
Abstract
The abnormalities found in human Down syndrome (trisomy 21) have been thought to result from increased expression of genes on chromosome 21 because of their higher gene dosage. Now, several groups have shown this to be generally the case, but some inter-individual variability and other exceptions were found.
Similar content being viewed by others

The gene-dosage hypothesis for Down syndrome
The Down syndrome (trisomy 21) phenotype is characterized by abnormalities affecting most organs and organ systems [1]. Although the extent and severity of the abnormalities is highly variable among individuals, all have some level of intellectual disability that is associated with specific brain regions and the performance of specific cognitive tasks [2, 3]. The incidence of Down syndrome remains at approximately 1 in 800 live births, and given the intellectual disabilities, it is a significant social and medical issue. Although it has been known for several decades that Down syndrome is caused by an extra, normal copy of the long arm of human chromosome 21 (21q), the molecular and cellular events linking the presence of an extra chromosome to the phenotypic features are unknown. The central hypothesis in Down-syndrome research is that gene dosage results in a 50% increase in expression of genes on chromosome 21q, and that this directly or indirectly alters the timing, pattern or extent of development. Accordingly, an essential question in Down-syndrome research is: are all trisomic genes overexpressed in all tissues and at all time points? If not, then which genes are overexpressed, and when and where? Answers to these questions are critical for determining which genes are relevant to phenotype development, for linking expression of specific genes to specific phenotypic features, and to account for phenotypic variability.
Chromosome 21 encodes over 300 genes and predicted genes [4]. These include approximately 170 protein-coding genes with clear orthologs in the mouse genome plus a significant number of gene models predicted on the basis of spliced expressed sequence tags (ESTs) with verified expression. Because analyses of cases of Down syndrome that are due to partial trisomies of chromosome 21q have not eliminated any significant segment of 21q from containing genes potentially impacting cognitive function [5], and because we understand so little about individual gene functions, essentially all genes within 21q need to be considered as candidates for relevance to the Down-syndrome phenotype. Given the large number of genes involved, determining which genes are overexpressed requires a large-scale approach, which is complicated by the small differences in expression level (50%) between normal and Down syndrome individuals predicted by gene dosage. Recently, several groups have contributed to progress in this area, by screening microarrays with RNA from brains of human fetuses with Down syndrome [6] or a trisomic mouse model [7], by screening a cDNA array containing mouse orthologs of human chromosome 21 genes with RNA from several tissues of a mouse model [8], and by using quantitative reverse-transcriptase-coupled (RT) PCR analysis of RNA from different tissues of a mouse model at different ages [9]. The results of all four studies support the hypothesis that gene-dosage effects exist in Down syndrome, but they also show that dosage effects may be specific to particular genes, alleles and/or tissues, and that background and stochastic or transient effects may be confounding factors.
Large-scale studies of gene-dosage effects in Down-syndrome fetuses and trisomic mice
Mao et al. [6] screened two Affymetrix oligonucleotide arrays (containing probes for approximately 12,000 and 22,000 human genes) with RNA from age-matched Down-syndrome and euploid control fetuses at 17-20 weeks gestation (see Table 1). They used RNA from four normal and four Down-syndrome cerebrums and from four normal and four Down-syndrome-derived astrocyte cell lines cultured from cerebral cortex. Samples were analyzed individually in order to detect variation between individuals. From all analyses, a global increase in expression level of 25 chromosome 21 genes was observed in the Down-syndrome samples compared to the euploid controls. The increase overall was consistent with predictions if gene expression followed gene dosage, but levels varied among individuals such that there were examples of individual genes showing no increases in comparisons of individual Down-syndrome-euploid pairs (see also below). Importantly, these variations in expression levels might be linked to phenotypic variations that would have been apparent at later developmental time points. Differences in expression levels of genes on other chromosomes - about 85 genes whose expression increased and 100 whose expression decreased - were also observed.
