
Abstract
Background
Androgens play a critical role in the development of prostate cancer-dysregulation of androgen-regulated growth pathways can led to hormone-refractory prostate cancer. A comprehensive understanding of androgen-regulated cellular processes has not been achieved to date. To this end, we have applied a large-scale proteomic approach to define cellular processes that are responsive to androgen treatment in LNCaP prostate cancer cells.
Results
Using isotope-coded affinity tags and mass spectrometry we identified and quantified the relative abundance levels of 1,064 proteins and found that distinct cellular processes were coregulated by androgen while others were essentially unaffected. Subsequent pharmacological perturbation of the cellular process for energy generation confirmed that androgen starvation had a profound effect on this pathway.
Conclusions
Our results provide evidence for the role of androgenic hormones in coordinating the expression of critical components involved in distinct cellular processes and further establish a foundation for the comprehensive reconstruction of androgen-regulated protein networks and pathways in prostate cancer cells.
Similar content being viewed by others


Background
Androgens are critical for the growth of normal and diseased prostate epithelium [1]. Androgenic ligands bind and activate the androgen receptor (AR) leading to the induction of prostate-specific genes associated with cell growth and the differentiated prostate epithelial phenotype [2]. The biological impact that androgens have on prostate epithelial cell function is underscored by the fact that androgen deprivation therapy is an established therapeutic regime in the treatment of advanced prostate cancer (PCa) [3]. However, PCa that emerges after androgen deprivation therapy routinely re-activates the androgen-response pathway during the development of hormone refractory PCa [4–9]. Large-scale transcript profiling studies have been initiated in order to identify androgen-regulated genes in human PCa cell lines with the intent that these targets will elucidate the pathways that result in hormone refractory PCa growth [10–13]. However, transcript-expression profiles do not always reflect changes that occur at the level of the protein [14]. As proteins are the biological effectors of cell function, an assessment of their quantity and activation state provides a more informative description of a cellular phenotype. Therefore, we have initiated a large-scale quantitative proteomic analysis to systematically identify and quantify androgen-mediated protein abundance changes in the human LNCaP prostate cancer cell line using the isotope-coded affinity tags (ICAT) and mass spectrometry [15, 16]. This investigation represents a first-step towards mapping a protein network regulated by androgens in neoplastic prostate epithelium.
Results and discussion
Identification and quantification of microsomal proteins
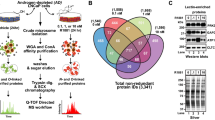
One of our primary research goals is to obtain a global view of androgen-mediated protein abundance changes in human prostate cancer cells in response to androgens. We have initiated studies to systematically quantify all androgen-mediated protein abundance changes that occur within the nuclear, membranous and cytosolic fractions of human prostate cancer cells using the ICAT reagent method. In this article we focused on characterizing the protein abundance changes found within the membranous fraction of LNCaP cells using a crude microsomal protein preparation methodology previously employed [16]. Total protein isolated from the microsomes of androgen-depleted (AD cells) and androgen-stimulated (AS cells, 10 nM R1881 for 72 hrs) LNCaP cells were subjected to proteomic analysis using the ICAT method. The ICAT-labeled peptide mixture (AD cells, d0-reagent (light ICAT isotope reagent); AS cells, d8-reagent (heavy ICAT isotope reagent)) was subjected to three-dimensional liquid chromatography and analyzed by tandem mass spectrometry (MS/MS), which resulted in the identification and quantification of 1,064 proteins (Figure 1a and panel 1, pages 1-58 in Additional data file 1) [15, 16]. A plot of the natural log of the d8-heavy/d0-light abundance ratios of the identified proteins revealed that 98.4% (1,047/1,064 proteins) changed less than four-fold and 81.2% (864/1,064) changed less than two-fold in response to androgen exposure (Figure 1a and panel 1, pages 1-58 in Additional data file 1).

Identified proteins derived from the microsomes of AD and AS cells ordered by ratios of abundance. (a) Plot of the natural log d8/d0 ratio of the total number of ICAT reagent-labeled proteins derived from microsomes of 72 h AD (d0, light ICAT isotope reagent) and AS (10 nM R1881(methyltrienolone)) (d8, heavy ICAT isotope reagent) cells. Red and green represent androgen-induced and repressed protein abundance changes. A total of 1,064 proteins were identified and quantified (detailed in Materials and methods). (A list of all proteins is contained in panel 1 (pages 1-58) in Additional data file 1.) (b) PSA polypeptide sequence. Bold italics denote tryptic, cysteine-containing peptides. (c) Observed ICAT reagent-labeled PSA peptides. The number after each peptide sequence denotes the number of times that a peptide was identified. (d) PSA expression in the microsomes of AS cells. (e-l) Comparative immunoblot analysis of AD (d0) and AS (d8) microsomal extracts probed with antibodies to PSA, TMPRS22, FAS, LDLR, β-catenin, HSP60, MST-3 and FasL, respectively. Mass spectrometric d0/d8 ratios as displayed by the XPRESS quantification software [16] are shown below each blot. The asterisk denotes the cleaved processed form of the TMPRS22 protease [19].
