
Abstract
Background
Published accounts of horizontally acquired genes in plant-parasitic nematodes have not been the result of a specific search for gene transfer per se, but rather have emerged from characterization of individual genes. We present a method for a high-throughput genome screen for horizontally acquired genes, illustrated using expressed sequence tag (EST) data from three species of root-knot nematode, Meloidogyne species.
Results
Our approach identified the previously postulated horizontally transferred genes and revealed six new candidates. Screening was partially dependent on sequence quality, with more candidates identified from clustered sequences than from raw EST data. Computational and experimental methods verified the horizontal gene transfer candidates as bona fide nematode genes. Phylogenetic analysis implicated rhizobial ancestors as donors of horizontally acquired genes in Meloidogyne.
Conclusions
High-throughput genomic screening is an effective way to identify horizontal gene transfer candidates. Transferred genes that have undergone amelioration of nucleotide composition and codon bias have been identified using this approach. Analysis of these horizontally transferred gene candidates suggests a link between horizontally transferred genes in Meloidogyne and parasitism.
Similar content being viewed by others


Background
Nematodes are the most abundant and speciose metazoans, and account for up to 80% of the kingdom's members [1]. Not surprisingly, nematodes have evolved to occupy diverse ecological niches. Like the well-studied Caenorhabditis elegans, most are free-living and graze on microbes or detritus, and as such, have no obvious direct impact on humans. Others, however, are adapted as parasites and are responsible for such widespread problems as human disease, debilitation of livestock and crop damage. Plant-parasitic forms are responsible for an estimated $100 billion in annual crop damage worldwide [2]. The most damaging family (the Heteroderidae) includes the root-knot (Meloidogyne spp.) and the cyst (Globodera and Heterodera spp.) nematodes. Root-knot nematodes penetrate plant hosts and migrate between the cells in roots, where they induce formation of large multinucleate cells called 'giant cells'. Galls form around the giant cells, and the roots become distorted, often leading to compromised root function and retardation of plant growth [3].
It is not clear which genetic differences between the plant parasitic and non-parasitic forms may be responsible for conferring parasitic ability. On the basis of phylogenetic analysis [4] it appears that plant parasitism arose independently at least three times over the course of nematode evolution. Consequently, one cannot be assured that any gene or set of genes that aid in the parasitic lifestyle in one nematode species will also exist in another. Conceptually, several mechanisms affecting evolution to parasitism can be envisioned. These include adaptation of pre-existing genes to encode new functions; changes in genes regulating metabolic or developmental pathways; gene duplication; gene loss; and acquisition of genes from other species (horizontal gene transfer, HGT). HGT has become a widely accepted mechanism of rapid evolution and diversification in prokaryotic populations [5–7]. Recent genome analyses of primitive eukaryotes, such as the sea squirt (Ciona intestinalis) [8] and single-celled parasitic diplomonads [9], implicate HGT events in early eukaryotic evolution. In contrast, the extent of horizontal transfer involving higher eukaryotes has been controversial, with many cases of hypothesized horizontally transferred genes [10–14] having been refuted by later studies [15, 16].
On the basis of biochemical and immunological criteria, genes have been identified in Globodera rostochiensis and Heterodera glycines that allow these nematodes to endogenously produce enzymes that can degrade cellulose and pectin, the two major components of plant cell walls. A possible ancient bacterial origin of these genes has been theorized [17–19]. A bacterial origin for a number of root-knot nematode (RKN) genes also has been proposed, although their possible role in parasitism is less clear. Some, such as a gene encoding chorismate mutase [20], were likewise identified on the basis of biochemical properties, whereas others, including a polygalacturonase gene [21], were identified from expressed sequence tag (EST) datasets, the latter from our data [22] using a keyword search. Veronico et al. [23] isolated a presumed polyglutamate synthetase gene with bacterial homology by sequencing neighboring regions of the M. artiellia chitin synthetase locus. We wished to determine whether other RKN genes might have been acquired by horizontal gene transfer, particularly as such genes might potentially be related to parasitism.
