
Abstract
One of the cellular defenses against virus infection is the silencing of viral gene expression. There is evidence that at least two gene-silencing mechanisms are used against the human immuno-deficiency virus (HIV). Paradoxically, this cellular defense mechanism contributes to viral latency and persistence, and we review here the relationship of viral latency to gene-silencing mechanisms.
Similar content being viewed by others


To succeed, all long-term relationships require some degree of compromise from both partners. This is no less true for persistent virus infections and their hosts. Unrestricted replication of the parasite may be to the detriment of the health of the host and shorten its life span, thus depriving the parasite of its niche. Equally, no replication at all is a dead end for the parasite. The pathogen thus constrains its replication, and the host, given that it has effectively lost the battle to eliminate the invader, makes the best of a bad job and controls it when it gets out of hand. Restricting replication quantitatively or temporally so that the virus reproduces just sufficiently, or at particularly strategic times (for example, pregnancy), to achieve transmission while remaining silent at other times (latency), are techniques used by several viral families, of which the herpesviruses are the best studied.
The human immunodeficiency virus (HIV), the causative agent of AIDS, is also postulated to become latent when it infects a T lymphocyte (T cell) that has ceased to divide, and where levels of transcription factors that both cell and virus need for gene expression are declining [1]. This may be an oversimplification, however, as latency may be effected by more than one process and it may occur in cells other than memory T cells. Latency in HIV is of immense practical importance because it provides a reservoir of virus that can reactivate years later and that is protected from immune clearance and the effects of antiviral drugs. Control of gene expression from an integrated retroviral genome, the provirus, also provides an insight into how the chromatin reacts to parasites invading the genome, a process that is thought to have occurred throughout evolution, as evidenced by the abundance of endogenous retroviruses [2, 3] and repeated elements [4] in the human genome.
With knowledge burgeoning about the role of chromatin in the control of gene expression it is timely to review HIV latency. It is the greatest barrier to virus eradication, and understanding it can only enhance our knowledge of cellular gene expression.
After entry into the cell, HIV, like other retroviruses, uncoats and reverse transcribes its dimeric RNA genome into first a complementary DNA (cDNA) and then a double-stranded DNA (dsDNA). The DNA duplex containing the viral genes flanked by non-coding repeat sequences (long terminal repeats, LTRs) is then integrated by the viral integrase enzyme into the host DNA. To complete the viral life cycle, the integrated genome, now termed a provirus, utilizes host machinery, including transcription factors and RNA polymerase II, to activate its genome. The viral 5'-LTR acts as an enhancer and a promoter, directing the transcription of viral mRNAs, which are translated to make viral components.
The proviral genome is, like the host DNA, associated with chromosomal proteins. Here we review some of the information on gene expression from the integrated retroviral genome in the context of gene-silencing mechanisms that might contribute to latency. On the basis of this evidence and the dynamics of proviral gene expression, we propose that the cell uses different forms of proviral silencing: one occurring soon after integration and related to the integration site of the provirus, and a second, delayed, location-independent mechanism affecting proviruses that initially had established transcriptional activity.
Proviral transcription is affected by its chromatin structure
Several observations point to the local state of chromatin being highly influential in the ability of the HIV provirus to overcome a major intracellular hurdle - transcription initiation. HIV-1 superinfecting already latently infected cell lines is expressed [5], implying that local factors and global cellular conditions influence gene expression independently. Cell lines harboring a single copy of a simple retroviral provirus, that of murine leukemia virus (MLV), with varying levels of expression do not differ in their capacity to support the transcription of a transiently transfected LTR-driven reporter gene [6]. This suggests that factors independent of those influencing the resident provirus can affect transcription. Another finding highlighting the influence of local conditions is that silenced, methylated endogenous retro-virus (ERV) DNA becomes infectious after cloning [7].
More recently, a wealth of experiments illustrating the role of chromatin in HIV gene expression has been reported. Protein complexes involved in chromatin remodeling, including histone acetyltransferases (HATs) [8] and late SV40 transcription factor (LSF), which recruits histone deacetylase complex (HDAC-1) [9], can alter HIV-1 gene expression both in vitro and in vivo. HIV-1 expression is also affected by pharmacological modification of histone tails. Agents such as polyamides, which bind to the proviral promoter and block HDAC-1 recruitment [10], and trichostatin A, a HDAC inhibitor [11], have striking effects. Histone modifications involved in HIV gene expression have also been demonstrated by chromatin histone immunoprecipitation (ChIP) assays. HIV-1 reactivation from latently infected cell lines by either the induction of cell-cycle arrest [12] or the application of phorbol ester [13] both require the recruitment of HATs, accompanied by acetylation of histones at the HIV-1 promoter. ATP-dependent chromatin-remodeling proteins, including members of the SWI/SNF complex, are also recruited during HIV reactivation by phorbol ester application, resulting in the disruption of nucleosomes at the LTRs [14]. Retinoic acid, rather specifically in HIV-1, can interfere with nucleosome remodeling at the LTRs, but not with histone acetylation, and inhibits HIV-1 transcription [15]. Thus, during activation, histones associated with the HIV-1 promoter are first acetylated and chromatin-remodeling complexes are then recruited to disrupt the resident nucleosome (see [16] for a review).