Androgenic hormones have been reported to regulate the expression of a large number of genes in the prostate [17]. One of the best-characterized androgen responsive targets is prostate specific antigen (PSA), a secreted serine protease that is commonly used to detect early stage PCa in men [17]. The PSA polypeptide sequence is predicted to generate six cysteine-containing tryptic peptides, all of which were identified and quantified by the ICAT method (Figure 1b,c). PSA was easily detectable in the microsomes of AS cells (Figure 1d). We extended the orthogonal verification of protein abundance ratios determined by quantitative mass spectrometry by subjecting a small group of proteins to semi-quantitative western blot analysis (Figure 1f-l). A four-fold increase in AS cell PSA expression was determined by the ICAT method (Figure 1e and panel 2, page 59 in Additional data file 1); a finding that was confirmed by Western analysis. An approximately eight-fold increase in the expression of the known androgen-regulated transmembrane serine protease 2 (TMPRSS2) level in AS cells was measured by the ICAT method, and Western analysis supported this result (Figure 1f) [18, 19]. Androgens are known to activate the lipogenic pathway in PCa cells [20–22]. The ICAT method demonstrated that fatty acid synthase (FAS) and low-density lipoprotein receptor (LDLR) proteins were increased by approximately three- and approximately 1.8-fold, respectively in AS cells (Figure 1g,h). β-catenin, a component of cellular adherens junctions [23] that also functions as a co-activator of AR mediated transcription [24–26], was increased approximately 1.3-fold in AS cells (Figure 1i), while the heat shock chaperonin 60 protein (HSP60), was increased approximately 1.7-fold in AD cells, as measured by the ICAT method (Figure 1j). The abundance ratios for these proteins were calculated from the assessments of at least three independent peptide quantifications, providing an opportunity to estimate the amount of error in the quantitative measurement (panel 2, page 59 in Additional data file 1). We also confirmed protein quantifications that were derived from single ICAT peptide ratios. For example, the human sterile 20 serine/threonine protein kinase 3 (mst-3) [27] and the Type II transmembrane protein Fas ligand (FasL) [28] were quantified based upon single ICAT peptide ratios (panel 3, page 60 in Additional data file 1). The ICAT method measured an approximately 1.8-fold increase in mst-3 levels in AS cells, while FasL levels were relatively unchanged between AD and AS cells, which was verified by immunoblot analysis (Figure 1k,l, respectively). While it is preferable to assess multiple ICAT-labeled peptides corresponding to a particular protein for optimal quantitative accuracy, these validation experiments demonstrate the validity of protein quantifications determined by single ICAT measurements.
Assignment of proteins into cellular process categories
Interpretation of large-scale cDNA microarray and proteomic analyses has traditionally been plagued by the inability to organize the vast amounts of data into a meaningful biological framework from which new hypotheses can be developed and tested experimentally [29]. Therefore, we sought to gain a deeper understanding of the functional pathways that were represented in the 1,064-protein dataset. Proteins were assigned to one or several cellular process categories (of a possible 44 categories) based upon their annotation at the Incyte Human Proteome Database [30]. These cellular process categories are a reflection of the biological process ontology adopted by the Gene Ontology hierarchical classification system [31, 32]. Non-annotated proteins were assigned to the cellular process referred to as 'unknown process', which allowed for the assignment of all 1,064 proteins to one or multiple cellular process categories (Figure 2 and panel 4, pages 61-238 in Additional data file 1). The unknown process category contained approximately 19% of the identified proteins (Figure 2), which is consistent with the observation that a significant fraction of genes encoded in the human genome have no defined function [33]. The category of 'signal transduction' contained the second largest number of identified proteins at approximately 9%, while cellular processes associated with cellular growth, ion transport and metabolism represented approximately 33% of all identified proteins (for example, protein synthesis, 5.38%; energy generation, 4.68%; vesicular transport, 4.30%; protein modification, 4.24%; lipid metabolism, 3.99%; small molecule transport, 3.86%; nucleotide metabolism, 3.48%; carbohydrate metabolism, 3.42%). Of 45 defined cellular process categories, 42 contained at least one protein observed in this experiment, while no proteins were assigned to the categories of 'asymmetric cell division', 'mitochondrial transcription' and 'Pol I transcription' (Figure 2).

Classification of microsomal proteins in prostate epithelial cells into cellular process categories. The 1,064 proteins identified and quantified in this study were assigned to one or several cellular process categories on the basis of their annotation at the Incyte Human Proteome Database [30]. Unassigned proteins were placed into the cellular process category referred to as 'unknown process'. The numbers in parentheses represent the total number of proteins assigned to that specific cellular process category.
Identification of androgen-sensitive and androgen-insensitive cellular processes
Assignment of the 1,064 proteins to a specific cellular process provided a unique opportunity to determine whether certain cellular processes are under the control of androgens in LNCaP cells. For example, androgens are known to regulate lipid metabolism in PCa cells [20–22], and a group of proteins in the 'lipid metabolism' cellular process contained ICAT ratios (d0:d8) that were consistent with the androgen-mediated stimulation of this cellular process (panel 5, pages 239-242 in Additional data file 1). This led us to investigate the distribution among cellular process categories of proteins showing expression change (up or down) greater than a predetermined threshold. For example, among the 1,064 proteins detected, 350 (33%) showed an androgen-response change greater than or equal to 1.6-fold (panel 6, page 243 in Additional data file 1). If these 350 proteins were distributed randomly among the cellular process categories, it would be expected that about one third of the proteins in each category would show expression change ≥1.6. Alternatively, there may be categories in which androgen-responsive proteins are over-represented, thus identifying androgen-responsive cellular processes. Also, there may be categories in which androgen-responsive proteins are under-represented, thus identifying cellular processes likely to be androgen-insensitive.