Claims of HGT have frequently pivoted on incongruencies between a particular gene tree and the assumed underlying species tree. Acquisition of new sequence data has often revealed that genes believed to be absent in a species were merely missing in the database rather than missing from the genome [16]. Obviously, because full genomes are not available for all plant and animal species, we are not able to make definitive statements about the presence or absence of a particular gene in every organism. However, with the completed C. elegans genome available as a reference 'model' nematode, it is now possible to examine the emerging genetic resources for Meloidogyne comprehensively, to begin to address the question of evolution of parasitism and, in particular, a possible role for HGT.
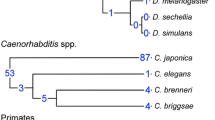
A similarity to a bacterial protein sequence is the simplest criterion for considering a nematode protein, and thus the gene that encodes it, as a possible HGT candidate. For that candidate truly to define an HGT event, its presence must be incongruent with nematode phylogeny (Figure 1). Nevertheless, the presence of a gene in one nematode species (such as Meloidogyne) but its absence in another (such as C. elegans) might merely reflect a gene loss in the latter lineage. In addition to C. elegans, several other invertebrate genomes have been completely sequenced, and at the time of this study, the best characterized of those was Drosophila melanogaster. Consequently, we chose this resource as a tool to identify genes which may be present in nematodes, but which are absent in C. elegans. A bacteria-like gene present in Meloidogyne and Drosophila, but absent in C. elegans, is unlikely to have experienced HGT, but may rather reflect a gene loss in the C. elegans lineage. We therefore developed a 'phylogenetic filter' based on these relationships to rapidly reveal Meloidogyne HGT candidates identified by sequence similarity to bacterial proteins. The intent of this filter is to efficiently eliminate spurious HGT candidates.

Schematic species tree indicating relationships between bacteria, Drosophila, C. elegans and plant-parasitic nematodes in the family Heteroderidae. The locations of three possible horizontal gene-transfer events that would pass through our initial phylogenetic filter are indicated by dotted lines. Transfer 'a' occurs after divergence of the lineages leading to C. elegans and Heteroderidae, transfer 'b' after divergence of root-knot nematodes and cyst nematodes, and transfer 'c' to the lineage leading to a specific Meloidogyne species. Adapted from [59].
Surprisingly, the relationship between the invertebrate phyla Nematoda and Arthropoda (which includes Drosophila) is controversial. The traditional view is that arthropods are more closely related to annelids than to nematodes, but some recent molecular phylogenies place nematodes and arthropods together in a high-level taxon named Ecdysozoa, which does not include annelids [24, 25]. Other molecular studies give conflicting results [26, 27]. Regardless of the evolutionary relationship between Nematoda and Arthropoda, C. elegans and Drosophila remain useful and valid models for our analyses, and the relationships shown in Figure 1 are consistent with both hypotheses.
Genes that were transferred from bacteria to nematodes would pass through our phylogenetic filter if the transfer event occurred subsequent to the divergence of the C. elegans and Meloidogyne lineages (Figure 1). Should a gene appear to be present in other closely related plant parasites, such as the cyst nematodes, the transfer event probably affected a common ancestor of the two families of parasitic nematodes (event 'a' in Figure 1). Alternatively, the transfer event may be more recent, such as to the progenitor of the Meloidogyne lineage since its divergence from the cyst nematodes (event 'b' in Figure 1), or in a lineage leading to a single Meloidogyne species (event 'c').
Although bacteria-like Meloidogyne genes that are not present in C. elegans and Drosophila comprise a preliminary pool of candidates, multiple gene loss may be responsible for the presence/absence pattern revealed by the filter. To test this more thoroughly, we established a screen to compare the now small pool of preliminary candidates with all other sequences in the public databases. The most parsimonious explanation to be drawn from candidates with no significant matches to any metazoan genes is that they arose by horizontal gene transfer from a non-metazoan pool, as opposed to multiple independent gene losses in the metazoan lineages. Candidates thus identified were subsequently validated through phylogenetic analysis of relationships between the most similar matches from our screening processes
We describe here a comprehensive two-step search for HGT candidates in M. incognita, M. javanica and M. hapla using EST data [22, 28, 29]. Genome-to-genome comparisons were made to discover patterns of presence and absence that would indicate horizontally acquired genes. Second, kingdom-wide comparisons further reduced the candidate pool; these genes were then examined from an evolutionary standpoint. Twelve Meloidogyne candidates were discovered and their potential role in plant pathogenicity is discussed.