Chromatin remodeling is also involved in repressing proviral gene expression. Protein factors associated with repression, such as c-Myc, occupy the HIV-1 promoter alongside HDAC-1 in a coordinated manner [17]. Recruitment of HDACs, the histone methyltransferase Suv39H1, the protein HP1 (which is typical of heterochromatin) and trimethylation of H3 lysine 9 at the HIV LTR have been shown to correlate with repression of gene expression in microglial cells [18]. Depleting Suv39H1 and the HP1γ subtype by siRNA increases the level of HIV gene expression [19].
The enigma of DNA methylation and viral gene silencing: correlation or causation?
Whereas the weight of evidence for the involvement of chromatin histone remodeling in the control of retroviral gene expression is compelling, the involvement of DNA methylation is more controversial. DNA methylation is associated with the recruitment of HP1, a marker of tight repression [20]. In vitro, the expression levels of transfected methylated LTR-driven reporter constructs based on HIV [21], human T-cell leukemia virus type 1 (HTLV-1) [22] or ERVs [23] have been assessed. Methylation-sensitive restriction enzymes were used to detect DNA methylation in silenced transfected HIV-1 constructs [24] or in ERV LTR-driven reporter constructs [23]. These studies established a clear link between DNA methylation and the absence of transcription.
Data from transiently transfected plasmids may not, however, accurately represent the behavior of integrated proviruses. For example, an integrated HTLV-1 responds differently from a transfected construct in response to extracellular stimuli [25], and in hepatocyte cell lines the promoter activity of a transfected HIV-based construct differs from that of an integrated vector [26]. A further example of episomes and chromosome-associated constructs behaving differently is seen in human papillomavirus gene expression. Associating a matrix-attachment region to a human papillomavirus gene has diametrically opposite effects on gene expression depending on whether the construct is transiently transfected or stably transfected (and presumably integrated) [27].
Despite these caveats, a latent MLV provirus with a methylated genome, confirmed by methylation-sensitive restriction enzymes, can be reactivated by 5-azacytidine, an inhibitor of DNA methylation [6]. In addition, methylation of the HTLV-1 proviral LTR has been assessed using methylation-sensitive restriction enzymes in cells from infected patients [28] or in transformed cell lines [29]. Methylation correlated inversely with the level of viral RNA [29] and the provirus could be reactivated by 5-azacytidine [29, 30]. Mutating the CpG sites (sites of DNA methylation) eliminates integrants that are completely silenced, strongly suggesting a role for DNA methylation in silencing [31].
In contrast, however, a methylated MLV provirus introduced into a defined site of the host-cell genome with Cre recombinase still lacks transcriptional activity after pharmacological inhibition of DNA methylation [32]. More problematically, bisulfite genomic sequencing analysis on HTLV-1-infected cells from patients and transformed cell lines showed that while the 5'-LTR is hypermethylated, the 3'-LTR is hypomethylated. In neither HTLV-1 infection [30] nor in its often-used animal model, bovine leukemia virus (BLV) infection [33], does the pattern of methylation of the LTR correspond to the clinical manifestation of the infection or to disease progression.
In cell lines latently infected with HIV-1, such as ACH-2, the 5'-LTR of the provirus is hypermethylated, whereas the 3'-LTR is hypomethylated. Activating the provirus with the cytokine tumor necrosis factor-α (TNF-α) partially relieves DNA methylation of the 5'-LTR [34]. However, in clones of cells derived after infection with a defective HIV-1 expressing green fluorescent protein (GFP) from the LTR, proviral expression did not correlate with DNA methylation, and bisulfite genome analysis showed most cytosine residues to be unmethylated [35]. Thus, in MLV, HTLV-1 and HIV, the influence of DNA methylation on proviral behavior is controversial. Stability of gene expression may correlate more with the density of DNA methylation, as opposed to there being a binary 'methylated' or 'demethylated' state [36].
What controls the behavior of the chromatin associated with the provirus?