Using the hypergeometric distribution [34], we calculated p-values for the observed number of proteins that changed by more than a specified ratio threshold compared with the number expected at random for each cellular process category. In Table 1, a low p-value indicates that the observed number of proteins with changes ≥1.6 deviates significantly from the number expected at random (panel 6, page 243 in Additional data file 1; see also panel 8 on page 244 and panel 9 on page 245, for p-values for ICAT ratio thresholds of ≥1.8 and ≥2.0, respectively). This deviation can be either greater (over-representation) or lesser (under-representation) than expected. Table 1 shows cellular process categories containing p-values less than 0.004. Each of these categories represents cellular processes likely to be either androgen-responsive (over-represented) or androgen-insensitive (under-represented). The androgen-responsive cellular processes included 'RNA splicing' (5.92 × 10-5), 'energy generation' (3.01 × 10-4), 'nucleotide metabolism' (5.20 × 10-4), 'lipid metabolism' (2.30 × 10-3) and 'RNA processing/modification' (2.32 × 10-3). The androgen-insensitive cellular processes included 'vesicular transport' (1.87 × 10-3) and 'protein synthesis' (3.1 × 10-3).
Coregulated protein expression associated with energy and lipid metabolism
The identification of both androgen-responsive and androgen-insensitive cellular processes prompted us to document the direction (either up or down) of those proteins with expression ratios ≥1.6 among these cellular processes. These proteins could display either discordant or concordant protein abundance changes. Concordant increases or decreases in protein levels would support the concept of androgen-mediated coregulated protein expression involving specific cellular process categories. The large number of proteins with expression ratios ≥1.6 provided the opportunity to sort these proteins based upon their expression ratio (d0/d8 and d8/d0) and determine the direction of change in response to androgen (panel 9, pages 246-269 in Additional data file 1). For example, inspection of the 'RNA splicing' cellular process (panel 9, pages 265-269 in Additional data file 1, category 38) revealed that 12 out of the 14 proteins containing ICAT ratios ≥1.6 were all increased in AD cells, suggesting a coordinated coregulated protein expression pattern influencing this cellular process. To highlight the connectivity of coregulated protein expression patterns displayed among the androgen-responsive cellular processes, we reconstructed the biochemical pathways associated with the 'energy generation' and 'lipid metabolism' biochemical processes (Figure 3 and panel 10, pages 270-274 in Additional data file 1). This included the tricarboxylic acid cycle (TCA), oxidative phosphorylation (Oxphos), glycolysis, fatty acid β-oxidation, glycogen metabolism, fatty acid synthesis and cholesterol synthesis pathways. As previously reported [20, 21], androgen stimulated many of the enzymes (for example, FAS and citrate synthase (CS)) involved in the anabolic processes of fatty acid and cholesterol synthesis (Figure 3). Androgen also stimulated glycogen catabolism [35], as both glycogen phosphorylase and glycogen transferase increased in AS cells, suggesting that androgens promote mobilization of glycogen stores for energy production. However, androgen also had a repressive effect on the catabolic pathways of glycolysis, TCA, Oxphos and fatty acid oxidation, as these pathways were coordinately decreased in AS cells (Figure 3). In addition, enzymes required for the breakdown of straight and branched-chain fatty acids in the mitochondria and peroxisome were specifically increased in AS cells (Figure 3). This suggests that these pathways were up-regulated to increase the synthesis of two carbon acetyl-CoA subunits for subsequent oxidation in the TCA cycle and the synthesis of ATP by the Oxphos pathway. This observation is clinically relevant to PCa, since α-methylacyl-CoA racemace (AMACR), the enzyme which catalyzes a required step for the β-oxidation pathway in peroxisomes [36], is specifically over-expressed in PCa and represents a new molecular marker for early stage PCa [37–39]. De-repression of the α and β branched-chain fatty acid oxidation pathways may represent an early androgen-deregulated step in PCa development [40]. The distinct increase in protein components of the glycolytic, TCA, and Oxphos pathways in AD cells also suggests that these pathways were specifically increased to meet a metabolic demand for ATP in AD cells (Figure 3). Interestingly, normal prostate epithelial cells are known to accumulate and secrete large amounts of citrate into the lumen of the prostate [41]; citrate, a required intermediate for the complete oxidation of glucose and fats, has been shown to decline dramatically in PCa [42]. A metabolic shift towards increased citrate oxidation has been proposed to represent an early step when a pre-neoplastic cell transforms into a malignant PCa cell [43]. Our results show that de-repression of energy related pathways could be a natural consequence of inactivating the androgen signaling pathway in PCa.