Results and discussion
Genome-to-genome comparisons act as a phylogenetic filter in candidate searching
Given the large number of sequences to examine and the expectation that most were not horizontally acquired, we developed a phylogenetic filter based on genome-to-genome sequence comparisons. Further, because the available data included raw ESTs from the National Center for Biotechnology Information (NCBI) GenBank (dbEST) as well as clustered ESTs from the Parasitic Nematode Sequencing Project [22, 28, 29], for which the data can be presumed to be significantly more reliable, we wished to compare the efficiency of reducing each dataset with this filter. Meloidogyne sequences from NCBI dbEST (M. incognita, M. javanica and M. hapla sequences, named NMi, NMj, and NMh respectively) were translated in six frames and individually compared to conceptual six-phase translations of the C. elegans and Drosophila genomes as well as all available bacterial sequences. This first filter, which makes no assumptions about gene annotation in the target genomes, and which employed the relatively error-prone raw ESTs, reduced the pool of HGT candidates by eliminating more than 99% of the original ESTs for all three species tested (Table 1). Using clustered ESTs (M. incognita and M. javanica sequences, named WMi and WMj) as queries to the worm, fly and bacterial protein databases (which are based on gene annotation) produced a similar degree of reduction (Table 1). Importantly, genes previously predicted to be the result of HGT events were identified by, and passed through, the phylogenetic filter (see below).
The main objective of the phylogenetic filter was to reduce the computational load necessary to screen HGT candidates against all metazoan proteins. A second filter, consisting of a BLAST analysis against the GenBank nonredundant (nr) protein database, served to eliminate genes that may have been independently lost in the C. elegans and Drosophila lineages, but are still representative of a more ancient animal gene (Table 1). This filter eliminated four candidates from the WMi data set. Examination of these showed a putative copper homeostatis protein and a protein of unknown function, both with significant matches to Homo sapiens (e-values of 1.10e-23 and 4.20e-18respectively), one aldehyde dehydrogenase with a significant match to Mus musculus (2.70e-26) and one asparaginyl-tRNA synthetase. Three of the four had best matches to bacteria (Table 2). Interestingly, manual inspection revealed that all four sequences did have significant matches to C. elegans, but passed through our initial phylogenetic filter because the bacterial matches were stronger than those for C. elegans or Drosophila. The 12 final candidates in WMi had no significant match to C. elegans or Drosophila in the preliminary screen. The best eukaryotic matches to these candidates from the BLAST search against nr are shown in Table 3. The second filter generated similar enrichment in WMj, reducing the number of candidates from eleven to seven.
The fact that more candidates from the raw datasets were eliminated during second-round filtering (for example, from 99 to 27 in NMi) reflects the redundancy in the datasets. If multiple EST sequences representing a single gene pass through the first filter, each of those EST sequences will be in the preliminary candidate pool. The second filter is likely to simultaneously remove more than one of these homologous sequences if it removes any at all. Therefore, searching with raw EST sequences is likely to result in a larger absolute decrease in the candidate number than will searching with clustered EST sequences.
The number of final candidates listed in Table 1 are candidate HGT genes after clustering. A smaller number of candidates was discovered from the raw EST datasets compared with the clustered sequences, which suggests that our method of HGT candidate searching is partially dependent on sequence quality. The lower number of final candidates obtained using raw EST data is principally due to filtering of areas of low complexity and tandem repeats, and uncertainty of similarity matching for shorter sequences during BLAST searches. Similarly, the size of the dataset influences the number of final candidates obtained. Thus, the absence of candidates in M. hapla is likely to be due to a combination of the small number of unique ESTs analyzed (because of redundancy in the data), and possibly overall quality of the raw ESTs, rather than to a lack of laterally acquired genes in the genome. Despite the lowered efficiency of candidate discovery when using the lower-quality, raw EST sequences, this tool was able to recover five candidates from the NMi dataset, compared to the 12 candidates identified from the higher-quality clustered sequences in the WMi dataset. The fact that candidates were discovered across disparate sequence-quality conditions not only provides additional validation of our methods, but also suggests a high degree of flexibility and robustness in the tool.