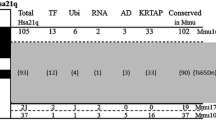
Regions of human chromosome 21 are orthologous to segments of three mouse chromosomes [10] (Figure 1): the centromere-proximal 30 megabase (Mb) region of chromosome 21 up to and including the ZNF295 gene is orthologous to the telomeric region of mouse chromosome 16 [10] and the next two approximately 1-2 Mb segments of chromosome 21 are orthologous to regions of mouse chromosomes 17 and 10, respectively. Because of the large region of homology with mouse chromosome 16, development of segmental trisomy mouse models for Down syndrome has focused on this region. Currently, the best mouse models of Down syndrome are the Ts65Dn mouse (reviewed in [11, 12]) and the Ts1Cje mouse [13]. Ts65Dn mice have three copies of 94 genes orthologous to human chromosome 21 genes, contained within chromosome 16 from the Gabpa/App gene cluster to the distal telomere [4]. Ts1Cje mice are trisomic for 71 orthologs of human chromosome 21 genes, within chromosome 16 distal to (and not including) the superoxide dismutase 1 (Sod1) gene to the telomere [13].

Human chromosome 21 and homologous regions in mouse models. Regions that are syntenic with mouse chromosomes are indicated on the left; those that are trisomic in the major mouse models are indicated on the right. See text for further details.
Amano et al. [7] screened Affymetrix oligonucleotide arrays representing about 11,000 mouse genes with RNA from whole brains of postnatal day zero Ts1Cje mice. Six trisomic females and six littermate controls were examined, and 38 genes within the trisomic segment showed detectable expression. Of these, 37 showed a mean increase in expression level of about 1.5-fold in the trisomic mice, consistent with gene-dosage effects. But out of all the possible trisomy:euploid comparisons (6 × 6 = 36), not all individual pairwise comparisons showed similar increases. Indeed, of the 37 genes, only 24 showed increases in 18 or more of the 36 possible pairs. Because these mice are maintained on an inbred background (C57BL/6J), the only genetic contribution to expression differences is the trisomic segment. Expression levels of ten trisomic genes were assayed by real-time RT-PCR using RNA from four additional trisomic and normal control mice. All showed relative expression increases of over 1.34-fold, including those with increases of about 1.2-fold in the array experiments, as well as the Prdm15 (Znf295) gene, which showed no increase in arrays. Also, similar to the results of Mao et al. [6], some non-trisomic genes showed altered expression levels: 59 showed levels under 0.7 times normal and 199 showed levels over 1.2 times normal, out of the 10,000 genes with detectable expression.
Kahlem et al. [8] created custom arrays containing cDNAs for 77 orthologs of human chromosome 21 genes that are trisomic in the Ts65Dn mouse. Arrays were screened with Ts65Dn RNA from nine tissues, including cerebellum, cortex and midbrain, in each case pooling RNAs from four individuals, aged 3-4 months. Expression of 66 trisomic genes was detected in at least one of the nine tissues. In eight tissues, overall levels of expression were consistent with gene dosage, with trisomy:euploid ratios ranging from about 1.63 and about 1.73 in cortex and heart, respectively, to about 1.23 in kidney. Only muscle, with ratios of 1.16, failed to show notable gene-dosage effects. A small number of specific gene-tissue combinations deviated from dosage effects; 10 combinations showed unchanged or decreased ratios and 15 showed ratios over 2.0. Quantitative RT-PCR analysis of several genes corroborated the array results in 78% of cases.
To circumvent the sensitivity limitations of microarrays, which do not always detect genes expressed at low levels, Lyle et al. [9] used real-time RT-PCR for experiments with Ts65Dn mice. RNA from brain, liver, kidney, heart, muscle and lung of postnatal day 30 mice, and brain, liver, kidney and heart of 11-month-old mice was used, in each case pooling material from four trisomic or euploid male mice. Assays of 78 trisomic genes showed an overall mean expression ratio of approximately 1.5. There were statistically significant variations, however. For example, 59 gene-tissue combinations (out of 666; 78 genes were tested in 10 tissues, and 114 gene-tissue combinations had no detectable expression) showed no significant increase in trisomic mice, and 26 gene-tissue combinations showed over 2.2-fold increases in trisomic mice.
Variability between individuals
Both Mao et al. [6] and Amano et al. [7] observed deviations from dosage predictions of trisomy: euploid RNA-expression ratios in some pairwise comparisons between individuals. Sources of such variation - apart from experimental artifacts and limitations - include allelic variation in trisomic genes or in background (euploid) genes that regulate trisomic genes and possible stochastic or environmental effects. Certainly, allelic variation has been observed in human gene-expression patterns [14–16]. Specifically for chromosome 21, promoter polymorphisms in six genes were reported that resulted in expression differences of 30-66% among alleles expressed in HEK293 embryonic kidney cells [17]. If an individual trisomic for a low-expressing allele is compared to a euploid individual disomic for a high-expressing allele, significant expression differences may be undetectable; conversely, trisomy for a high-expressing allele could result in much more than a 50% difference compared with a euploid for low-expressing alleles. Similar arguments apply to background variation (in euploid genes) in humans and in Ts65Dn mice (because the latter are maintained as F1 hybrids of two strains).
