatory information at the insertion position as YFP expression. We review here a number of insertional hotspots across the loci of well known developmental regulatory genes, including some in which the inserted vector assumes the expression pattern of a neighboring gene rather than the one into which the insertion occurred, and we demonstrate - with the help of comparative genomics - that these areas identify a feature common to all vertebrate genomes [13]. These so-called genomic regulatory blocks (GRBs) are protected from evolutionary breakpoints, can be identified through establishing minimal conserved human/teleost conserved synteny, and are a useful tool for explaining human genetic disease resulting from position effect mutations.
Insertional screens in zebrafish
Although both mouse and zebrafish, among vertebrates, have been used for insertional screens [1, 14, 15], the latter to date is the only vertebrate species in which large numbers of random insertions have been made to generate transgenic lines that mimic expression patterns driven by endogenous enhancers [2, 12]. Both transposons and viral vectors have been used as insertional agents, the main difference being that although transposon vectors are much easier to handle, they yield lower numbers of insertions per germline [16, 17]. However, transposons are now being used by many laboratories, they are excellent vehicles for transgenesis [18], and will be vital for the large-scale testing of vertebrate cis regulatory elements.
Both retroviruses and transposons have distinct integration preferences (for review [19]). The retroviral vector used in our screen, namely the Moloney mouse leukemia virus (MLV), has been extensively tested for integration preference by identifying 903 nonselected insertion sites in human HeLa cells (because at the time only the human genome sequence was sufficiently annotated to allow unequivocal mapping of integrations) [20]. Wu and coworkers [20] found a significant bias of MLV to insert near to transcription start sites, but no obvious hotspots for integrations. The insertion preference for MLV in the zebrafish genome was recently found to be similar to that in the human genome (Burgess SM, personal communication). For the first round of mapping, using zebrafish genome release zv4, we were able to map 35 out of 95 insertions unambiguously [12]. With the release of zv6 (March 2006) we were able to map 95% of insertions or more.
In our screen, we tested an estimated 15,000 insertions in the zebrafish genome and found several loci that were markedly enriched for insertions; we found five insertions in a 159 kilobase (kb) interval upstream of sox11b [4, 12] (Rinkwitz S, et al., unpublished data), as compared with about 1.5 expected (1,600 megabases/15,000 insertions is about 1 insertion/100 kb). Likewise, we found four insertions in an interval of 150 kb around fgf8, and nine insertions in a 50 kb area around id1 [13], the latter number being 18 times greater than expected by chance. Thus, MLV has a distinct insertion preference [20], and insertion hotspots are found with high numbers of insertions, even when applying the stringent criteria proposed by Wu and coworkers [21].
Although they skew the overall outcome of an enhancer detection screen, hotspots allow us to assay how expression patterns change with respect to exact insertion position. In the cases listed above, all but one insertion (far upstream of fgf8) took on the global expression pattern of the developmental regulatory gene [13], suggesting that in these cases exact insertion position within a chromosomal domain is not critical for the (global) gene expression pattern, although detailed differences in expression were not assayed. Furthermore, there was a number of cases in which an insertion with a specific pattern had occurred into a gene neighboring the gene with that same pattern, and the gene into which the insertion had occurred exhibited a different pattern of expression, for example in the neighborhood of pax6.2 and rx3 [13]. There are different ways to determine which gene in the area is the target gene. In the simplest scenario, there is a single gene in the midst of a gene desert, and there is a good agreement between the transcriptional pattern of this gene, as determined by in situ hybridization, with the enhancer detection pattern, such as in the case of sox11b or id1. In other cases, the expression patterns of all genes in the area were determined by in situ hybridization, and only one - that of the developmental regulator - agreed with the pattern of the transgene, whereas neighboring genes often had near ubiquitous expression patterns. Examples of these are pax6.2, fgf8, and rx3, mentioned above.
Both the analysis of hotspots and the cases of insertions into neighboring genes paint a picture that is substantially different from what has been reported from the large-scale Drosophila enhancer detection screens (for instance [22]), namely that it is the nearest gene relative to where the insertion has landed whose expression pattern is mimicked. Instead, there appear to be substantial numbers of large chromosomal segments around developmental regulatory genes in the zebrafish (and, by extension, vertebrate) genome. Enhancer detection insertions into such segments exhibit expression patterns resembling that of the regulatory gene, often independent of precise insertion location [12, 13]. All of the loci listed above turn out to have extended regions containing highly conserved noncoding elements (HCNEs) as well as extensive conserved synteny around them. To explain the above findings, we should like to turn first to knowledge gained about the chromosomal architecture of vertebrate genomes through recent comparative genomic analyses, multispecies alignments, and transgenesis with individual regulatory elements.
Genome-wide discovery of highly conserved noncoding elements
Detection of HCNEs in vertebrate genomes is relatively straightforward compared with procedures required to detect other known functional elements. To detect them with high reliability, one requires at least two aligned genomic sequences from suitably distant species and a reasonably complete set of transcribed or coding parts of the sequence in at least one of them. At the time of this writing, whole genome alignments are available for a collection of 17 vertebrate species [23], which allows the extraction of highly conserved regions by different methods, from the simple sliding window approach to more sophisticated ones [24]. The transcriptome coverage of the human and mouse genomes is becoming deep, and their annotation allows for simple separation of transcribed from nontranscribed, or coding from noncoding sequences. Transcriptomes of other organisms are not as complete, but they can be used in combination with the more complete ones to ensure that there is as thorough filtering of coding regions as possible. There are several web-based tools that can be used for HCNE extraction, including the following: the ECR browser, which can be used to extract HCNEs from whole genome or user-submitted alignments; the UCSC browser, which provides the PhastCons tracks that can be filtered against transcript; and the Table browser, which allows detection of elements overlapping coding sequence. Methods for large-scale extraction of HCNEs were summarized by Bejerano and coworkers [25].
In 2004 and 2005 several groups reported their analyses of genome-wide distribution of conserved noncoding elements [5, 6, 26, 27]. The current terminological confusion about those elements dating from that period resulted in a number of similar terms and an alphabet soup of abbreviations, which we summarize in Table 1. The first of the studies [26] looked at noncoding regions with extreme conservation (≥200 identical base pairs [bp]) between human and mouse.
The investigators retrieved 481 segments, which they divided into two categories: transcribed ultraconserved regions (type I UCR; mostly in 3' untranslated region of protein coding genes) and nontranscribed ultraconserved regions (type II UCR; intronic and intergenic). For type II UCRs they demonstrated a high tendency to co-localize with genes for developmental transcription factors. The latter has been observed by other research groups as well. By using a lower conservation threshold between human and mouse, as well as additional evidence for conservation in fish (fugu), Sandelin and coworkers [5] retrieved 3,583 conserved non-coding elements, which enabled them to paint a clear picture of genomic organization of those elements - especially the tendency for clusters of HCNEs to span megabase-size regions centered on their target genes. The same was shown by Woolfe and coworkers [6] by using 1,373 regions extracted from a direct comparison of the genomes of human and fish. The same report also provided the first systematic experimental evidence of enhancer activity for a subset of these elements in zebrafish (see below).