Identification of previously hypothesized HGT candidates
The literature reports seven genes postulated to have been horizontally acquired by M. incognita, M. hapla or M. javanica during evolution of plant-parasitic nematodes [17–21]; our search algorithm revealed six of these genes. The notable exception is Mj-CM, which is postulated to encode chorismate mutase in M. javanica [20]. To examine why this gene was not identified by our filtering process, we used both Mj-CM sequences found in GenBank (AF095949, AF095950) in a series of BLASTX queries. No significant matches were found in the Drosophila, C. elegans or bacterial databases, nor in the Meloidogyne datasets used in this study. Recent BLAST searches at nematode.net [30] against all Meloidogyne ESTs, including sequences not available when our analyses were first conducted confirm that the chorismate mutase gene is absent from WMi and WMj, although a single, significant match to an M. arenaria chorismate mutase EST was revealed. Another RKN gene also postulated to have been acquired by HGT, and which encodes polyglutamate synthetase, was previously identified in M. artiellia [23]. Significantly, hybridization data showed that this particular gene is absent from both the M. javanica and G. rostochiensis genomes [23]. We speculate that acquisition of this gene by M. artiellia is a recent HGT event (event 'c', Figure 1), and thus it is truly absent from the Meloidogyne genomes from which our datasets were derived. In other words, failure to 'discover' this gene was not a failure of our screening process, but is likely to be a correct reflection of the biology.
The most extensively studied HGT candidates are four genes encoding β-1,4-endoglucanase, initially identified in the cyst nematodes G. rostochiensis and H. glycines [18, 19]. These four genes (NemaGene Contig IDs MI00537, MI01011, MI01381 and MI01842) [30] appear to define two sets of paralogs formed before divergence of the cyst and root-knot nematodes. As noted [18, 19], β-1,4-endoglucanases presumably equip these nematodes with the ability to endogenously degrade the most abundant component of cell walls, namely cellulose. Similarly, the second most abundant component of cell walls (pectin) is the assumed target of nematode-encoded pectate lyase and exo-polygalacturonase, both functions also postulated to have been acquired by HGT. The pectate lyase gene (MI00592) was identified in G. rostochiensis and H. glycines [17] and the exo-polygalacturonase (MI00252) was identified in our M. incognita data [21, 22]. Because of the obvious role of nematode genes that allow endogenous production of cell-wall degrading enzymes in attacking a plant host, it has been hypothesized that their acquisition by HGT may have been key steps in the evolution of plant-parasitic nematodes from ancestral free-living forms [3]. In that model, an intermediate, symbiotic association of a soil-dwelling (but free-living) nematode with a soil bacterium possessing these enzymes is postulated before the HGT event. It was suggested [3] that acquisition of these new functions (either by symbiosis or HGT) permitted previously free-living nematodes to expand their range into a new ecological niche (the plant) as a prelude to speciation into parasitic forms.
Also revealed by our tool were six new HGT candidates, including homologs for glutamine synthetase, L-threonine aldolase and nodL, and three to which function could not be unequivocally ascribed.
Rhizobial origin of Meloidogynegenes
Of the six newly identified HGT candidates, four have highest similarity to genes in the nitrogen-fixing soil bacteria that nodulate plant roots and which are collectively termed rhizobia. Meloidogyne and rhizobia are sympatric (that is, they share an ecological niche in the soil [3], and arguably in the plant too [31]), satisfying the minimal requirement for an HGT to occur, namely physical proximity. Interestingly, models of bacterial evolution suggest HGT as a mechanism of adaptation into either symbiosis or parasitism [32]. This is specifically thought to be the case for divergent species of rhizobia, such as the symbiont Sinorhizobium meliloti and the pathogen Rhizobium radiobacter (formerly known as Agrobacterium tumefaciens), where differential selection and gene maintenance is likely to be responsible for different lifestyle strategies [33].
Two of the Meloidogyne genes revealed by our filters, which encode an L-threonine aldolase gene (MI01644) and a deduced protein of unknown function (MI00109), exhibit striking amino-acid identity to rhizobial proteins (48% and 51% respectively), but a complete absence of meaningful homology with any eukaryotic sequence (Table 3). Consequently, these genes are strong candidates for having entered nematodes via HGT, presumably from a rhizobial ancestor.