The link between chromatin remodeling and proviral activity seems incontrovertible, but how is it controlled? One possible factor is the site of integration. A study of 35 HIV-1-infected clones found heterogeneity in their individual levels of gene expression. There was no correlation between the expression level of the integrated provirus and a second transfected construct in the same clone, again implicating the local environment in controlling proviral expression [37]. Analyzing the accessibility of restriction enzymes to DNA as a guide to nucleosome remodeling showed that this did correlate with the level of gene expression, despite a lack of correlation between gene expression and promoter methylation [37]. In addition, latent HIV-1 proviruses are frequently found integrated near alphoid repeats, which are frequently found in heterochromatin [38, 39]. This association with heterochromatin was further supported by a large-scale study of 971 HIV-1 integration sites that revealed that proviruses with inducible gene expression - presumably representing latent provirus - had integrated near gene deserts and centromeres, which are rich in alphoid repeats [40]. Others, however, were near very highly expressed genes [40] and a study of 74 HIV-1 integration sites of latent proviruses obtained from resting CD4 T cells from 16 patients found that most of them were in actively expressed regions [41]. Integration sites may, therefore, be influential, but other factors are also in operation. Consistent with the notion that viral gene expression is influenced by local factors set up at the time of integration, gene-therapy vectors containing DNA elements that can shield the provirus from the effects of adjacent chromatin, such as an MLV-based vector with a locus control region [42] or lentiviral vectors with a matrix-attachment region [43], establish high-level, position-independent gene expression. Overall, these studies strongly imply a significant role for the position of integration.
Does the integration site program gene expression indefinitely?
The time frame of silencing in a number of experiments is the major piece of evidence suggesting that factors other than integration site affect proviral expression. The 'site' effect on proviral chromatin configuration and gene expression would presumably be imposed soon after integration and, if it were the overriding influence, be permanent. Attenuation of gene expression has, however, been observed in longer-term culture in several experiments, mostly conducted using long-term cell clones infected with MLV or its derived vectors [32, 44–46], sometimes with the transgene driven by a heterologous promoter rather than the viral LTR.
Even more telling, within the cell clones with provirus integrated at the same sites, variation of gene expression was observed in a number of different retroviral vectors [31, 44, 47–49]. Where variation exists, the level of proviral gene expression between mother and daughter cells shows some degree of correlation [49, 50]. Silencing, where it occurred, was often linked to DNA methylation, as determined by digestion with restriction enzymes [46, 51] or bisulfite genomic sequencing [51]. This was not universally the case, however, and variation was possible even in cells devoid of de novo methyltransferases [47, 48]. Attempts to reactivate these proviruses using 5-azacytidine or trichostatin A after they had been silenced in long-term culture were at best only partially successful [44–47, 49]. Variation in gene expression and the same difficulty in reversing the repression are also observed in HIV-1-infected cell clones [52]. Arguably, the required strength of the reactivating stimulus may be a dose-response effect. Lorincz et al. [51] reported that more efficient reactivation could be achieved by applying trichostatin to cells pretreated with 5-azacytidine, which presumably led to the removal of the methylation mark on DNA and relieved the transgene from tight repression. Thus, the overall picture is that, whereas the chromosomal position certainly contributes to the chromatin configuration of a provirus, the level of gene expression is modulated by other factors. One possibility is that variation emerges as cell division alters existing epigenetic marks on the provirus [50]. Intriguingly, there are at least two reports of upregulation of a DNA methyltransferase after HIV-1 infection [53, 54], suggesting the possibility of an active mechanism that silences integrated provirus.
More than one form of proviral silencing may exist
A model for retroviral gene expression thus has to accommodate several observations. First is the strong evidence of the involvement of chromatin. Second is the correlation between DNA methylation and proviral behavior, which is not consistent in all studies: there may be a gradation of silencing from strongly repressed to unstably expressed. Thirdly, the position of integration is important. Finally, proviral shutdown can and does occur over time. Even within cell clones, variation in gene expression is common.
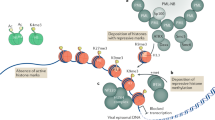
One possible hypothesis is that the degree of proviral gene expression reflects the permissiveness of the chromatin at the site of integration. Thus, when integration occurs in repressed chromatin, the provirus is heavily repressed, which is probably correlated with DNA methylation. Where the density of DNA methylation is less, the provirus enters a state of unstable gene expression, manifesting as variation of expression from cell to cell (variegation; Figure 1). Although this is an attractive model, it cannot completely explain all the observations listed in Box 1. Another hypothesis is that the behavior of the provirus at an early stage after infection is primarily governed by the site of integration [37, 55] and is only partially related to the local chromatin configuration [35, 38]. Proviruses silenced at this point are probably more amenable to reactivation: indeed, the rate-limiting step in initiation of HIV-1 gene expression is the recruitment of the general transcription factor TFIIH [56], a relatively late step in the derepression of local chromatin. Of the proviruses that are not silenced initially, a proportion undergo shutdown with time. Proviruses silenced in this manner are tightly repressed and can be difficult to reactivate using external stimuli (Figure 2). Indeed, given the complexity of the genome, it is entirely possible that the two models are complementary: depending on the site of integration, one or other of the mechanisms depicted in Figures 1 and 2 is at work.

Variation in the level of proviral expression within cell clones might be accounted for by the degree of methylation. In this model the degree of repression, set up at the time of integration, is critical to the degree of gene expression. Studies supporting this model include [36,47,49,50]. DNA methylation is an attractive candidate as a molecular correlate of repression and is depicted as such here and in Figure 2. There is, however, evidence suggesting that other molecular mechanism may be involved (see text). (a) Provirus integrated into repressive chromatin is stably repressed, probably correlating with a high degree of proviral DNA methylation. (b) Provirus in partially repressed chromatin is unstable and may proceed to become tightly repressed, or continue to be expressed but could be induced to a higher degree of expression. The change in epigenetic mark could arise from cell division. (c) Integration into permissive chromatin leads to high-level gene expression.