Over-representation analysis of gene ontology terms or protein domains associated with genes that co-localize with clusters of HCNEs exhibits a high enrichment for genes that encode transcription factors involved in embryonic development and tissue differentiation. A smaller but possibly important group of genes spanned by clusters of HCNEs are the nontranscription factor genes involved in neuronal specialization and growth, including axon guidance [28], or, for instance, genes in the hedgehog or fgf pathways [6]. Because embryonic development and axonal guidance are probably among the processes with the most complex spatiotemporal pattern of gene activity, it seems plausible that the genes controlling them should have a large set of regulatory inputs necessary for achieving the appropriate complexity and precision.
Recognition and testing of individual highly conserved noncoding elements
There is mounting evidence for the regulatory role of HCNEs. Soon after their genome wide discovery, it was realized that a significant number of previously characterized developmental enhancers overlap with HCNEs. Generally, there are several methods to test cis regulatory activity of HCNEs, and they do so at different levels of fidelity. For instance, the transient method devised by Muller and coworkers [29] (in which a plasmid carrying the HCNE plus basal promoter-reporter gene is injected into fertilized eggs and regulatory activity assayed 1 day later) is rapid, but in some cases it leads to identification of expression domains not seen with the endogenous gene (for example [6]). A more advanced but more time consuming approach is to generate transgenes in zebrafish, mice, or Xenopus [7, 10, 30], and these are probably the methods of choice for future large-scale approaches to evaluating HCNE function.
Göttgens and coworkers [31] reported the discovery of enhancers that are highly conserved between human and mouse, and some in chicken. One of those enhancers was situated 23 kb upstream of the mouse Scl gene and was shown to drive neural expression of a reporter gene in a transgenic Xenopus assay.
Bagheri-Fam and coworkers [32] identified several such elements conserved between human and fugu near the SOX9 gene, of which they considered at least three candidate enhancers. These are often broken off from their target genes in the translocation breakpoints present in patients with campomelic dysplasia, which is a skeletal malformation syndrome with XY sex reversal - clearly a developmental disorder (see below).
By studying conserved noncoding elements in a gene desert next to the DACH/Dach gene, Nobrega and coworkers [33] were the first to show that a functional long-range enhancer can reside more than a megabase away from its target gene. Milewski and coworkers [34] isolated two HCNEs upstream of the mouse Pax3 gene as enhancers driving Pax3 expression in neural crest. Pax3 itself is expressed in a number of other embryonic structures, indicating that non-neural crest regulatory inputs are located elsewhere in the regulatory sequence. Interestingly, the investigators characterized an apparently functional Tead2 binding site, which is immediately adjacent to one of the HCNEs, but is not itself highly conserved across mammals. This again begs the question of whether the conservation is essential for the transcription factor binding properties of those enhancers.
Kimura-Yoshida and coworkers [35] convincingly demonstrated that separate, evolutionarily conserved enhancers of the fugu otx2 gene drive spatiotemporally distinct subdomains of otx2 expression during head specification in mouse and zebrafish. This was a convincing demonstration that different HCNEs serve as separate regulatory inputs for the target gene(s), implying that the genes with the highest number of HCNEs were those with most complex spatiotemporal expression patterns. It is therefore not surprising that those genes include transcription factors involved in the acquisition of unique neuronal identities. Similarly, Uemura and coworkers [10] demonstrated that, near the zebrafish isl1 and the mouse Isl1 genes, there are separate enhancers that specify expression of the gene in sensory versus motor neuron populations. Furthermore, these enhancers (more than 300 kb away from the coding region in human and mouse) diversified in their specificities between mammals and teleosts. Inoue and coworkers [9] tested individual HCNEs around the zebrafish fgf8 gene and could assign specific expression patterns to most of them.
Woolfe and coworkers [6] were the first to test a subset of HCNEs in the light of their genome-wide organization. Using the transient plasmid assay reported by Muller and coworkers [29], they studied the enhancer activity a set of 25 HCNEs around four genes already known to be regulated by this kind of element (SOX21, PAX6, HLXB9, and SHH); 23 exhibited enhancer activity, although some of them in areas where the respective regulatory gene is not expressed. Their general conclusion was also that separate HCNEs drive spatiotemporally distinct (but often overlapping) components of the overall expression pattern of the target gene. A similar approach was used by Shin and coworkers [36], who drew similar conclusions. Likewise, a large number of HCNEs from the vertebrate iroquois clusters were tested in zebrafish and Xenopus, and the majority of them were shown to act as specific enhancers [7]. The largest scale functional screen undertaken thus far is that reported by Pennacchio and coworkers [37], who tested 167 HCNEs in a mouse transgenic enhancer assay. Seventy-five (43%) of the tested elements exhibited enhancer activity in a wide spectrum of embryonic structures, and most were expressed in parts of the developing nervous system.
Turnover of long-range regulatory elements
Even though they exhibit extraordinary levels of conservation, the HCNEs also undergo evolutionary divergence as a result of accumulation of mutations whose extent, for the most part, corresponds to the evolutionary distance of the organisms in a canonical tree of life [24]. Drake and coworkers [38] investigated the intraspecies and interspecies variation in HCNEs and demonstrated that they are undergoing purifying selection - indicating the functional importance of conserving their sequence - rather than the alternative explanation of being mutational cold spots. However, HCNEs do change over evolutionary time. For instance, in the case of id1 mentioned above, only two HCNEs could be discerned when human and zebrafish genomes were compared, whereas multiple elements were found when comparing two teleost genomes (see [13]). Thus, regulatory elements tend to become 'invisible', yet they retain function with increasing evolutionary distance [30]. There are a couple of interesting cases dealing with two specific branches in the tree of life that have recently been observed.
The first of these cases is the increased divergence in teleosts relative to other vertebrates. Teleosts (bony fish) appear to exhibit greater divergence of HCNEs from those of tetrapod vertebrates than does the more distant elephant shark, which (at the time of writing) is the only cartilaginous fish with sequenced genome, albeit at low coverage [1.4×], and is an outgroup species to all other vertebrates with sequenced genomes [39, 40]. A possible explanation is the fact that teleosts underwent an additional whole genome duplication early in the lineage [41–43]. Many of the HCNE arrays and the associated target genes survived in two copies [13].
The second case is that of the increased divergence in hominids versus other mammals. Human and chimpanzee were recently shown to have undergone accelerated evolution at a subset of their HCNEs [44]. The subset was enriched for genes involved in neuronal cell adhesion, probably reflecting the accelerated evolution of regulatory elements responsible for the differentiation of hominid specific brain circuitry involved in human-specific cognitive traits.
One would expect to see little sequence variation within species inside UCRs, given their high conservation. This was indeed observed; Bejerano and coworkers [26] reported that the regions in question are devoid of validated single nucleotide polymorphisms. On the other hand, there is some striking evidence to the contrary. Chen and colleagues [45] reported an analysis of a small set of single nucleotide polymorphisms found in the ultraconserved regions by Bejerano and coworkers [26], which by their classification are 100% identical between human and mouse across 200 bp or more. They also demonstrated a surprising number of fixed differences between human and chimpanzee in those regions. It appears, at least in those cases, that there exists only a weak purifying selection of those elements - weaker than that on essential protein-coding sequences - which seemingly contradicts the high observed level of conservation between human and rodents. Another unexpected result was the demonstration by Fisher and coworkers [30] that two equivalent regions of a genomic sequence in human and zebrafish are both able to act as an enhancer in zebrafish, even though sequence level similarity was lost. (It should be noted that the RET gene, the apparent target of the nonconserved enhancer region from the report by Fisher and coworkers [30], encodes a tyrosine kinase, which does not correspond to the molecular function of 'typical' target genes of HCNE arrays [developmental transcription factors, micro-RNAs, morphogens, and neuronal connection regulators]). Thus, although many more genes than the loci enriched for HCNEs [5] have specific expression patterns (and, by extension, specific regulatory elements), in many the sequence similarity is not discernible over large evolutionary distances, such as that between human and teleost.