The deduced product of a third M. incognita gene (MI00426) has striking sequence similarity to glutamine synthetase (GS). Glutamine synthetases fall into two structurally and functionally distinct classes. GSI, which to date appears restricted to prokaryotes [34], is involved in ammonium assimilation as part of the nitrogen-fixation pathway in rhizobia [35]. The ability to be reversibly adenylylated at Tyr397 of the active site is a characteristic of GSI. The second class, GSII, is found in all eukaryotes and a small number of prokaryotes, and appears to be involved in purine synthesis [35]. Unlike GSI, GSII is not adenylylated (and lacks the conserved tyrosine). On the basis of both amino-acid sequence similarity (Table 3) and a Pfam [36] HMM search (e-value 4.3e-24), it is clear that the RKN glutamine synthetase is a GSI homolog, implying a prokaryotic origin. Strikingly, the nematode protein has greatest similarity (56% amino-acid identity) to GSI from the rhizobial bacterium, Mesorhizobium loti, including conservation of Tyr397. The best match to a eukaryotic glutamine synthetase (GSII) is substantially lower (Table 3), strongly implicating the RKN gene as a robust candidate for an HGT event.
The fourth rhizobial-like HGT candidate (MI01045) identified by our filter has 58% amino-acid identity (8.8e-54) to NodL from Rhizobium leguminosarum (Table 3). This protein encodes an N-acetyltransferase previously thought to be present only in rhizobia [37], where it functions in the biosynthesis of Nod factor. Nod factors are a rhizobial species-specific family of lipo-chito-oligosaccharides which function in signal exchange between the bacterium and its symbiotic partner plant [38]. The first visible signs of nodule formation (root-hair deformation) as part of the symbiotic pathway are triggered by Nod factors [39], and although the specific mechanisms of Nod factor function remain unknown, it is clear that it has a central role in initiation of cell division and possibly also nodule differentiation in the root [40]. For most rhizobia, the product of nodD acts as a transcriptional activator and induces expression of a set of nod genes. Experimental evidence [39] shows that lack of either nodABC or nodD in rhizobia results in a Nod- phenotype (that is, a strain unable to initiate nodule formation on the host plant). By contrast, R. radiobacter, which forms a parasitic relationship with plants by producing a crown gall rather than nodules, lacks these genes, and appears to possess only nodL, nodX and nodN, suggesting these three nod genes are sufficient to affect root growth and are involved in a parasitic lifestyle rather than being specific to symbiosis [33].
To examine further the relationship between putative nodL candidates found in M. incognita and M. javanica with the cognate genes in rhizobia, we undertook a phylogenetic analysis and found that the two nematode genes fall squarely within the rhizobial nodL clade (Figure 2). This analysis further grouped other sequences with significant similarity to the deduced Meloidogyne NodL protein. Not surprisingly, these enzymes clustered according to specific enzymatic function of the different classes of acetyltransferase. Significantly, the solitary significant match of the Meloidogyne NodL sequences to a eukaryote is to a yeast serine-acetyltransferase, an enzyme clearly separated from the RKN by function as well as in our phylogeny (Figure 2).

Cladogram of NodL-like proteins. The unrooted tree is generated by protein-distance and neighbor-joining methods and shows relationships of the deduced, putative Meloidogyne NodL proteins with similar enzymes, color-coded according to known function. Numbers indicate percent support from 1,000 non-parametric bootstrap replicates [53]. The scale bar represents 0.1 amino-acid replacements per site across the length of a given branch.
Bayesian analysis of the amino-acid alignment confirms this grouping. The posterior probability of the two Meloidogyne sequences being most closely related to the one other eukaryotic sequence, from Saccharomyces cerevisiae, is estimated to be 0. Instead, a group consisting of the two Meloidogyne sequences along with the Sinorhizobium meliloti and Rhizobium leguminosarum sequences is estimated to have a posterior probability of 1.0. For the clade consisting solely of the four rhizobial and the two Meloidogyne sequences, the posterior probability is estimated to be about 0.657, and almost all of the remaining posterior probability is accounted for by adding the Streptomyces coelicolor sequence to this clade of rhizobial and Meloidogyne sequences.