Androgen-responsive metabolic protein network in LNCaP cells. Specific biochemical pathways represented by the 'energy generation' and 'lipid metabolism' cellular processes, which included the TCA cycle, oxidative phosphorylation, glycolysis, glucogenesis, cholesterol synthesis, glycogen catabolism, fatty acid synthesis and fatty acid oxidation are shown to demonstrate the coordinated coregulated protein expression pattern in response to androgen in LNCaP cells. Black roman font represents proteins not identified using the ICAT reagent method. Black italic font represents proteins that did not change with androgen exposure (d0/d8 ratios > 0.62 and < 1.60). Red represents proteins whose abundance increased with androgen treatment (d0/d8 ratios ≤0.62). Green represents proteins whose abundance decreased following androgen treatment (d0/d8 ratios ≥1.6).
Chemical perturbations of energy metabolism in prostate cancer cells
AD cells increased the levels of proteins associated with the 'energy generation' cellular process, suggesting that these cells were metabolically different in the context of mitochondrial function compared with AS cells (Figure 3). We observed that androgen starvation caused LNCaP cells to adopt a distinct neuronal morphology (Figure 4a, middle panel) when compared to steady-state (SS) or AS cells (Figure 4a, left and right panel, respectively), a finding also reported by other investigators [44]. The cellular distribution of mitochondria in AD cells was quite distinct as the mitochondria traversed the length of the long dendritic-like processes (Figure 4b, middle panel), whereas SS and AS cells displayed a more perinuclear mitochondrial localization pattern (Figure 4b, left and right panels, respectively). The coordinated increase in proteins of the 'energy generation' cellular process in AD cells led us to hypothesize that these cells were stressed at the level of the mitochondria. To test whether AD cells had an increased sensitivity to pharmacological agents that specifically disrupt different steps of the Oxphos pathway when compared with AS cells, we incubated SS, AD and AS cells with rotenone (10 μM), carbonyl cyanide m-chlorophenylhydrazone (CCCP) (50 μM), antimycin A (10 μg/ml) or oligomycin (5 μg/ml) and determined cell viability 24 hours later (Figure 4c). As shown in Figure 4c, a 24 h exposure to rotenone, which specifically blocks the NADH dehydrogenase of complex I [45] elicited a slightly higher level of cell death in AD (38.1 ± 3.0%) cells when compared to AS (31.8 ± 2.0%) and SS cells (27.2 ± 1.9%). CCCP, a small aliphatic agent that uncouples electron transport and ATP production [46], resulted in higher levels of cell death in AD cells (53.7 ± 1.9%) when compared to AS (41.5 ± .79%) and SS cells (29.0 ± 2.9%). Antimycin, which specifically blocks complex III of the respiratory chain between cytochrome b and cytochrome c1 [47], induced higher levels of cell death in AD cells (50.9 ± 2.4%) when compared with AS cells (31.1 ± 2.6%) or SS cells (19.9 ± 1.1%). Lastly, oligomycin, which blocks ATP synthesis by binding the F0F1-ATPase of complex V of the respiratory chain [48], also induced higher levels of cell death in AD cells (48.5 ± 1.7%) than in AS (24.1 ± 6.6%) and SS cells (15.8 ± 2.3%). These results clearly show that androgen depletion sensitizes LNCaP cells to cell death induced by drugs that perturb the respiratory chain pathway. A number of plausible explanations could explain the increased cell death response of AD cells to mitochondrial poisons. For example, the neuronal phenotype of AD cells, which resembles the neuroendocrine phenotype of some advanced, hormone refractory PCa that arise after long-term androgen ablation therapy [49, 50], may utilize different carbon sources that predispose them to activation of the cell death pathway if mitochondrial function is compromised. Although the biochemical mechanism(s) responsible for inducing higher levels of death in AD cells has yet to be defined, we have established a novel relationship between androgen and energy metabolism in LNCaP cells that will require further scientific inquiry. Whether or not this relationship is maintained in other androgen-dependent prostate cancer cell lines is unknown. However, in the context of clinical therapy for prostate carcinoma, applying traditional androgen-ablation methods in combination with drugs that specifically target the mitochondria may increase the therapeutic efficacy of existing treatment approaches.

Respiratory chain inhibitors induce higher levels of cell death in AD cells. (a) Light photomicrographs of SS, AD and AS cells after 72 h. (b) Distribution of mitochondria in SS (left), AD (center) and AS (right) cells. (c) Respiratory chain inhibitors rotenone (10 μM), CCCP (50 μM), antimycin A (10 μg/ml) and oligomycin (5 μg/ml) induce higher levels of cell death in AD cells, as assessed by a cell counting assay after a 24 h incubation with the specified drugs. Data are representative of three independent experiments.