The experiments with Ts65Dn mice used RNAs pooled from multiple individuals [8, 9]. Variation between individuals is expected because of the history of the strain: in the Ts65Dn mice, the trisomic segment originated from the DBA/2J strain, but because of extensive early breeding to C57BL/6JEi, there may be no DBA/2J alleles remaining except immediately around the breakpoint of the rearrangement. The Ts65Dn strain is now maintained as F1 hybrids of C57BL/6JEi and C3H/HeSnJ [18]. Thus, the three alleles in individual mice may all derive from C57BL/6JEi or be combinations of C57BL/6JEi and C3H/HeSnJ, and inter-individual variation will be found.
Genetic variation does not exist, however, in the experiments with Ts1Cje mice, because they are maintained on the inbred C57BL/6J strain. The reported inter-individual variation in expression levels [7] must therefore have other causes, such as stochastic processes or subtle environmental effects, possibly fluctuating in time but frozen in these snapshots of expression patterns. This is consistent with reports of variable expression levels of 37 genes in the hippocampus of inbred rats (the Sprague-Dawley strain); 2-3-fold variations in average expression level were seen among 20 individuals, with as much as 10-fold variation seen with some genes [19].
Deviation from the expected 1.5-fold increase and variations in non-trisomic gene expression
Gene-tissue combinations with ratios of trisomy:euploid expression significantly greater or less than 1.5-fold were reported in the Ts65Dn experiments [8, 9]. Notably, Kahlem et al. [8] reported that expression of trisomic genes in skeletal muscle was increased only 1.16-fold, but Lyle et al. [9] reported that expression of only 28 of 78 genes was increased significantly less than 1.5-fold. Is this difference due to the different techniques used (arrays versus RT-PCR), or to age effects (day 30 versus 3-4-month-old mice)? Also, while the former study [8] reported only 5 gene-tissue combinations with levels increased over 1.5-fold (excluding testes), and 15 increased under 1.5-fold (out of 594 combinations), the latter [9] reported 123 combinations with over 1.5-fold increases and 298 with under 1.5-fold increases (out of 666). Although these differences are reported as statistically significant, in particular for data from Lyle et al. [9], the biological significance remains to be determined. For example, it is not possible to state that the cell will recognize 1.43-fold as different from i.1.50-fold for the hormonally upregulated Neu-associated kinase (Hunk) gene in the postnatal day 30 brain, or 1.41-fold as not different from normal for the GABPA transcription factor gene in the kidney [9].
Alterations in expression levels of many non-trisomic genes were reported by two of the studies [6, 7]. Currently, there is no known functional link between the euploid genes and the trisomic genes that would predict such dysregulation. Furthermore, 19 of the euploid genes found to have altered expression by Mao et al. [6] were reported to be expressed by Amano et al. [7], but none showed altered expression in the latter study. Such inconsistencies could be due to differences in the organism, tissue, or developmental time studied, or to subtle environmental effects.
From the results of these experiments [6–9], it is reasonable to conclude that expression of trisomic mRNAs in human Down syndrome and mouse chromosome 16 segmental trisomies is governed by gene dosage. Additional large-scale experiments along these lines, therefore, seem unnecessary and are unlikely to further illuminate the field. The inter-individual variation, and the possible specific effects on regulation of genes in different tissues at different times, however, argue that dosage still needs to be examined carefully, but this will be more productive when applied to specific genes, specific tissues or cell types, among individuals, and in relation to phenotypic variability. The preference, of course, is to examine expression at the protein level.
References
Epstein CJ: Down syndrome (trisomy 21). In Metabolic and Molecular Bases of Inherited Disease. Edited by: Scriver CA, Beaudet AL, Sly WS, Valle D. 1995, New York: McGraw Hill, 749-794.
Pennington BF, Moon J, Edgin J, Stedron J, Nadel L: The neuropsychology of Down syndrome: evidence for hippocampal dysfunction. Child Dev. 2003, 74: 75-93. 10.1111/1467-8624.00522.