Variation in the expression of proviruses integrated at the same position in different cells might be accounted for by a delayed mechanism leading to proviral silencing. Soon after infection the behavior of the provirus depends on the site of integration. (a) Integration into a chromosome position that is nonpermissive for gene expression results in a silent provirus. Note that although the environment may be nonpermissive for gene expression, the provirus itself need not be tightly repressed and is amenable to reactivation by various stimuli. (b) Integration into permissive chromatin permits viral gene expression. In HIV-1 this stage is prolonged because of the stability conferred by the Tat-TAR positive-feedback axis. (c) With time the provirus is silenced. At present, the trigger leading to the collapse of proviral activity is not known. (d) Once silenced, the provirus is tightly repressed and cannot be easily reactivated.
The additional transcriptional control mechanisms of complex retroviruses like HIV add a further level of control. Unlike simple retroviruses, where attenuation of gene expression is often observed, once transcription of an HIV-1 provirus has been established, it is extremely stable [52]. This is probably due to the viral protein Tat and its response element TAR [39]. Tat exerts positive feedback and enhances proviral gene expression by several mechanisms (reviewed in [57, 58]; see also [59–61]). This positive-feedback axis leads to remarkably durable HIV-1 gene expression, which persists for more than 18 months once established [52]. The virus-encoded transactivator Tat may be an evolutionary development by the virus to counter the cellular silencing mechanisms.
In summary, retroviral gene expression is influenced by more than one mechanism involving chromatin (Figure 2). The location of integration probably crucially affects the initial level of proviral activity. With time, the provirus may be silenced. These silencing mechanisms are likely to affect most integrated constructs, accounting for the silencing observed in simple retroviruses, HIV-1 and retroviral vectors. In HIV-1, however, silencing can be counteracted or delayed by the powerful Tat-TAR positive-feedback axis [39]. The trigger event that leads to silencing is not clear. One possibility is that all proviruses are susceptible to silencing mediated by epigenetic changes through a direct mechanism, possibly via the upregulated DNA methyltransferases [53, 54]. Such a mechanism must act early after infection.
Formation of heterochromatin is known to spread to adjacent genetic regions unless it is stopped by an insulator [62–64]. In the first model (Figure 1), proviral gene silencing observed in long-term cultures is one end of the spectrum of the process that relates the site of integration to proviral activity, the provirus being silenced by spreading hetero-chromatinization. Alternatively, a microRNA (miRNA)-based mechanism may be involved [65]. Another possibility is that silencing is related to the cyclical expression of transcription factors, such as NFκB for HIV-1 [66]. In a cell infected with HIV-1, the level of NFκB dips intermittently, the production of Tat would not be maintained and the Tat-TAR positive-feedback axis would collapse. The now vulnerable provirus would be further repressed by chromatin changes that cannot be easily reversed (see Box 1). In simple retroviruses and retroviral vectors, this positive-feedback axis is absent, and therefore silencing is more frequent [32, 44–46, 51, 67].
We can begin to draw some tentative conclusions. The 'site' effect, whereby an identical provirus behaves differently according to its point of integration, argues for powerful regional control mechanisms for gene expression independent of the gene itself, providing a general defense against insertional elements. The effects of known influences on chromatin configuration clearly affect proviral gene expression, and study of individual genes in the context of proviral insertions may be illuminating to dissect out the individual contributions of methylation, acetylation and so on. Lastly, intrinsic promoter properties of the provirus have an effect that can potentially hold back the effect of regional silencing influences as long as the promoter is functioning. Ironically, the cellular silencing mechanisms actually contribute to the persistence of HIV by facilitating its evasion of drug and immunological attack.
Knowledge about the mechanism of proviral silencing and how to reverse it may lead to clinical applications in HIV infection. There have been attempts at clearing HIV from an infected patient by reactivating latent virus using interleukin-2 [68, 69] or the histone deacetylase inhibitor valproic acid [70], thus rendering the virus susceptible to anti-retroviral therapy and immune clearance. These have been, at best, only partially successful. Understanding proviral silencing will be instrumental in devising further strategies. New insights into the control of gene expression by miRNA, which is possibly another defense against invading molecular parasites [71], will undoubtedly also have an impact [72, 73].
The oldest endogenous retroviruses in the human genome entered the genomes of our mammalian ancestors at around the time of the extinction of the dinosaurs [74]. It might be expected that, with such a long history of coexistence between mammalian genomes and transposable elements, there would exist well developed defenses against these molecular parasites, most probably chromatin dependent. It is tempting to speculate that the silencing of retroviruses, and possibly of cellular genes, originates from such defensive mechanisms.