This leads to the following questions. First, are there many more genes whose loci are spanned by arrays of long range enhancers, but that have diverged beyond the ability of sequence alignment programs to match them? Second, is the need to conserve regulatory function the only mechanism (or indeed even a significant one) underlying the strong evolutionary pressure to keep these genes conserved?
Distance of highly conserved noncoding elements to the genes they regulate
An interesting study conducted by Sun and coworkers [46] showed that the spacing between HCNEs in mammals is much more conserved than the spacing between other genomic elements. From the study, it is not obvious whether this is the consequence of the existence of other functional, nonconserved elements between the HCNEs (which might be in a transition toward the state of nonconserved, but functionally equivalent to those described by Fisher and coworkers [30] for the RET gene) or, alternatively, that the distance from the target gene(s) itself is a functional determinant with purifying selection acting upon it. The latter mechanism has also previously been suggested to account for the observation that many HCNE arrays correspond to transposon-free regions (TFRs) in mammalian genomes [47], but Sun and coworkers show that the relationship between conservation of spacing between HCNEs and TFRs is far from straightforward.
Sun and coworkers also demonstrated that the spacing conservation between humans and nonmammalian vertebrates exhibited a bimodal distribution, in which one subset of HCNE spacings was also highly conserved, whereas the other peak of the distribution corresponded more to the ratio of total genome size of the nonmammal species to the human genome. In the cases studied by Kikuta and coworkers [13] there appear to exist cases of both types of HCNE arrays; the 'conserved size' ones between human and zebrafish appear to contain most of the protein coding genes, whereas in some cases, in which the bystander genes (see below) in zebrafish were lost (as in one copy of the OTP HCNE array), there has been a significant contraction of the corresponding HCNE array size. Additionally, some regions corresponding to TFRs in mammals as well as teleosts were found in zebrafish to tolerate viral insertions with no apparent phenotypic effect [13].
Mechanism of enhancer activity of highly conserved noncoding elements
We still have no adequate explanation for the mechanism by which HCNEs act as enhancers, and of the origin of the evolutionary pressure for keeping them conserved across large phylogenetic distances. A typical view of an enhancer is as a DNA region that contains one or more context-specific transcription factor binding sites. However, virtually all known vertebrate transcription factors have degenerate binding specificities, and bind stretches of 5 to (in extreme cases) 30 bp. Clusters of binding sites (cis regulatory modules) are never as long as the longest HCNEs, and the binding sites in them are practically never so tightly strung together as not to allow for any sequence variability between them. A genome-wide study of the transcription factor binding site content of HCNEs [48] showed that the sequence signatures are indeed associated with tissue-specific signatures in a statistically significant manner; however, the specificity and sensitivity of their models is still largely inadequate for functional characterization of individual candidate enhancers.
Other biological mechanisms are emerging for the action of individual enhancers. For example, a HCNE from the Dlx5-Dlx6 bigene cluster was recently shown to be transcribed into a noncoding RNA (Evf-2). Evf-2 functions as a transcriptional co-activator of the Dlx2 gene, suggesting a mechanism for regulatory crosstalk of different Dlx clusters [49]. However, the evidence for the transcription of other HCNEs is rather scarce, and it is probable that the Evf-2 mechanism is but one of a number of mechanisms whereby HCNEs regulate their target genes.
Likewise, the evolutionary origin of HCNEs does not appear to be exclusively of one kind. Bejerano and coworkers [50] presented the case of one HCNE that originated from a short interspersed repeat element (SINE) at the base of the tetrapod lineage. One copy of that element was shown to serve as enhancer for the ISL1 gene, whereas several others encode alternatively spliced exons of several other genes. However, most other HCNEs are present in only one copy per haploid tetrapod genome [5, 6], making their relationship with repeats and transposable elements either very ancient or very unlikely. A recent report of the existence of parallel sets of HCNEs in the Caenorhabditis genus might suggest that those elements occurred early in the evolutionary history of Metazoa (or even earlier) and continue to co-evolve with their target genes and, in the case of GRBs, functionally unrelated bystander genes in the GRBs. Whole-genome duplication was shown to be an evolutionary avenue for bystander genes to escape the lock-in into the GRB. It remains to be seen whether this opportunity was used frequently in the evolution of metazoan genomes, as well as whether tandem duplications of individual chromosomal loci offer the same escape mechanism to bystander genes.
Gene complexes and tandem duplications of regulatory genes
In contrast to single regulatory genes with extended regulatory regions, some genome regions encompass two or more phylogenetically related developmental regulatory genes within relatively short distances of each other. In contrast to the GRBs described above, these clustered genes have long been thought to be kept together because sequences essential for their proper (co-)regulation are harbored within their compact intergenic regions or outside of these clusters. The best researched among these complexes are the Hox clusters. Mammalian genomes contain four Hox clusters (A, B, C, and D) that originated from a single ancestral cluster in chordates through two rounds of whole genome duplication [51, 52]. An extra round of genome duplication in the teleost lineage created a total of eight hox clusters in zebrafish [53, 54], one of which has been reduced to a single micro-RNA by loss of all of its Hox genes after duplication [55]. The 11 genes in the human HoxA cluster span only about 109 kb, densely populated by protein coding and gene regulatory sequences.
The temporal and spatial collinear activation of the hox genes along the body axis of the developing embryo was for a long time seen as the primary force maintaining their chromosomal order during evolution [56, 57]. However, the precise mechanisms keeping hox genes together are still unclear, and several instances of 'broken' hox clusters have recently been described in invertebrate genomes [58–60]. In the fruit fly Drosophila melanogaster, hox gene regulation can function outside of the context of an intact gene complex [61]. Thus, there appears to be no absolute requirement for hox cluster continuity in invertebrates, and it was recently suggested that although evolutionary breakups within fly hox clusters can be demonstrated, the extremely compact intergenic regions between the genes make these breakups exceedingly rare [62]. In vertebrates the identification of a global control region far upstream of the mouse HoxD cluster, which controls transcription of six genes (namely Lnp, Evx2, and Hoxd13 to Hoxd10) in the developing digits and central nervous system, may provide an explanation for Hox cluster maintenance [63]. Sharing of enhancers also has an important role in ensuring overlapping activity of genes [64, 65]. In contrast to the long-range control elements outside clusters that form 'regulatory landscapes' [66], enhancers within the Hox cluster appear to act on the gene closest by in a competitive manner [67, 68]. Zebrafish hoxb3a and hoxb4a share an exon with a 5' adjacent master promoter that controls a 37 kb transcription unit including all exons of both genes [8] and further the micro-RNA miR-10b [69]. These structural features and regulatory mechanisms appear poised to ensure overlapping expression and tissue specific activity of both hox genes as well as the micro-RNA gene and further contribute to the maintenance of precisely clustered gene order.