Nadel L: Down's syndrome: a genetic disorder in biobehavioral perspective. Genes Brain Behav. 2003, 2: 156-166. 10.1034/j.1601-183X.2003.00026.x.
Gardiner K, Fortna A, Bechtel L, Davisson MT: Mouse models of Down syndrome: how useful can they be? Comparison of the gene content of human chromosome 21 with orthologous mouse genomic regions. Gene. 2003, 318: 137-147. 10.1016/S0378-1119(03)00769-8.
Korenberg JR, Chen XN, Schipper R, Sun Z, Gonsky R, Gerwehr S, Carpenter N, Daumer C, Dignan P, Disteche C, et al: Down syndrome phenotypes: the consequences of chromosomal imbalance. Proc Natl Acad Sci USA. 1994, 91: 4997-5001.
Mao R, Zielke CL, Zielke HR, Pevsner J: Global up-regulation of chromosome 21 gene expression in the developing Down syndrome brain. Genomics. 2003, 81: 457-467. 10.1016/S0888-7543(03)00035-1.
Amano K, Sago H, Uchikawa C, Suzuki T, Kotliarova SE, Nukina N, Epstein CJ, Yamakawa K: Dosage-dependent over-expression of genes in the trisomic region of Ts1Cje mouse model for Down syndrome. Hum Mol Genet. 2004, 13: 1333-1340. 10.1093/hmg/ddh154.
Kahlem P, Sultan M, Herwig R, Steinfath M, Balzereit D, Eppens B, Saran NG, Pletcher MT, South ST, Stetten G, et al: Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of Down syndrome. Genome Res. 2004, 14: 1258-1267. 10.1101/gr.1951304.
Lyle R, Gehrig C, Neergaard-Henrichsen C, Deutsch S, Antonarakis SE: Gene expression from the aneuploid chromosome in a trisomy mouse model of Down syndrome. Genome Res. 2004, 14: 1268-1274. 10.1101/gr.2090904.
Davisson MT, Bechtel LJ, Akeson EC, Fortna A, Slavov D, Gardiner K: Evolutionary breakpoints on human chromosome 21. Genomics. 2001, 78: 99-106. 10.1006/geno.2001.6639.
Davisson MT, Costa ACS: Mouse models of Down syndrome. In Mouse Models in the Study of Genetic Neurological Disorders. Volume 9 of Advances in Neurochemistry. Edited by: Popko B. 1999, New York: Plenum Press, 297-327.
Crnic LS, Pennington BF: Down syndrome: neuropsychology and animal models. Progr Infancy Res. 2000, 1: 69-111.
Sago H, Carlson EJ, Smith DJ, Kilbridge J, Rubin EM, Mobley WC, Epstein CJ, Huang TT: Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc Natl Acad Sci USA. 1998, 95: 6256-6261. 10.1073/pnas.95.11.6256.
Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW: Allelic variation in human gene expression. Science. 2002, 297: 1143-10.1126/science.1072545.
Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen KY, Morley M, Spielman RS: Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet. 2003, 33: 422-425. 10.1038/ng1094.
Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS, Cheung VG: Genetic analysis of genome-wide variation in human gene expression. Nature. 2004, 430: 743-747. 10.1038/nature02797.
Buckland PR, Coleman SL, Hoogendoorn B, Guy C, Smith SK, O'Donovan MC: A high proportion of chromosome 21 promoter polymorphisms influence transcriptional activity. Gene Expr. 2004, 11: 233-239.
Davisson MT, Schmidt C, Reeves RH, Irving NG, Akeson EC, Harris BS, Bronson RT: Segmental trisomy as a mouse model for Down syndrome. Prog Clin Biol Res. 1993, 384: 117-133.
Alfonso J, Pollevick GD, Castensson A, Jazin E, Frasch AC: Analysis of gene expression in the rat hippocampus using real time PCR reveals high inter-individual variation in mRNA expression levels. J Neurosci Res. 2002, 67: 225-234. 10.1002/jnr.10105.
Acknowledgements
This work was supported by the Fondation Jerome Lejeune.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Gardiner, K. Gene-dosage effects in Down syndrome and trisomic mouse models. Genome Biol 5, 244 (2004). https://doi.org/10.1186/gb-2004-5-10-244
Published:
DOI: https://doi.org/10.1186/gb-2004-5-10-244