Box 1 Two contrasting phenotypes of retroviral provirus silencing
Immediately after infection
The site of integration critically affects the level of proviral gene expression of HIV-1 [37].
DNA methylation does not correlate with proviral gene expression in cells infected with HIV-1 [35, 38] or HTLV-1 [29].
HIV-1 gene expression can be augmented by trichostatin A [11] and/or Tat [37].
At late times after infection
Variation in proviral gene expression is observed in clonal populations of cells infected with MLV [51], with MLV-based vectors [44–46] and with HIV-1-based vectors [39, 52].
Local factors (chromatinization) correlate with proviral expression in HIV-1 [52]. The degree of DNA methylation correlates with behavior of the proviruses of MLV [51] and MLV-based vectors [44–46].
Variable reactivation by trichostatin A on HIV [39], by 5-azacytidine and/or trichostatin A on MLV [44–46].
Depending on the study, the time frame required for the 'late times' phenotype to arise can range from weeks [44–46] to months [39, 52].
References
Persaud D, Zhou Y, Siliciano JM, Siliciano RF: Latency in human immunodeficiency virus type 1 infection: no easy answers. J Virol. 2003, 77: 1659-1665. 10.1128/JVI.77.3.1659-1665.2003.
Sverdlov ED: Retroviruses and primate evolution. BioEssays. 2000, 22: 161-171. 10.1002/(SICI)1521-1878(200002)22:2<161::AID-BIES7>3.0.CO;2-X.
Stoye JP: Endogenous retroviruses: still active after all these years?. Curr Biol. 2001, 11: R914-916. 10.1016/S0960-9822(01)00553-X.
Batzer MA, Deininger PL: Alu repeats and human genomic diversity. Nat Rev Genet. 2002, 3: 370-379. 10.1038/nrg798.
Chen BK, Saksela K, Andino R, Baltimore D: Distinct modes of human immunodeficiency virus type 1 proviral latency revealed by superinfection of nonproductively infected cell lines with recombinant luciferase-encoding viruses. J Virol. 1994, 68: 654-660.
Hoeben RC, Migchielsen AA, van der Jagt RC, van Ormondt H, van der Eb AJ: Inactivation of the Moloney murine leukemia virus long terminal repeat in murine fibroblast cell lines is associated with methylation and dependent on its chromosomal position. J Virol. 1991, 65: 904-912.
Harbers K, Schnieke A, Stuhlmann H, Jahner D, Jaenisch R: DNA methylation and gene expression: endogenous retroviral genome becomes infectious after molecular cloning. Proc Natl Acad Sci USA. 1981, 78: 7609-7613. 10.1073/pnas.78.12.7609.
Steger DJ, Eberharter A, John S, Grant PA, Workman JL: Purified histone acetyltransferase complexes stimulate HIV-1 transcription from preassembled nucleosomal arrays. Proc Natl Acad Sci USA. 1998, 95: 12924-12929. 10.1073/pnas.95.22.12924.
Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM: The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol. 2000, 74: 6790-6799. 10.1128/JVI.74.15.6790-6799.2000.
Ylisastigui L, Coull JJ, Rucker VC, Melander C, Bosch RJ, Brodie SJ, Corey L, Sodora DL, Dervan PB, Margolis DM: Polyamides reveal a role for repression in latency within resting T cells of HIV-infected donors. J Infect Dis. 2004, 190: 1429-1437. 10.1086/423822.
Quivy V, Adam E, Collette Y, Demonte D, Chariot A, Vanhulle C, Berkhout B, Castellano R, de Launoit Y, Burny A, et al: Synergistic activation of human immunodeficiency virus type 1 promoter activity by NF-kappaB and inhibitors of deacetylases: potential perspectives for the development of therapeutic strategies. J Virol. 2002, 76: 11091-11103. 10.1128/JVI.76.21.11091-11103.2002.
Thierry S, Marechal V, Rosenzwajg M, Sabbah M, Redeuilh G, Nicolas JC, Gozlan J: Cell cycle arrest in G2 induces human immunod-eficiency virus type 1 transcriptional activation through histone acetylation and recruitment of CBP, NF-kappaB, and c-Jun to the long terminal repeat promoter. J Virol. 2004, 78: 12198-12206. 10.1128/JVI.78.22.12198-12206.2004.
Lusic M, Marcello A, Cereseto A, Giacca M: Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003, 22: 6550-6561. 10.1093/emboj/cdg631.
Henderson A, Holloway A, Reeves R, Tremethick DJ: Recruitment of SWI/SNF to the human immunodeficiency virus type 1 promoter. Mol Cell Biol. 2004, 24: 389-397. 10.1128/MCB.24.1.389-397.2004.
Kiefer HL, Hanley TM, Marcello JE, Karthik AG, Viglianti GA: Retinoic acid inhibition of chromatin remodeling at the human immunodeficiency virus type 1 promoter. Uncoupling of histone acetylation and chromatin remodeling. J Biol Chem. 2004, 279: 43604-43613. 10.1074/jbc.M408069200.