Using polymerase chain reaction (PCR) primers designed from the Meloidogyne sequence we have attempted to amplify nodL from a range of nematode species. For each of the Meloidogyne species tested (including M. hapla), we have been able to confirm the presence of the gene. However, similar experiments do not yield amplification products from the cyst nematodes we tested. Although other interpretations can be made, these results are consistent with nodL being acquired by an 'event b' HGT (see Figure 1).
Meloidogyne nodLtruly is a nematode gene
A question that arises in analyzing eukaryotic sequences with strong matches to bacterial proteins, especially when the match is unique, is whether the gene in question truly was isolated from a eukaryote, or whether it represents a prokaryotic contaminant (any nucleic acid matches of ESTs to prokaryotes, which probably would be contaminants, were removed before database submission [28]). Claims of nematode genes having been acquired by HGT [17–19, 23] have addressed this issue in a number of ways. To provide experimental evidence that the Meloidogyne nodL sequences represent nematode loci, we cloned and sequenced a full-length transcript from M. incognita (Mi-NodL). Identification of the SL1 trans-splice leader at the 5' end of the message [41], and a poly(A) tail at the 3' end, confirmed that this is a bona fide nematode gene (Figure 3). Analysis of genomic Mi-NodL sequences revealed an intron (Figure 3), further reinforcing the notion that this gene is integrated within the M. incognita genome.

Structure of Meloidogyne incognita NodL and its deduced translation product. Features of the genomic sequence were established by comparison with that of a full-length cDNA clone, and are indicated by arrows in the following order: addition site of SL-1 trans-splice leader; beginning of intron; end of intron; and site of poly(A) tail.
In cases of a recent HGT, it has been suggested that the nucleotide composition of the transferred gene might reflect that of the donor species rather than the recipient species [42]. To establish a baseline nucleotide composition of M. incognita transcripts, we calculated the average G+C content for our entire M. incognita (WMi) sequence dataset, obtaining a value of 34.3%. By contrast, the average G+C content of rhizobial species ranges from 57 to 65% [43]. Consistent with the average for M. incognita, the G+C content of Mi-NodL is 36%. This value is strikingly different for the nodL genes in Rhizobium leguminosarum (57% G+C) and Mesorhizobium loti (68% G+C). We similarly examined the G+C content of all 12 HGT candidates, and found the values to be consistently representative of Meloidogyne.
Another way to consider nucleotide composition is through codon usage. In particular, we considered how similar the Meloidogyne codon usage is to that of a 'typical' rhizobial protein by using the codon adaptation index (CAI) [44]. From an R. leguminosarum codon-usage table, we calculated the CAI for those amino acids precisely conserved between Mi-NodL and the rhizobial NodL protein to be 0.621 and 0.703 respectively. To evaluate the null hypothesis that the expected codon usage between the two nodL genes is identical, the difference in CAI values was adopted as a test statistic. The observed value of this test statistic was 0.082 and its null distribution was approximated by simulating 10,000 datasets as described in Materials and methods. Because the absolute value of the test statistic calculated from the simulated datasets exceeded 0.082 only 62 of 10,000 times, we reject the null hypothesis of identical expected codon usage in the M. incognita and R. leguminosarum nodL genes and conclude that codon usage in these genes is significantly different between the species. Collectively, comparison of the nematode and rhizobial nodL genes suggests that each is adapted for function in the organism in which it resides, and despite the high degree of similarity between the amino-acid sequences of these genes, the DNA sequences are strikingly different.
From the Lawrence and Ochman model [42], in which differences in G+C and codon bias are diagnostic for HGT events, it might be argued that our findings on the base composition of bacterial and nematode sequences are inconsistent with HGT. However, analyses in which synteny and phylogenetic information were also considered suggest that codon bias and G+C content are poor indicators of HGT [45]. A role for 'amelioration', whereby structural characteristics of the foreign gene are eventually homogenized to resemble those of the recipient species, has been assumed, but the rate was postulated to be the same as the rate of random, forward mutation [42]. In addition to alterations in codon usage (as reflected in G+C content), for a bacterial gene to function efficiently in a nematode presumably requires acquisition of regulatory elements (including a promoter) and structural elements (including a poly(A) tail and, optionally, a trans-spliced leader). Other elements (such as introns) might also be acquired. It is possible that a careful phylogenetic analysis comparing rates of evolution of Meloidogyne genes acquired by HGT with those present in the more ancient nematode lineage, might shed light on the rate of amelioration of gene structure following inter-kingdom HGT.