Conclusions
In this article we have applied a large-scale quantitative mass spectrometry approach to reconstruct the androgen-responsive protein network in human prostate cancer cells. This study represents the largest dataset of identified and quantified proteins from human cells to date. Our primary goal was to construct an androgen-regulated protein abundance map from the microsomal fraction of prostate cancer cells. Future analyses that quantify androgen-mediated protein abundance changes from the nuclear and cytoplasmic fractions will provide a more global perspective of the androgen-responsive protein networks in prostate cancer cells. From the current analysis we were able to identify both androgen-sensitive and androgen-insensitive cellular processes. Androgen-responsive cellular processes displayed a coregulated protein expression pattern in response to androgen, suggesting that these cellular processes were coordinately controlled and share a common gene expression regulatory network (for example, common transcription factor binding sites in their promoters) [51]. The androgen-responsive proteins of the 'energy generation' cellular process were specifically increased in AD cells, suggesting these cells were metabolically stressed. Pharmacological perturbation with drugs that specifically blocked mitochondrial respiration induced higher levels of cell death in AD cells when compared with AS cells, thus validating the utility of applying a large-scale quantitative proteomic approach to uncover biological differences between two cellular states. Systematic analysis of the other androgen-responsive cellular processes may increase our understanding of androgen and AR signaling in the development of androgen-refractory PCa. For example, proteins comprising the 'RNA splicing' and 'RNA processing/modification' cellular processes were specifically increased (panel 9, pages 262-269 in Additional data file 1, categories 37 and 38) in AD cells. Interestingly, steroid receptors can coordinate transcription and splicing through their association with specific nuclear receptor coregulatory proteins [52]. AR may coordinate a similar activity in prostate epithelial cells, as AR can physically interact with FLH2 [53], a tissue-specific AR co-activator, which can bind the polypyrimidine tract binding protein-associated splicing factor [54]. Future studies will be required to elucidate the relationship, if any, between androgen and RNA splicing/metabolism in prostate cancer cells, given that many cancers are associated with abnormalities in alternative splicing [55].
We also identified several generally androgen-insensitive cellular processes, including 'vesicular transport' and 'protein synthesis'. However, androgens may still influence these pathways by regulating the expression of a few critical modulators. These processes may also be regulated through alternative regulatory mechanisms that involve post-translational modifications (for example, phosphorylation and ubiquitination). Future analyses will be required to substantiate and test these hypotheses more directly. The androgen coregulated protein expression patterns described in this study are consistent with the coregulated gene expression patterns detected by large-scale cDNA microarray analyses [56]. Coregulated gene and protein networks may represent a common theme for coordinating cellular processes in the cell. Targeting and blocking the activity of specific regulators through RNA interference or chemical inhibitors will help uncover the biological consequences of these specific coregulated cellular processes found in this analysis. Robust characterization of existing cellular processes and the creation of new cellular process categories may uncover the function of unknown coregulated genes and proteins. Overall, utilizing a large-scale quantitative proteomic approach via the ICAT reagent method may elucidate analogous growth factor and hormone responsive pathways in other model systems.
Materials and methods
Preparation of microsomal fractions, ICAT reagent labeling and mass spectrometric analysis
Microsomes were prepared and isolated from androgen-depleted and androgen-stimulated LNCaP cells (109). Cells were exposed to either phenol-red deficient RPMI 1640 (Invitrogen, Carlsbad, CA) media supplemented with 10% charcoal-stripped fetal bovine serum (androgen-depleted, AD cells) (Hyclone Laboratories Inc., Logan, UT) or androgen-depleted media supplemented with 10 nM R1881 (methyltrienolone, Perkin Elmer Life Sciences, Boston, MA) (androgen-stimulated, AS cells) for 72 h. A total of 5.0 mg of microsomal proteins, 2.5 mg from AD cells and 2.5 mg from AS cells, were ICAT reagent labeled (AD, d0-ICAT reagent, AS, d8-ICAT reagent) as previously described [16] with the following modifications: microsomes were reduced with 5 mM tributylphosphine (TBP) for 3 h, and TBP reduced extracts were ICAT reagent labeled for 3 h and digested with trypsin overnight at 37°C. The 5 mg trypsin digested ICAT-reagent labeled peptide mixture was subjected to cation exchange, and then 53 cation-exchanged and avidin-purified ICAT-reagent labeled fractions were analyzed by microcapillary liquid chromatography electrospray ionization tandem mass spectrometry (μLC-ESI-MS/MS) [16].
SEQUEST, INTERACT and XPRESS scoring criterion
Uninterpreted MS/MS spectra were searched against a database of approximately 73,600 human sequences generated as a subset of a non-redundant amino acid database downloaded from the National Cancer Institute's Advanced Biomedical Computing Center [57]. Tandem mass spectra were analyzed using the SEQUEST database search software. The database search criteria included a static modification of cysteine residues of 503 Da (mass of cysteine plus light ICAT reagent tag) and a variable modification of 8 Da for cysteines (for the heavy ICAT reagent tag). Additionally, searches were performed with no enzyme-constraint on the peptides analyzed from the sequence database. SEQUEST output was analyzed using the INTERACT program where peptides containing the following values were considered to be correct identifications: Xcorr > 1.5, Δ correlation scores ≥ 0.1, Sp rank ≤ 8, singly tryptic, contain at least one cysteine residue. This generated a list that contained 5,450 MS/MS spectra. All peptide identifications were confirmed by manual inspection of the data. Comparing these criteria to those generated from the PeptideProphet software [58], 93% of the entries had a probability of 0.99 or higher and 97% of the entries in the list had a probability of 0.90 or higher. All d0/d8 peptide ratios were analyzed and validated/corrected using the XPRESS [16] quantification program. Quantitative ratios are generated by taking the ratios of the calculated heavy and light elution peak areas for ICAT-reagent labeled peptides. As we have calculated ratios only for those ions that we have acquired and confirmed the identification of by MS/MS, we can also quantify what are termed 'singleton' peaks. This is because we always start with a positive identification for the singleton elution peak so the presence of a peak for the counterpart ion is not required.