Van Lint C: Role of chromatin in HIV-1 transcriptional regulation. Adv Pharmacol. 2000, 48: 121-160.
Jiang G, Espeseth A, Hazuda DJ, Margolis DM: c-Myc and Sp1 contribute to proviral latency by recruiting HDAC1 to the human immunodeficiency virus type 1 promoter. J Virol. 2007, 81: 10914-10923. 10.1128/JVI.01208-07.
Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, Aunis D, Rohr O: Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26: 412-423. 10.1038/sj.emboj.7601516.
Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, et al: Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007, 26: 424-435. 10.1038/sj.emboj.7601517.
Henikoff S: Heterochromatin function in complex genomes. Biochim Biophys Acta. 2000, 1470: O1-O8.
Bednarik DP, Cook JA, Pitha PM: Inactivation of the HIV LTR by DNA CpG methylation: evidence for a role in latency. EMBO J. 1990, 9: 1157-1164.
Saggioro D, Forino M, Chieco-Bianchi L: Transcriptional block of HTLV-I LTR by sequence-specific methylation. Virology. 1991, 182: 68-75. 10.1016/0042-6822(91)90649-V.
Feenstra A, Fewell J, Lueders K, Kuff E: In vitro methylation inhibits the promotor activity of a cloned intracisternal A-particle LTR. Nucleic Acids Res. 1986, 14: 4343-4352. 10.1093/nar/14.10.4343.
Bednarik DP, Mosca JD, Raj NB: Methylation as a modulator of expression of human immunodeficiency virus. J Virol. 1987, 61: 1253-1257.
Moriuchi M, Moriuchi H: Seminal fluid enhances replication of human T-cell leukemia virus type 1: implications for sexual transmission. J Virol. 2004, 78: 12709-12711. 10.1128/JVI.78.22.12709-12711.2004.
Nash KL, Jamil B, Maguire AJ, Alexander GJ, Lever AM: Hepatocyte-specific gene expression from integrated lentiviral vectors. J Gene Med. 2004, 6: 974-983. 10.1002/jgm.591.
Stunkel W, Huang Z, Tan SH, O'Connor MJ, Bernard HU: Nuclear matrix attachment regions of human papillomavirus type 16 repress or activate the E6 promoter, depending on the physical state of the viral DNA. J Virol. 2000, 74: 2489-2501. 10.1128/JVI.74.6.2489-2501.2000.
Saggioro D, Panozzo M, Chieco-Bianchi L: Human T-lymphotropic virus type I transcriptional regulation by methylation. Cancer Res. 1990, 50: 4968-4973.
Clarke MF, Trainor CD, Mann DL, Gallo RC, Reitz MS: Methylation of human T-cell leukemia virus proviral DNA and viral RNA expression in short- and long-term cultures of infected cells. Virology. 1984, 135: 97-104. 10.1016/0042-6822(84)90120-X.
Koiwa T, Hamano-Usami A, Ishida T, Okayama A, Yamaguchi K, Kamihira S, Watanabe T: 5'-long terminal repeat-selective CpG methylation of latent human T-cell leukemia virus type 1 provirus in vitro and in vivo. J Virol. 2002, 76: 9389-9397. 10.1128/JVI.76.18.9389-9397.2002.
Swindle CS, Kim HG, Klug CA: Mutation of CpGs in the murine stem cell virus retroviral vector long terminal repeat represses silencing in embryonic stem cells. J Biol Chem. 2004, 279: 34-41. 10.1074/jbc.M309128200.
Lorincz MC, Schubeler D, Groudine M: Methylation-mediated proviral silencing is associated with MeCP2 recruitment and localized histone H3 deacetylation. Mol Cell Biol. 2001, 21: 7913-7922. 10.1128/MCB.21.23.7913-7922.2001.
Tajima S, Tsukamoto M, Aida Y: Latency of viral expression in vivo is not related to CpG methylation in the U3 region and part of the R region of the long terminal repeat of bovine leukemia virus. J Virol. 2003, 77: 4423-4430. 10.1128/JVI.77.7.4423-4430.2003.
Ishida T, Hamano A, Koiwa T, Watanabe T: 5' long terminal repeat (LTR)-selective methylation of latently infected HIV-1 provirus that is demethylated by reactivation signals. Retrovirology. 2006, 3: 69-10.1186/1742-4690-3-69.
Pion M, Jordan A, Biancotto A, Dequiedt F, Gondois-Rey F, Rondeau S, Vigne R, Hejnar J, Verdin E, Hirsch I: Transcriptional suppression of in vitro-integrated human immunodeficiency virus type 1 does not correlate with proviral DNA methylation. J Virol. 2003, 77: 4025-4032. 10.1128/JVI.77.7.4025-4032.2003.
Lorincz MC, Schubeler D, Hutchinson SR, Dickerson DR, Groudine M: DNA methylation density influences the stability of an epigenetic imprint and Dnmt3a/b-independent de novo methylation. Mol Cell Biol. 2002, 22: 7572-7580. 10.1128/MCB.22.21.7572-7580.2002.