The basis of conserved synteny in hox clusters is likely to be complicated, with no single explanation for cluster maintenance. Rather, a number of different evolutionary constraints, such as proximity of genes to each other, sharing of exon sequences, and global control regions, appear to act simultaneously on these enigmatic gene clusters. Nevertheless, that hox clusters are actively kept together can be gleaned from their divergence in gene content after teleost whole genome duplication, or even breakup, as has recently been reported for a parahox cluster as well [70].
Gene clusters and bigene arrays probably resulted from tandem duplication events during evolution. In these events, short segments of the chromosomes were duplicated by unequal crossing over during meiosis, causing not only protein coding sequences but also transcription factor binding sites to be present twice. Newly duplicated genes and related regulatory sequences are free to evolve, and they may acquire a selective function (subfunctionalization; retrograde evolution theory) or they may take on a new task (neofunctionalization; patchwork evolution theory) [71], giving rise to morphologic diversity in vertebrates [72]. Increasing the number of target genes for transcription factors by gene duplication played a key role in establishing regulatory networks [73]. Bigene arrays are pairs of structurally highly similar genes that resulted from tandem duplication and that are located in close proximity to each other, as is observed for members of the Dlx [74, 75], Zic [76], Hmx [77], and Myf [78] gene families. These genes are often co-expressed at specific sites in the developing embryo and may also act redundantly. The tandem duplication of a single ancestral Dlx gene probably occurred in early chordates, because a single bigene cluster was identified in the urochordate Ciona intestinalis [79]. Genome duplications in the vertebrate lineage have then led to three bigene clusters, Dlx1-Dlx2, Dlx3-Dlx7 (Dlx7 later renamed to Dlx4), and Dlx5-Dlx6, in the mammalian genome [80]. In zebrafish, because of a further genome duplication followed by gene loss events, two clusters (dlx1a/2a and dlx5a/6a) and a further three single genes (dlx2b, dlx3b, dlx4a) have thus far been described. Fundamental structural and regulatory principles became obvious by analyzing the mouse Dlx3-Dlx7 bigene cluster [81]. Despite some differential expression, Dlx3 and Dlx7 are both co-expressed in the visceral arches and the developing limbs. Five conserved noncoding sequences (>80% human-mouse identity) were identified within the 17 kb intergenic sequence. That some of these enhancers are shared and require clustering of the genes was shown in transgenic Dlx3-lacZ mice, in which visceral arch expression of Dlx3 was lost when sequences distant from the gene (close to Dlx7) are missing [81, 82].
Long-range gene regulation and synteny
Other than gene clusters, areas that are spanned by multiple HCNEs often contain only a single developmental regulatory gene, the target gene, and also frequently contain additional genes, whose molecular function and expression pattern are unrelated to those of the presumed target gene. This would imply that they somehow do not respond to the enhancers in their vicinity, even when they are physically closer to them than the target gene is. Limited functional evidence for this arrangement exists. For instance, a sonic hedgehog regulatory element whose mutation causes pre-axial polydactyly has been shown to reside in an intron of the functionally unrelated neighboring gene LMBR1 [83]. This is further corroborated by the analysis of zebrafish enhancer detection insertion sites [13], in which a number of enhancer detection insertions often gave the expression of a target gene of a GRB, even though their integration sites were inside or beyond genes with unrelated function and expression patterns. The need to preserve the integrity of GRBs is indicated by, thus far, a few human position effect mutations (see below), but it is expected that their numbers will rise, given the common occurrence of GRBs in all vertebrate genomes (Table 2).
Genomic regulatory blocks: vertebrate chromosomes are subdivided into functional segments
It was noted early that the HCNEs cluster around and within their target genes, but it is not unusual for the cluster to spill into the introns of neighboring genes and beyond [5, 28]. In many instances the neighboring genes have expression patterns that are less specific than those of the gene apparently targeted by the HCNE cluster. Those neighboring genes are kept in synteny with the target genes much more often than can be expected to occur at random. Indeed, in the case of HCNEs conserved across all vertebrates, it has been shown that the longest conserved synteny blocks (with the notable exception of very large genes) are almost all defined by clusters of HCNEs, and contain the presumptive target gene as well as the surrounding genes they inhabit. If those genes are indeed functionally unrelated to the target gene, then the only reason for maintaining synteny is that the HCNEs they harbor must remain within the reach of their target genes.
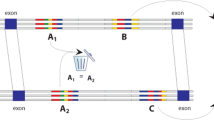
Kikuta and coworkers [13] used the whole genome duplication event in teleosts to show that this is indeed the case, by clearly demonstrating several cases in which there was a separation of the HCNE cluster from the gene that contains it after the duplication of the locus. In these cases the HCNEs, without exception, remained next to one of the two duplicates of the target gene, whereas the neighboring gene survived in one copy or sometimes 'broke free' from the synteny with the target gene by chromosomal rearrangement and lost its HCNEs. (Figure 1).

Human GRB encompassing PAX6 and bystander genes (middle track) and the two duplicated zebrafish loci. The human locus spans >1 megabase (Mb) and contains the PAX6 target gene (in red) and five bystander genes (green). Highly conserved noncoding elements (HCNEs) conserved from human to zebrafish are denoted as blue ovals. Note that some HCNEs are conserved in both zebrafish loci, but that most are only conserved in one locus, leading to subfunctionalization. Bystander genes are usually conserved in one copy only. In the upper part of the figure, two insertions in zebrafish pax6 genomic regulatory blocks (GRBs) take on the correct expression pattern of the orthologs, although CLGY825 is 120 kb downstream of pax6.1, next to the bystander zcsl3, whereas CLGY954 is inserted inside the bystander gene elp4, downstream of pax6.2. Note also the complementarity of reporter expression. Although pax6.1 is strong in diencephalon and hindbrain, pax6.2 retains stronger expression in retina, pineal, and spinal cord. For more details, see Kikuta and coworkers [13].
Therefore, the chromosomes of vertebrates contain territories of long-range regulation defined by HCNEs that we termed 'genomic regulatory blocks' (GRBs). These GRBs contain 'target genes' (genes that respond to long-range regulation by the conserved elements) and 'bystander genes', which are functionally unrelated, frequently broadly expressed, and kept in synteny by the HCNEs contained in their introns or beyond. Interestingly, the whole genome duplication in teleosts appears to have opened a way for some of the bystander genes to escape the synteny with the target gene (for an example, see Figure 2). In addition, as mentioned above, the teleost genome duplication appears also to have relaxed the sequence conservation requirements of HCNEs. In many cases regulatory elements, although still functional [30], have mutated beyond recognition, and only the conserved order of coding sequence indicates that these regions are under purifying selection. It remains to be seen whether there is any evidence that this mechanism was also at work in previous whole-genome duplications in Metazoa and thus helped shape the evolution of animal body plan.

The fate of duplicated teleost GRBs. If the target gene (red) is retained in both copies, then the two loci/genomic regulatory blocks (GRBs) undergo degenerative changes. This occurs either through chromosomal breaks (left), removing the bystander gene, which may land elsewhere in the genome and which loses the highly conserved noncoding elements (HCNEs); or by loss, through neutral evolution, of the bystander gene and some HCNEs (which are both retained in the intact other copy of the GRB).