Immunoblotting, immunofluorescence and microscopy
Microsomal protein lysates derived from 72 h AD and AS cells (16 μg) were subjected to immunoblot analysis using 1 μg/ml mouse anti-PSA (Research Diagnostics Inc. RDI-PSA85abm/RDI-PSA85abm-1), 1:1,500 dilution of rabbit anti-TMPRSS2, 1:200 dilution of rabbit anti-LDLR (Research Diagnostics Inc.), 1 μg/ml mouse anti-MST-3 (BD Transduction Laboratories clone 47), 1 μg/ml mouse anti-FAS (BD Transduction Laboratories clone 23), 2 μg/ml mouse anti-β-catenin (BD Transduction Laboratories clone 14), 1 μg/ml mouse anti-Fas ligand (BD Transduction Laboratories clone 13) and 0.05 μg/ml mouse anti-Hsp-60 (Stressgen clone LK1). 72 h AD, AS and LNCaP cells grown in phenol-red deficient RPMI 1640 containing 10% fetal bovine serum (steady-state cells, SS cells) were labeled with the Mitotracker® Red CMXRos and Alexa Fluor 488 phalloidin and DAPI (4',6-diamidino-2-phenylindole, dihydrochloride) conjugates (Molecular Probes, Eugene, OR) according to the manufacturers instructions [59]. Fluorescent images were acquired on an DeltaVision microscope (Applied Precision, Inc., Issaquah, WA) and image stacks were deconvolved using SoftWoRx 2.5 (API). All images were processed at the Scientific Imaging Lab, Fred Hutchinson Cancer Research Center. Light microscope photographs of cells were imaged with Fujifilm Super HQ 35 mm film (100 speed film) at 10X on an Olympus IX70 inverted microscope.
Cell death assay
LNCaP cells were seeded and exposed to three growth conditions for 72 hours prior to a 24 hour incubation with the respiratory chain inhibitors. These three growth conditions included normal growth medium (steady-state, SS; androgen-depleted, AD and androgen-stimulated, AS). 72 h AD, AS and SS cells were incubated with the respiratory chain inhibitors rotenone (10 μM), carbonyl cyanide m-chlorophenylhydrazone (CCCP) (50 μM), antimycin A (10 μg/ml), oligomycin (5 μg/ml), for 24 h. Untreated cells were incubated with vehicle (ethanol) for 24 h. SS cells were seeded at a density of 1 × 103 cells/cm2 while AD and AS cells were seeded at a density of 3 × 103 cells/cm2 to generate equivalent cell densities at the time of drug addition. Cell death was determined using the trypan-blue dye assay [60]. At least 200 cells were counted in triplicate for all experiments. All drugs were purchased from Sigma.
Assignment of identified proteins into cellular process categories
All identified proteins that contained a Swiss-Prot annotation were searched at the Incyte Human Proteome Database [30] and assigned to one or several cellular process categories if applicable. All remaining proteins that contained a Genbank GenPept accession number were searched at the Incyte Human Proteome Database and assigned to a cellular process category if applicable. Non-annotated proteins were assigned to the cellular process category described as 'unknown process'.
References
Balk SP: Androgen receptor as a target in androgen-independent prostate cancer.Urology 2002,60(3 Suppl 1):132–138. discussion 138–139.
Prins GS: Molecular biology of the androgen receptor.Mayo Clin Proc 2000, 75:S32-S35.
Huggins C, Hodges CV: Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate.CA Cancer J Clin 1972, 22:232–240.
Culig Z, Hobisch A, Cronauer MV, Cato AC, Hittmair A, Radmayr C, Eberle J, Bartsch G, Klocker H: Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone.Mol Endocrinol 1993, 7:1541–1550.
Suzuki H, Sato N, Watabe Y, Masai M, Seino S, Shimazaki J: Androgen receptor gene mutations in human prostate cancer.J Steroid Biochem Mol Biol 1993, 46:759–765.
Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, Balk SP: Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer.N Engl J Med 1995, 332:1393–1398.
Elo JP, Kvist L, Leinonen K, Isomaa V, Henttu P, Lukkarinen O, Vihko P: Mutated human androgen receptor gene detected in a prostatic cancer patient is also activated by estradiol.J Clin Endocrinol Metab 1995, 80:3494–3500.
Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B, Balk SP: Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist.Cancer Res 1999, 59:2511–2515.
Marcelli M, Ittmann M, Mariani S, Sutherland R, Nigam R, Murthy L, Zhao Y, DiConcini D, Puxeddu E, Esen A, et al.: Androgen receptor mutations in prostate cancer.Cancer Res 2000, 60:944–949.
Waghray A, Feroze F, Schober MS, Yao F, Wood C, Puravs E, Krause M, Hanash S, Chen YQ: Identification of androgen-regulated genes in the prostate cancer cell line LNCaP by serial analysis of gene expression and proteomic analysis.Proteomics 2001, 1:1327–1338.