Jordan A, Defechereux P, Verdin E: The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J. 2001, 20: 1726-1738. 10.1093/emboj/20.7.1726.
Jordan A, Bisgrove D, Verdin E: HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22: 1868-1877. 10.1093/emboj/cdg188.
Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV: Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005, 122: 169-182. 10.1016/j.cell.2005.06.006.
Lewinski MK, Bisgrove D, Shinn P, Chen H, Hoffmann C, Hannenhalli S, Verdin E, Berry CC, Ecker JR, Bushman FD: Genome-wide analysis of chromosomal features repressing human immuno-deficiency virus transcription. J Virol. 2005, 79: 6610-6619. 10.1128/JVI.79.11.6610-6619.2005.
Han Y, Lassen K, Monie D, Sedaghat AR, Shimoji S, Liu X, Pierson TC, Margolick JB, Siliciano RF, Siliciano JD: Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. J Virol. 2004, 78: 6122-6133. 10.1128/JVI.78.12.6122-6133.2004.
Osborne CS, Pasceri P, Singal R, Sukonnik T, Ginder GD, Ellis J: Amelioration of retroviral vector silencing in locus control region beta-globin-transgenic mice and transduced F9 embryonic cells. J Virol. 1999, 73: 5490-5496.
Lutzko C, Senadheera D, Skelton D, Petersen D, Kohn DB: Lentivirus vectors incorporating the immunoglobulin heavy chain enhancer and matrix attachment regions provide position-independent expression in B lymphocytes. J Virol. 2003, 77: 7341-7351. 10.1128/JVI.77.13.7341-7351.2003.
Zentilin L, Qin G, Tafuro S, Dinauer MC, Baum C, Giacca M: Variegation of retroviral vector gene expression in myeloid cells. Gene Ther. 2000, 7: 153-166. 10.1038/sj.gt.3301057.
McInerney JM, Nawrocki JR, Lowrey CH: Long-term silencing of retroviral vectors is resistant to reversal by trichostatin A and 5-azacytidine. Gene Ther. 2000, 7: 653-663. 10.1038/sj.gt.3301155.
Kuriyama S, Sakamoto T, Kikukawa M, Nakatani T, Toyokawa Y, Tsujinoue H, Ikenaka K, Fukui H, Tsujii T: Expression of a retrovirally transduced gene under control of an internal housekeeping gene promoter does not persist due to methylation and is restored partially by 5-azacytidine treatment. Gene Ther. 1998, 5: 1299-1305. 10.1038/sj.gt.3300738.
Yao S, Sukonnik T, Kean T, Bharadwaj RR, Pasceri P, Ellis J: Retrovirus silencing, variegation, extinction, and memory are controlled by a dynamic interplay of multiple epigenetic modifications. Mol Ther. 2004, 10: 27-36. 10.1016/j.ymthe.2004.04.007.
Pannell D, Osborne CS, Yao S, Sukonnik T, Pasceri P, Karaiskakis A, Okano M, Li E, Lipshitz HD, Ellis J: Retrovirus vector silencing is de novo methylase independent and marked by a repressive histone code. EMBO J. 2000, 19: 5884-5894. 10.1093/emboj/19.21.5884.
Katz RA, Jack-Scott E, Narezkina A, Palagin I, Boimel P, Kulkosky J, Nicolas E, Greger JG, Skalka AM: High-frequency epigenetic repression and silencing of retroviruses can be antagonized by histone deacetylase inhibitors and transcriptional activators, but uniform reactivation in cell clones is restricted by additional mechanisms. J Virol. 2007, 81: 2592-2604. 10.1128/JVI.01643-06.
Ramunas J, Montgomery HJ, Kelly L, Sukonnik T, Ellis J, Jervis EJ: Real-time fluorescence tracking of dynamic transgene variegation in stem cells. Mol Ther. 2007, 15: 810-817.
Lorincz MC, Schubeler D, Goeke SC, Walters M, Groudine M, Martin DI: Dynamic analysis of proviral induction and de novo methylation: implications for a histone deacetylase-independent, methylation density-dependent mechanism of transcriptional repression. Mol Cell Biol. 2000, 20: 842-850. 10.1128/MCB.20.3.842-850.2000.
Mok HP, Javed S, Lever A: Stable gene expression occurs from a minority of integrated HIV-1-based vectors: transcriptional silencing is present in the majority. Gene Ther. 2007, 14: 741-751. 10.1038/sj.gt.3302923.
Fang JY, Mikovits JA, Bagni R, Petrow-Sadowski CL, Ruscetti FW: Infection of lymphoid cells by integration-defective human immunodeficiency virus type 1 increases de novo methylation. J Virol. 2001, 75: 9753-9761. 10.1128/JVI.75.20.9753-9761.2001.