The possible future application of genomic regulatory blocks in the study of human disease
Many of the target genes of GRBs are known to be involved in different types of cancer or genetic malformations. A number of examples are listed in Table 2. Although bona fide position effect mutations in humans have been found only in a subset of these disease loci (for instance, MAF, SOX9, POU3F4, SHH, PAX6, and possibly DLX5/DLX6), Table 2 shows that the probable regulatory domains of these disease genes, as estimated through determining minimal conserved gene order by human-chicken and human-teleost alignments, are very large. For 20 disease genes, the combined length of conserved gene order (and the extent of the underlying regulatory domains) covers 33 megabases when comparing human and teleost genomes, which is equivalent to 1% of the human genome. The same characteristic is evident for genes in which position effect breakpoints have been identified. Many loci of this list contain large gene deserts inhabited by multiple HCNEs, and it appears probable that position effect mutations will eventually be found in most if not all of them. Virtually all of the human position effect mutations found in these regions thus far are chromosomal rearrangements rather than point mutations [84–88], with the notable exception of a long-range enhancer of the human SHH gene [83]. Ahituv and coworkers [84] identified more than 2,100 blocks of conserved synteny between human and chick. They found that slightly less than half of the human genome aligns with the chicken genome, and 25% with the frog genome, suggesting that large parts of the vertebrate genome are located in GRBs. These authors also noted a strong tendency of gene deserts to be located in blocks of conserved synteny. A few lesions in bystander genes have already been identified and recognized as position effect mutations in the human genome (see Table 2). The year 2007 has already seen the publication of a large number of whole genome association studies of common single nucleotide polymorphisms with common diseases, such as prostate cancer, type 2 diabetes, obesity, and heart disease. It will be interesting to see the extent to which the concept of GRBs will aid in the identification of the underlying genetic defects. The existence of GRBs around key regulatory target genes that are active during vertebrate embryogenesis, and possibly into adulthood, suggests that the identification of human position effect mutations has only just begun.
Conclusion
Much of what has been known for three decades as 'junk DNA' can now be seen as containing precisely ordered collinear regulatory elements around their target genes. The fact that whole chromosomal segments have been preserved over hundreds of millions of years throughout all vertebrates suggests that selective pressure has acted upon them and that their architecture is probably important in shaping the vertebrate body plan. Only after the whole genome duplication that occurred in the teleost lineage about 250 million years ago could the order into which genomic regulatory blocks have evolved in the tetrapods degenerate to a degree without a loss of fitness.
References
Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N: Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci USA. 2004, 101: 12792-12797. 10.1073/pnas.0403929101.
Amsterdam A, Becker TS: Transgenes as screening tools to probe and manipulate the zebrafish genome. Dev Dyn. 2005, 234: 255-268. 10.1002/dvdy.20541.
Amsterdam A, Hopkins N: Mutagenesis strategies in zebrafish for identifying genes involved in development and disease. Trends Genet. 2006, 22: 473-478. 10.1016/j.tig.2006.06.011.
Gomez-Skarmeta JL, Lenhard B, Becker TS: New technologies, new findings, and new concepts in the study of vertebrate cis-regulatory sequences. Dev Dyn. 2006, 235: 870-885. 10.1002/dvdy.20659.
Sandelin A, Bailey P, Bruce S, Engstrom PG, Klos JM, Wasserman WW, Ericson J, Lenhard B: Arrays of ultraconserved non-coding regions span the loci of key developmental genes in vertebrate genomes. BMC Genomics. 2004, 5: 99-10.1186/1471-2164-5-99.
Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, Smith SF, North P, Callaway H, Kelly K, et al: Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005, 3: e7-10.1371/journal.pbio.0030007.
de la Calle-Mustienes E, Feijoo CG, Manzanares M, Tena JJ, Rodriguez-Seguel E, Letizia A, Allende ML, Gomez-Skarmeta JL: A functional survey of the enhancer activity of conserved non-coding sequences from vertebrate Iroquois cluster gene deserts. Genome Res. 2005, 15: 1061-1072. 10.1101/gr.4004805.
Hadrys T, Punnamoottil B, Pieper M, Kikuta H, Pezeron G, Becker TS, Prince V, Baker R, Rinkwitz S: Conserved co-regulation and promoter sharing of hoxb3a and hoxb4a in zebrafish. Dev Biol. 2006, 297: 26-43. 10.1016/j.ydbio.2006.04.446.
Inoue F, Nagayoshi S, Ota S, Islam ME, Tonou-Fujimori N, Odaira Y, Kawakami K, Yamasu K: Genomic organization, alternative splicing, and multiple regulatory regions of the zebrafish fgf8 gene. Dev Growth Differ. 2006, 48: 447-462. 10.1111/j.1440-169X.2006.00882.x.
Uemura O, Okada Y, Ando H, Guedj M, Higashijima S, Shimazaki T, Chino N, Okano H, Okamoto H: Comparative functional genomics revealed conservation and diversification of three enhancers of the isl1 gene for motor and sensory neuron-specific expression. Dev Biol. 2005, 278: 587-606. 10.1016/j.ydbio.2004.11.031.
Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC: Green fluorescent protein as a marker for gene expression. Science. 1994, 263: 802-805. 10.1126/science.8303295.
Ellingsen S, Laplante MA, Konig M, Kikuta H, Furmanek T, Hoivik EA, Becker TS: Large-scale enhancer detection in the zebrafish genome. Development. 2005, 132: 3799-3811. 10.1242/dev.01951.
Kikuta H, Laplante M, Navratilova P, Komisarczuk AZ, Engstrom PG, Fredman D, Akalin A, Caccamo M, Sealy I, Howe K, et al: Genomic regulatory blocks encompass multiple neighboring genes and maintain conserved synteny in vertebrates. Genome Res. 2007, 17: 545-555. 10.1101/gr.6086307.
Keng VW, Yae K, Hayakawa T, Mizuno S, Uno Y, Yusa K, Kokubu C, Kinoshita T, Akagi K, Jenkins NA, et al: Region-specific saturation germline mutagenesis in mice using the Sleeping Beauty transposon system. Nat Methods. 2005, 2: 763-769. 10.1038/nmeth795.
Starr TK, Largaespada DA: Cancer gene discovery using the Sleeping Beauty transposon. Cell Cycle. 2005, 4: 1744-1748.
Balciunas D, Davidson AE, Sivasubbu S, Hermanson SB, Welle Z, Ekker SC: Enhancer trapping in zebrafish using the Sleeping Beauty transposon. BMC Genomics. 2004, 5: 62-10.1186/1471-2164-5-62.
Parinov S, Kondrichin I, Korzh V, Emelyanov A: Tol2 transposon-mediated enhancer trap to identify developmentally regulated zebrafish genes in vivo. Dev Dyn. 2004, 231: 449-459. 10.1002/dvdy.20157.
Kawakami K: Transposon tools and methods in zebrafish. Dev Dyn. 2005, 234: 244-254. 10.1002/dvdy.20516.
Wu X, Burgess SM: Integration target site selection for retroviruses and transposable elements. Cell Mol Life Sci. 2004, 61: 2588-2596. 10.1007/s00018-004-4206-9.
Wu X, Li Y, Crise B, Burgess SM: Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003, 300: 1749-1751. 10.1126/science.1083413.
Wu X, Luke BT, Burgess SM: Redefining the common insertion site. Virology. 2006, 344: 292-295. 10.1016/j.virol.2005.08.047.
Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, et al: The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004, 167: 761-781. 10.1534/genetics.104.026427.