Waghray A, Schober M, Feroze F, Yao F, Virgin J, Chen YQ: Identification of differentially expressed genes by serial analysis of gene expression in human prostate cancer.Cancer Res 2001, 61:4283–4286.
DePrimo SE, Diehn M, Nelson JB, Reiter RE, Matese J, Fero M, Tibshirani R, Brown PO, Brooks JD: Transcriptional programs activated by exposure of human prostate cancer cells to androgen.Genome Biol 2002, 3:research0032.1-research0032.12.
Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, Hood L, Lin B: The program of androgen-responsive genes in neoplastic prostate epithelium.Proc Natl Acad Sci USA 2002, 99:11890–11895.
Gygi SP, Rochon Y, Franza BR, Aebersold R: Correlation between protein and mRNA abundance in yeast.Mol Cell Biol 1999, 19:1720–1730.
Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R: Quantitative analysis of complex protein mixtures using isotope-coded affinity tags.Nat Biotechnol 1999, 17:994–999.
Han DK, Eng J, Zhou H, Aebersold R: Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry.Nat Biotechnol 2001, 19:946–951.
Cunha GR, Donjacour AA, Cooke PS, Mee S, Bigsby RM, Higgins SJ, Sugimura Y: The endocrinology and developmental biology of the prostate.Endocr Rev 1987, 8:338–362.
Lin B, Ferguson C, White JT, Wang S, Vessella R, True LD, Hood L, Nelson PS: Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2.Cancer Res 1999, 59:4180–4184.
Afar DE, Vivanco I, Hubert RS, Kuo J, Chen E, Saffran DC, Raitano AB, Jakobovits A: Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia.Cancer Res 2001, 61:1686–1692.
Swinnen JV, Esquenet M, Goossens K, Heyns W, Verhoeven G: Androgens stimulate fatty acid synthase in the human prostate cancer cell line LNCaP.Cancer Res 1997, 57:1086–1090.
Swinnen JV, Ulrix W, Heyns W, Verhoeven G: Coordinate regulation of lipogenic gene expression by androgens: evidence for a cascade mechanism involving sterol regulatory element binding proteins.Proc Natl Acad Sci USA 1997, 94:12975–12980.
Swinnen JV, Verhoeven G: Androgens and the control of lipid metabolism in human prostate cancer cells.J Steroid Biochem Mol Biol 1998, 65:191–198.
Hulsken J, Behrens J, Birchmeier W: Tumor-suppressor gene products in cell contacts: the cadherin-APC-armadillo connection.Curr Opin Cell Biol 1994, 6:711–716.
Pawlowski JE, Ertel JR, Allen MP, Xu M, Butler C, Wilson EM, Wierman ME: Liganded androgen receptor interaction with beta-catenin: nuclear co-localization and modulation of transcriptional activity in neuronal cells.J Biol Chem 2002, 277:20702–20710.
Yang F, Li X, Sharma M, Sasaki CY, Longo DL, Lim B, Sun Z: Linking beta-catenin to androgen signaling pathway.J Biol Chem 2002, 277:11336–11344.
Song LN, Herrell R, Byers S, Shah S, Wilson EM, Gelmann EP: Beta-catenin binds to the activation function 2 region of the androgen receptor and modulates the effects of the N-Terminal domain and TIF2 on ligand-dependent transcription.Mol Cell Biol 2003, 23:1674–1687.
Schinkmann K, Blenis J: Cloning and characterization of a human STE20-like protein kinase with unusual cofactor requirements.J Biol Chem 1997, 272:28695–28703.
Suda T, Takahashi T, Golstein P, Nagata S: Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family.Cell 1993, 75:1169–1178.
Brazhnik P, de la Fuente A, Mendes P: Gene networks: how to put the function in genomics.Trends Biotechnol 2002, 20:467–472.
Incyte[http://www.incyte.com/]
Ashburner M, Lewis S: On ontologies for biologists: the Gene Ontology-untangling the web.Novartis Found Symp 2002, 247:66–80.
Gene Ontology™ Consortium[http://www.geneontology.org/]
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al.: Initial sequencing and analysis of the human genome.Nature 2001, 409:860–921.
Smith JJ, Marelli M, Christmas RH, Vizeacoumar FJ, Dilworth DJ, Ideker T, Galitski T, Dimitrov K, Rachubinski RA, Aitchison JD: Transcriptome profiling to identify genes involved in peroxisome assembly and function.J Cell Biol 2002, 158:259–271.
Gomez-Munoz A, Hales P, Brindley DN, Sancho MJ: Rapid activation of glycogen phosphorylase by steroid hormones in cultured rat hepatocytes.Biochem J 1989, 262:417–423.
Ferdinandusse S, Denis S, L IJ, Dacremont G, Waterham HR, Wanders RJ: Subcellular localization and physiological role of alpha-methylacyl-CoA racemase.J Lipid Res 2000, 41:1890–1896.
Jiang Z, Woda BA, Rock KL, Xu Y, Savas L, Khan A, Pihan G, Cai F, Babcook JS, Rathanaswami P, et al.: P504S: a new molecular marker for the detection of prostate carcinoma.Am J Surg Pathol 2001, 25:1397–1404.