Mikovits JA, Young HA, Vertino P, Issa JP, Pitha PM, Turcoski-Corrales S, Taub DD, Petrow CL, Baylin SB, Ruscetti FW: Infection with human immunodeficiency virus type 1 upregulates DNA methyltransferase, resulting in de novo methylation of the gamma interferon (IFN-gamma) promoter and subsequent downregulation of IFN-gamma production. Mol Cell Biol. 1998, 18: 5166-5177.
Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, Ecker JR, Bushman FD: Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2: E234-10.1371/journal.pbio.0020234.
Kim YK, Bourgeois CF, Pearson R, Tyagi M, West MJ, Wong J, Wu SY, Chiang CM, Karn J: Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency. EMBO J. 2006, 25: 3596-3604. 10.1038/sj.emboj.7601248.
Karn J: Tackling Tat. J Mol Biol. 1999, 293: 235-254. 10.1006/jmbi.1999.3060.
Emerman M, Malim MH: HIV-1 regulatory/accessory genes: keys to unraveling viral and host cell biology. Science. 1998, 280: 1880-1884. 10.1126/science.280.5371.1880.
Raha T, Cheng SW, Green MR: HIV-1 Tat stimulates transcription complex assembly through recruitment of TBP in the absence of TAFs. PLoS Biol. 2005, 3: e44-10.1371/journal.pbio.0030044.
Kumar PP, Purbey PK, Ravi DS, Mitra D, Galande S: Displacement of SATB1-bound histone deacetylase 1 corepressor by the human immunodeficiency virus type 1 transactivator induces expression of interleukin-2 and its receptor in T cells. Mol Cell Biol. 2005, 25: 1620-1633. 10.1128/MCB.25.5.1620-1633.2005.
Calzado MA, Sancho R, Munoz E: Human immunodeficiency virus type 1 Tat increases the expression of cleavage and polyadenylation specificity factor 73-kilodalton subunit modulating cellular and viral expression. J Virol. 2004, 78: 6846-6854. 10.1128/JVI.78.13.6846-6854.2004.
West AG, Huang S, Gaszner M, Litt MD, Felsenfeld G: Recruitment of histone modifications by USF proteins at a vertebrate barrier element. Mol Cell. 2004, 16: 453-463. 10.1016/j.molcel.2004.10.005.
Goetze S, Baer A, Winkelmann S, Nehlsen K, Seibler J, Maass K, Bode J: Performance of genomic bordering elements at predefined genomic loci. Mol Cell Biol. 2005, 25: 2260-2272. 10.1128/MCB.25.6.2260-2272.2005.
Burgess-Beusse B, Farrell C, Gaszner M, Litt M, Mutskov V, Recillas-Targa F, Simpson M, West A, Felsenfeld G: The insulation of genes from external enhancers and silencing chromatin. Proc Natl Acad Sci USA. 2002, 99 (Suppl 4): 16433-16437. 10.1073/pnas.162342499.
Klase Z, Kale P, Winograd R, Gupta MV, Heydarian M, Berro R, McCaffrey T, Kashanchi F: HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol Biol. 2007, 8: 63-10.1186/1471-2199-8-63.
Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, et al: Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004, 306: 704-708. 10.1126/science.1099962.
He J, Yang Q, Chang LJ: Dynamic DNA methylation and histone modifications contribute to lentiviral transgene silencing in murine embryonic carcinoma cells. J Virol. 2005, 79: 13497-13508. 10.1128/JVI.79.21.13497-13508.2005.
Chun TW, Davey RT, Engel D, Lane HC, Fauci AS: Re-emergence of HIV after stopping therapy. Nature. 1999, 401: 874-875. 10.1038/44755.
Davey RT, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, Natarajan V, Lempicki RA, Adelsberger JW, Miller KD, et al: HIV-1 and T cell dynamics after interruption of highly active anti-retroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci USA. 1999, 96: 15109-15114. 10.1073/pnas.96.26.15109.
Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, Wiegand A, Landay AL, Coombs RW, Richman DD, Mellors JW, et al: Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet. 2005, 366: 549-555. 10.1016/S0140-6736(05)67098-5.
Kumar A: The silent defense: micro-RNA directed defense against HIV-1 replication. Retrovirology. 2007, 4: 26-10.1186/1742-4690-4-26.
Jackson RJ, Standart N: How do microRNAs regulate gene expression?. Sci STKE. 2007, 2007: re1-10.1126/stke.3672007re1.
Bartel DP: MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004, 116: 281-297. 10.1016/S0092-8674(04)00045-5.
Tristem M: Identification and characterization of novel human endogenous retrovirus families by phylogenetic screening of the human genome mapping project database. J Virol. 2000, 74: 3715-3730. 10.1128/JVI.74.8.3715-3730.2000.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Mok, HP., Lever, A.M. Chromatin, gene silencing and HIV latency. Genome Biol 8, 228 (2007). https://doi.org/10.1186/gb-2007-8-11-228
Published:
DOI: https://doi.org/10.1186/gb-2007-8-11-228