Kuhn RM, Karolchik D, Zweig AS, Trumbower H, Thomas DJ, Thakkapallayil A, Sugnet CW, Stanke M, Smith KE, Siepel A, et al: The UCSC genome browser database: update 2007. Nucleic Acids Res. 2007, D668-D673. 10.1093/nar/gkl928. 35 Database
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al: Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15: 1034-1050. 10.1101/gr.3715005.
Bejerano G, Siepel AC, Kent WJ, Haussler D: Computational screening of conserved genomic DNA in search of functional noncoding elements. Nat Methods. 2005, 2: 535-545. 10.1038/nmeth0705-535.
Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D: Ultraconserved elements in the human genome. Science. 2004, 304: 1321-1325. 10.1126/science.1098119.
Plessy C, Dickmeis T, Chalmel F, Strahle U: Enhancer sequence conservation between vertebrates is favoured in developmental regulator genes. Trends Genet. 2005, 21: 207-210. 10.1016/j.tig.2005.02.006.
Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC, et al: Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005, 438: 803-819. 10.1038/nature04338.
Muller F, Chang B, Albert S, Fischer N, Tora L, Strahle U: Intronic enhancers control expression of zebrafish sonic hedgehog in floor plate and notochord. Development. 1999, 126: 2103-2116.
Fisher S, Grice EA, Vinton RM, Bessling SL, McCallion AS: Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science. 2006, 312: 276-279. 10.1126/science.1124070.
Gottgens B, Barton LM, Gilbert JG, Bench AJ, Sanchez MJ, Bahn S, Mistry S, Grafham D, McMurray A, Vaudin M, et al: Analysis of vertebrate SCL loci identifies conserved enhancers. Nat Biotechnol. 2000, 18: 181-186. 10.1038/72635.
Bagheri-Fam S, Ferraz C, Demaille J, Scherer G, Pfeifer D: Comparative genomics of the SOX9 region in human and Fugu rubripes: conservation of short regulatory sequence elements within large intergenic regions. Genomics. 2001, 78: 73-82. 10.1006/geno.2001.6648.
Nobrega MA, Ovcharenko I, Afzal V, Rubin EM: Scanning human gene deserts for long-range enhancers. Science. 2003, 302: 413-10.1126/science.1088328.
Milewski RC, Chi NC, Li J, Brown C, Lu MM, Epstein JA: Identification of minimal enhancer elements sufficient for Pax3 expression in neural crest and implication of Tead2 as a regulator ofPax3. Development. 2004, 131: 829-837. 10.1242/dev.00975.
Kimura-Yoshida C, Kitajima K, Oda-Ishii I, Tian E, Suzuki M, Yamamoto M, Suzuki T, Kobayashi M, Aizawa S, Matsuo I: Characterization of the pufferfish Otx2 cis-regulators reveals evolutionarily conserved genetic mechanisms for vertebrate head specification. Development. 2004, 131: 57-71. 10.1242/dev.00877.
Shin JT, Priest JR, Ovcharenko I, Ronco A, Moore RK, Burns CG, MacRae CA: Human-zebrafish non-coding conserved elements act in vivo to regulate transcription. Nucleic Acids Res. 2005, 33: 5437-5445. 10.1093/nar/gki853.
Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al: In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006, 444: 499-502. 10.1038/nature05295.
Drake JA, Bird C, Nemesh J, Thomas DJ, Newton-Cheh C, Reymond A, Excoffier L, Attar H, Antonarakis SE, Dermitzakis ET, et al: Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nat Genet. 2006, 38: 223-227. 10.1038/ng1710.
Venkatesh B, Kirkness EF, Loh YH, Halpern AL, Lee AP, Johnson J, Dandona N, Viswanathan LD, Tay A, Venter JC, et al: Ancient non-coding elements conserved in the human genome. Science. 2006, 314: 1892-10.1126/science.1130708.
Venkatesh B, Kirkness EF, Loh YH, Halpern AL, Lee AP, Johnson J, Dandona N, Viswanathan LD, Tay A, Venter JC, et al: Survey sequencing and comparative analysis of the elephant shark (Callorhinchus milii) genome. PLoS Biol. 2007, 5: e101-10.1371/journal.pbio.0050101.
Postlethwait JH, Yan YL, Gates MA, Horne S, Amores A, Brownlie A, Donovan A, Egan ES, Force A, Gong Z, et al: Vertebrate genome evolution and the zebrafish gene map. Nat Genet. 1998, 18: 345-349. 10.1038/ng0498-345.
Woods IG, Kelly PD, Chu F, Ngo-Hazelett P, Yan YL, Huang H, Postlethwait JH, Talbot WS: A comparative map of the zebrafish genome. Genome Res. 2000, 10: 1903-1914. 10.1101/gr.10.12.1903.
Jaillon O, Aury JM, Brunet F, Petit JL, Stange-Thomann N, Mauceli E, Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A, et al: Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004, 431: 946-957. 10.1038/nature03025.
Prabhakar S, Poulin F, Shoukry M, Afzal V, Rubin EM, Couronne O, Pennacchio LA: Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res. 2006, 16: 855-863. 10.1101/gr.4717506.
Chen CT, Wang JC, Cohen BA: The strength of selection on ultraconserved elements in the human genome. Am J Hum Genet. 2007, 80: 692-704. 10.1086/513149.
Sun H, Skogerbo G, Chen R: Conserved distances between vertebrate highly conserved elements. Hum Mol Genet. 2006, 15: 2911-2922. 10.1093/hmg/ddl232.
Simons C, Pheasant M, Makunin IV, Mattick JS: Transposon-free regions in mammalian genomes. Genome Res. 2006, 16: 164-172. 10.1101/gr.4624306.
Pennacchio LA, Loots GG, Nobrega MA, Ovcharenko I: Predicting tissue-specific enhancers in the human genome. Genome Res. 2007, 17: 201-211. 10.1101/gr.5972507.
Feng J, Bi C, Clark BS, Mady R, Shah P, Kohtz JD: The Evf-2 non-coding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 2006, 20: 1470-1484. 10.1101/gad.1416106.
Bejerano G, Lowe CB, Ahituv N, King B, Siepel A, Salama SR, Rubin EM, Kent WJ, Haussler D: A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature. 2006, 441: 87-90. 10.1038/nature04696.
Brooke NM, Garcia-Fernandez J, Holland PW: The ParaHox gene cluster is an evolutionary sister of the Hox gene cluster. Nature. 1998, 392: 920-922. 10.1038/31933.
Garcia-Fernandez J, Holland PW: Archetypal organization of the amphioxus Hox gene cluster. Nature. 1994, 370: 563-566. 10.1038/370563a0.
Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang YL, et al: Zebrafish hox clusters and vertebrate genome evolution. Science. 1998, 282: 1711-1714. 10.1126/science.282.5394.1711.
Hoegg S, Meyer A: Hox clusters as models for vertebrate genome evolution. Trends Genet. 2005, 21: 421-424. 10.1016/j.tig.2005.06.004.
Woltering JM, Durston AJ: The zebrafish hoxDb cluster has been reduced to a single microRNA. Nat Genet. 2006, 38: 601-602. 10.1038/ng0606-601.
Kmita M, Duboule D: Organizing axes in time and space; 25 years of colinear tinkering. Science. 2003, 301: 331-333. 10.1126/science.1085753.
Krumlauf R, Marshall H, Studer M, Nonchev S, Sham MH, Lumsden A: Hox homeobox genes and regionalisation of the nervous system. J Neurobiol. 1993, 24: 1328-1340. 10.1002/neu.480241006.