Luo J, Zha S, Gage WR, Dunn TA, Hicks JL, Bennett CJ, Ewing CM, Platz EA, Ferdinandusse S, Wanders RJ, et al.: Alpha-methylacyl-CoA racemase: a new molecular marker for prostate cancer.Cancer Res 2002, 62:2220–2226.
Rubin MA, Zhou M, Dhanasekaran SM, Varambally S, Barrette TR, Sanda MG, Pienta KJ, Ghosh D, Chinnaiyan AM: alpha-Methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer.JAMA 2002, 287:1662–1670.
Zhou M, Chinnaiyan AM, Kleer CG, Lucas PC, Rubin MA: Alpha-methylacyl-CoA racemase: a novel tumor marker over-expressed in several human cancers and their precursor lesions.Am J Surg Pathol 2002, 26:926–931.
Costello LC, Littleton G, Franklin RB: Regulation of citrate related metabolism in normal and neoplastic prostate New York: Raven Press 1978.
Huggins C: The prostate secretion.In 1946; Harvey Lectures 1946, 148–193.
Costello LC, Franklin RB: The intermediary metabolism of the prostate: a key to understanding the pathogenesis and progression of prostate malignancy.Oncology 2000, 59:269–282.
Burchardt T, Burchardt M, Chen MW, Cao Y, de la Taille A, Shabsigh A, Hayek O, Dorai T, Buttyan R: Transdifferentiation of prostate cancer cells to a neuroendocrine cell phenotypein vitroandin vivo.J Urol 1999, 162:1800–1805.
Fukami JI, Yamamoto I, Casida JE: Metabolism of rotenonein vitroby tissue homogenates from mammals and insects.Science 1967, 155:713–716.
Neumcke B: The action of uncouplers on lipid bilayer membranes.Membranes 1975, 3:215–253.
Pietrobon D, Azzone GF, Walz D: Effect of funiculosin and antimycin A on the redox-driven H+-pumps in mitochondria: on the nature of 'leaks'.Eur J Biochem 1981, 117:389–394.
Nakakita Y, Nakagawa M, Sakai H: Isolation of oligomycin A as a result of screening for antagonists of lipids.J Antibiot (Tokyo) 1980, 33:514–516.
Abrahamsson PA, Lilja H: Partial characterization of a thyroid-stimulating hormone-like peptide in neuroendocrine cells of the human prostate gland.Prostate 1989, 14:71–81.
Guate JL, Escaf S, Menendez CL, del Valle M, Vega JA: Neuroendocrine cells in benign prostatic hyperplasia and prostatic carcinoma: effect of hormonal treatment.Urol Int 1997, 59:149–153.
Roth FP, Hughes JD, Estep PW, Church GM: Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation.Nat Biotechnol 1998, 16:939–945.
Auboeuf D, Honig A, Berget SM, O'Malley BW: Coordinate regulation of transcription and splicing by steroid receptor coregulators.Science 2002, 298:416–419.
Muller JM, Isele U, Metzger E, Rempel A, Moser M, Pscherer A, Breyer T, Holubarsch C, Buettner R, Schule R: FHL2, a novel tissue-specific coactivator of the androgen receptor.EMBO J 2000, 19:359–369.
Dye BT, Patton JG: An RNA recognition motif (RRM) is required for the localization of PTB-associated splicing factor (PSF) to subnuclear speckles.Exp Cell Res 2001, 263:131–144.
Grabowski PJ, Black DL: Alternative RNA splicing in the nervous system.Prog Neurobiol 2001, 65:289–308.
Eisen MB, Spellman PT, Brown PO, Botstein D: Cluster analysis and display of genome-wide expression patterns.Proc Natl Acad Sci USA 1998, 95:14863–14868.
National Cancer Institute Advanced Biomedical Computing Center[ftp://ftp.ncifcrf.gov/pub/nonredun/protein.nrdb.Z]
Keller A, Nesvizhskii AI, Kolker E, Aebersold R: Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search.Anal Chem 2002, 74:5383–5392.
Molecular Probes[http://www.probes.com/]
Freshney RI: Culture of animal cells: a manual of basic technique4 Edition New York: Wiley 2000.
Acknowledgements
We thank our colleague David K Han for helpful discussions and critical reading of the manuscript. We also thank Adrian Quintanilla for assistance with the scientific imaging, and Masazumi Matsumara for rabbit TMPRS22 antibodies. This work was supported by grants from the National Institutes of Health to R.A. (1R33CA-93302) and the Damon Runyon Cancer Research Foundation to P.S. N. A gift from Merck and Co. to support research at the Institute for Systems Biology is gratefully acknowledged. M.E.W. was supported in part by a NIH Postdoctoral Genome Training Fellowship Grant. M.E.W. is a United Negro College Fund/Merck postdoctoral research fellow.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
13059_2003_808_MOESM1_ESM.pdf
Additional data file 1: All ICAT protein quantifications and statistical analyses pertaining to Figures 1,2,3,4 and Table 1 presented in this study (PDF 1 MB)
Rights and permissions
About this article
Cite this article
Wright, M.E., Eng, J., Sherman, J. et al. Identification of androgen-coregulated protein networks from the microsomes of human prostate cancer cells. Genome Biol 5, R4 (2003). https://doi.org/10.1186/gb-2003-5-1-r4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/gb-2003-5-1-r4