Edvardsen RB, Seo HC, Jensen MF, Mialon A, Mikhaleva J, Bjordal M, Cartry J, Reinhardt R, Weissenbach J, Wincker P, et al: Remodelling of the homeobox gene complement in the tunicate Oikopleura dioica. Curr Biol. 2005, 15: R12-R13. 10.1016/j.cub.2004.12.010.
Negre B, Casillas S, Suzanne M, Sanchez-Herrero E, Akam M, Nefedov M, Barbadilla A, de Jong P, Ruiz A: Conservation of regulatory sequences and gene expression patterns in the disintegrating Drosophila Hox gene complex. Genome Res. 2005, 15: 692-700. 10.1101/gr.3468605.
Seo HC, Edvardsen RB, Maeland AD, Bjordal M, Jensen MF, Hansen A, Flaat M, Weissenbach J, Lehrach H, Wincker P, et al: Hox cluster disintegration with persistent anteroposterior order of expression in Oikopleura dioica. Nature. 2004, 431: 67-71. 10.1038/nature02709.
Tiong SY, Whittle JR, Gribbin MC: Chromosomal continuity in the abdominal region of the bithorax complex of Drosophila is not essential for its contribution to metameric identity. Development. 1987, 101: 135-142.
Negre B, Ruiz A: HOM-C evolution in Drosophila: is there a need for Hox gene clustering?. Trends Genet. 2007, 23: 55-59. 10.1016/j.tig.2006.12.001.
Spitz F, Gonzalez F, Duboule D: A global control region defines a chromosomal regulatory landscape containing the HoxD cluster. Cell. 2003, 113: 405-417. 10.1016/S0092-8674(03)00310-6.
Gould A, Morrison A, Sproat G, White RA, Krumlauf R: Positive cross-regulation and enhancer sharing: two mechanisms for specifying overlapping Hox expression patterns. Genes Dev. 1997, 11: 900-913. 10.1101/gad.11.7.900.
Kmita M, Kondo T, Duboule D: Targeted inversion of a polar silencer within the HoxD complex re-allocates domains of enhancer sharing. Nat Genet. 2000, 26: 451-454. 10.1038/82593.
Spitz F, Herkenne C, Morris MA, Duboule D: Inversion-induced disruption of the Hoxd cluster leads to the partition of regulatory landscapes. Nat Genet. 2005, 37: 889-893. 10.1038/ng1597.
Kmita M, Fraudeau N, Herault Y, Duboule D: Serial deletions and duplications suggest a mechanism for the collinearity of Hoxd genes in limbs. Nature. 2002, 420: 145-150. 10.1038/nature01189.
Sharpe J, Nonchev S, Gould A, Whiting J, Krumlauf R: Selectivity, sharing and competitive interactions in the regulation of Hoxb genes. EMBO J. 1998, 17: 1788-1798. 10.1093/emboj/17.6.1788.
Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH: MicroRNA expression in zebrafish embryonic development. Science. 2005, 309: 310-311. 10.1126/science.1114519.
Mulley JF, Chiu CH, Holland PW: Breakup of a homeobox cluster after genome duplication in teleosts. Proc Natl Acad Sci USA. 2006, 103: 10369-10372. 10.1073/pnas.0600341103.
Roth C, Rastogi S, Arvestad L, Dittmar K, Light S, Ekman D, Liberles DA: Evolution after gene duplication: models, mechanisms, sequences, systems, and organisms. J Exp Zoolog B Mol Dev Evol. 2007, 308: 58-73. 10.1002/jez.b.21124.
Ohno S: Patterns in genome evolution. Curr Opin Genet Dev. 1993, 3: 911-914. 10.1016/0959-437X(93)90013-F.
Teichmann SA, Babu MM: Gene regulatory network growth by duplication. Nat Genet. 2004, 36: 492-496. 10.1038/ng1340.
Ghanem N, Jarinova O, Amores A, Long Q, Hatch G, Park BK, Rubenstein JL, Ekker M: Regulatory roles of conserved intergenic domains in vertebrate Dlx bigene clusters. Genome Res. 2003, 13: 533-543. 10.1101/gr.716103.
Stock DW, Ellies DL, Zhao Z, Ekker M, Ruddle FH, Weiss KM: The evolution of the vertebrate Dlx gene family. Proc Natl Acad Sci USA. 1996, 93: 10858-10863. 10.1073/pnas.93.20.10858.
Aruga J, Kamiya A, Takahashi H, Fujimi TJ, Shimizu Y, Ohkawa K, Yazawa S, Umesono Y, Noguchi H, Shimizu T, et al: A wide-range phylogenetic analysis of Zic proteins: implications for correlations between protein structure conservation and body plan complexity. Genomics. 2006, 87: 783-792. 10.1016/j.ygeno.2006.02.011.
Wang W, Lo P, Frasch M, Lufkin T: Hmx: an evolutionary conserved homeobox gene family expressed in the developing nervous system in mice and Drosophila. Mech Dev. 2000, 99: 123-137. 10.1016/S0925-4773(00)00488-3.
Maak S, Neumann K, Swalve HH: Identification and analysis of putative regulatory sequences for the MYF5/MYF6 locus in different vertebrate species. Gene. 2006, 379: 141-147. 10.1016/j.gene.2006.05.007.
Di Gregorio A, Spagnuolo A, Ristoratore F, Pischetola M, Aniello F, Branno M, Cariello L, Di Lauro R: Cloning of ascidian homeobox genes provides evidence for a primordial chordate cluster. Gene. 1995, 156: 253-257. 10.1016/0378-1119(95)00035-5.
Zerucha T, Ekker M: Distal-less-related homeobox genes of vertebrates: evolution, function, and regulation. Biochem Cell Biol. 2000, 78: 593-601. 10.1139/bcb-78-5-593.
Sumiyama K, Irvine SQ, Stock DW, Weiss KM, Kawasaki K, Shimizu N, Shashikant CS, Miller W, Ruddle FH: Genomic structure and functional control of the Dlx3-7 bigene cluster. Proc Natl Acad Sci USA. 2002, 99: 780-785. 10.1073/pnas.012584999.
Sumiyama K, Ruddle FH: Regulation of Dlx3 gene expression in visceral arches by evolutionarily conserved enhancer elements. Proc Natl Acad Sci USA. 2003, 100: 4030-4034. 10.1073/pnas.0530119100.
Lettice LA, Horikoshi T, Heaney SJ, van Baren MJ, van der Linde HC, Breedveld GJ, Joosse M, Akarsu N, Oostra BA, Endo N, et al: Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc Natl Acad Sci USA. 2002, 99: 7548-7553. 10.1073/pnas.112212199.
Ahituv N, Prabhakar S, Poulin F, Rubin EM, Couronne O: Mapping cis-regulatory domains in the human genome using multispecies conservation of synteny. Hum Mol Genet. 2005, 14: 3057-3063. 10.1093/hmg/ddi338.
de Kok YJ, Vossenaar ER, Cremers CW, Dahl N, Laporte J, Hu LJ, Lacombe D, Fischel-Ghodsian N, Friedman RA, Parnes LS, et al: Identification of a hot spot for microdeletions in patients with X-linked deafness type 3 (DFN3) 900 kb proximal to the DFN3 gene POU3F4. Hum Mol Genet. 1996, 5: 1229-1235. 10.1093/hmg/5.9.1229.
Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, et al: Mutations in SOX2 cause anophthalmia. Nat Genet. 2003, 33: 461-463. 10.1038/ng1120.
Kano H, Kurosawa K, Horii E, Ikegawa S, Yoshikawa H, Kurahashi H, Toda T: Genomic rearrangement at 10q24 in non-syndromic split-hand/split-foot malformation. Hum Genet. 2005, 118: 477-483. 10.1007/s00439-005-0074-0.
Pfeifer D, Kist R, Dewar K, Devon K, Lander ES, Birren B, Korniszewski L, Back E, Scherer G: Campomelic dysplasia translocation breakpoints are scattered over 1 Mb proximal to SOX9: evidence for an extended control region. Am J Hum Genet. 1999, 65: 111-124. 10.1086/302455.
Bejerano G, Haussler D, Blanchette M: Into the heart of darkness: large-scale clustering of human non-coding DNA. Bioinformatics. 2004, 20 (suppl 1): I40-I48. 10.1093/bioinformatics/bth946.
Boffelli D, Weer CV, Weng L, Lewis KD, Shoukry MI, Pachter L, Keys DN, Rubin EM: Intraspecies sequence comparisons for annotating genomes. Genome Res. 2004, 14: 2406-2411. 10.1101/gr.3199704.
Bailey PJ, Klos JM, Andersson E, Karlen M, Kallstrom M, Ponjavic J, Muhr J, Lenhard B, Sandelin A, Ericson J: A global genomic transcriptional code associated with CNS-expressed genes. Exp Cell Res. 2006, 312: 3108-3119. 10.1016/j.yexcr.2006.06.017.
Dermitzakis ET, Kirkness E, Schwarz S, Birney E, Reymond A, Antonarakis SE: Comparison of human chromosome 21 conserved nongenic sequences (CNGs) with the mouse and dog genomes shows that their selective constraint is independent of their genic environment. Genome Res. 2004, 14: 852-859. 10.1101/gr.1934904.
Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, et al: Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996, 85: 841-851. 10.1016/S0092-8674(00)81268-4.
Haber DA, Buckler AJ, Glaser T, Call KM, Pelletier J, Sohn RL, Douglass EC, Housman DE: An internal deletion within an 11p13 zinc finger gene contributes to the development of Wilms' tumor. Cell. 1990, 61: 1257-1269. 10.1016/0092-8674(90)90690-G.
Jamieson RV, Perveen R, Kerr B, Carette M, Yardley J, Heon E, Wirth MG, van Heyningen V, Donnai D, Munier F, et al: Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum Mol Genet. 2002, 11: 33-42. 10.1093/hmg/11.1.33.
Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, et al: Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004, 36: 955-957. 10.1038/ng1407.
Crackower MA, Scherer SW, Rommens JM, Hui CC, Poorkaj P, Soder S, Cobben JM, Hudgins L, Evans JP, Tsui LC: Characterization of the split hand/split foot malformation locus SHFM1 at 7q21.3-q22.1 and analysis of a candidate gene for its expression during limb development. Hum Mol Genet. 1996, 5: 571-579. 10.1093/hmg/5.5.571.
Leipoldt M, Erdel M, Bien-Willner GA, Smyk M, Theurl M, Yatsenko SA, Lupski JR, Lane AH, Shanske AL, Stankiewicz P, et al: Two novel translocation breakpoints upstream of SOX9 define borders of the proximal and distal breakpoint cluster region in campomelic dysplasia. Clin Genet. 2007, 71: 67-75. 10.1111/j.1399-0004.2007.00736.x.
Nishimura DY, Swiderski RE, Alward WL, Searby CC, Patil SR, Bennet SR, Kanis AB, Gastier JM, Stone EM, Sheffield VC: The fork-head transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat Genet. 1998, 19: 140-147. 10.1038/493.
Fang J, Dagenais SL, Erickson RP, Arlt MF, Glynn MW, Gorski JL, Seaver LH, Glover TW: Mutations in FOXC2 (MFH-1), a fork-head family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet. 2000, 67: 1382-1388. 10.1086/316915.
Beysen D, Raes J, Leroy BP, Lucassen A, Yates JR, Clayton-Smith J, Ilyina H, Brooks SS, Christin-Maitre S, Fellous M, et al: Deletions involving long-range conserved nongenic sequences upstream and downstream of FOXL2 as a novel disease-causing mechanism in blepharophimosis syndrome. Am J Hum Genet. 2005, 77: 205-218. 10.1086/432083.
Vortkamp A, Gessler M, Grzeschik KH: GLI3 zinc-finger gene interrupted by translocations in Greig syndrome families. Nature. 1991, 352: 539-540. 10.1038/352539a0.
Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, et al: Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996, 14: 392-399. 10.1038/ng1296-392.
Wallis DE, Roessler E, Hehr U, Nanni L, Wiltshire T, Richieri-Costa A, Gillessen-Kaesbach G, Zackai EH, Rommens J, Muenke M: Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet. 1999, 22: 196-198. 10.1038/9718.
Howard TD, Paznekas WA, Green ED, Chiang LC, Ma N, Ortiz de Luna RI, Garcia Delgado C, Gonzalez-Ramos M, Kline AD, Jabs EW: Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997, 15: 36-41. 10.1038/ng0197-36.
Engels S, Kohlhase J, McGaughran J: A SALL1 mutation causes a branchio-oto-renal syndrome-like phenotype. J Med Genet. 2000, 37: 458-460. 10.1136/jmg.37.6.458.
Crolla JA, van Heyningen V: Frequent chromosome aberrations revealed by molecular cytogenetic studies in patients with aniridia. Am J Hum Genet. 2002, 71: 1138-1149. 10.1086/344396.
Laumonnier F, Ronce N, Hamel BC, Thomas P, Lespinasse J, Raynaud M, Paringaux C, Van Bokhoven H, Kalscheuer V, Fryns JP, et al: Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am J Hum Genet. 2002, 71: 1450-1455. 10.1086/344661.
Sabherwal N, Bangs F, Roth R, Weiss B, Jantz K, Tiecke E, Hinkel GK, Spaich C, Hauffa BP, van der Kamp H, et al: Long-range conserved non-coding SHOX sequences regulate expression in developing chicken limb and are associated with short stature phenotypes in human patients. Hum Mol Genet. 2007, 16: 210-222. 10.1093/hmg/ddl470.
Acknowledgements
We thank Dr Shawn M Burgess for communicating results prior to publication. This work was supported by funds from the Sars Centre, the University of Bergen, and by the Functional Genomics Program (FUGE) in the Research Council of Norway to TSB and BL and by the European Commission as part of the ZF-Models integrated project in the 6th framework programme (Contract No. LSHG-CT-2003-503469) to TSB.
This article has been published as part of Genome Biology Volume 8, Supplement 1, 2007: Transposons in vertebrate functional genomics. The full contents of the supplement are available online at http://genomebiology.com/supplements/8/S1.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
About this article
Cite this article
Kikuta, H., Fredman, D., Rinkwitz, S. et al. Retroviral enhancer detection insertions in zebrafish combined with comparative genomics reveal genomic regulatory blocks - a fundamental feature of vertebrate genomes. Genome Biol 8 (Suppl 1), S4 (2007). https://doi.org/10.1186/gb-2007-8-s1-s4
Published:
DOI: https://doi.org/10.1186/gb-2007-8-s1-s4