Patterns of HGT from rhizobia
In the absence of an assembled genome sequence for Meloidogyne, it is not yet possible to examine conserved, genome-wide gene order of HGT candidates between nematodes and the hypothesized bacterial donor. Nevertheless, because the origin of many of the nematode HGT candidates appeared to be rhizobial, we wished to investigate the organization of the bacterial homologs. Unlike many prokaryotes, in which the genome resides largely on a single, circular chromosome, with varying numbers of small episomes, rhizobial genomes are typically organized in a manner conceptually more like eukaryotes. Sinorhizobium meliloti, for example, has three large, single-copy plasmids [43], and the primary Mesorhizobium loti chromosome is linear. Rhizobia have the ability to transfer genes horizontally to other bacteria, and M. loti carries a 'symbiosis island' which spans approximately 9% of its genome and has been shown to have a role in rhizobial evolution via HGT [46]. This symbiosis island contains certain genes involved in nodulation and nitrogen-fixation functions, but none of these is a homolog of the nematode HGT candidates we have identified. However, four of these genes do map to the same M. loti linear chromosome (Figure 4), including nodL and the glutamine synthetase gene, both of which are involved in nodulation/nitrogen fixation in rhizobia. Together with the L-threonine aldolase homolog candidate, these three genes are found within 257 kb of each other, a distance that represents only 3.65% of the M. loti chromosome, which is less than half the size of the symbiosis island. The fourth candidate, of unknown function, lies approximately 149 kb from the opposite side of the symbiosis island from the other three (Figure 4). Interestingly, examination of the colinearity and gene arrangements between S. meliloti, R. radiobacter and M. loti indicates that the location of the genes in M. loti probably represents a more primitive state [33] and is therefore more likely to reflect the proximity of these genes in rhizobial ancestral species. Although it cannot be known if these genes were acquired in a single transfer event between a rhizobial ancestor and an ancestor to Meloidogyne, remnants of the HGT event (other than the already identified genes) may remain, and candidates are currently being mapped into the M. incognita genome to examine possible synteny with M. loti. BLAST analysis of the genes in the intervening span of chromosome indicates only three significant matches to the M. incognita (WMi) data set, all with significant matches to C. elegans, that is, they are not HGT candidates.

Schematic map (not to scale) of four genes on the Mesorhizobium loti linear chromosome with putative homologs in M. incognita, encoding NodL, L-threonine aldolase, glutamine synthetase and an unknown function. Also indicated is the 612 kb transferable M. loti symbiosis island.
Conclusions
We have demonstrated that a high-throughput bioinformatics approach based on EST sequences is an efficient and effective way to identify possible HGT candidates in plant-parasitic nematodes. Previous reports of horizontally acquired genes have been based mainly on biochemical or immunological criteria. Using an informatics approach, we rediscovered previously identified candidates (thus validating our method), and were able to identify new candidates for HGT. Strikingly, a common theme underpinning the HGT candidates is their apparent direct relationship to the parasitic lifestyle of Meloidogyne [3]. Also striking was our finding that phylogenetically, rhizobia appear to be the predominant group of 'donor' bacteria. This is significant for two reasons. First, root-knot nematodes and rhizobia occupy similar niches in the soil and in roots, and thus the opportunity for HGT may be omnipresent. Second, both organisms establish intimate developmental interactions with host plants, and mounting evidence suggests that the mechanisms for these interactions are also shared [31]. It seems a reasonable hypothesis that the origin of parasitism in Meloidogyne may have been facilitated by acquisition of genetic material from soil bacteria through horizontal transfer. Indeed, such events may have represented key steps in speciation of plant-parasitic nematodes
Materials and methods
Available data
Sequences were obtained from the Parasitic Nematode Sequencing Project (PNSP) [30] including clustered Meloidogyne ESTs built with the NemaGene approach [28]. We analyzed 1,799 M. incognita (WMi) sequences and 3,119 M. javanica (WMj) sequences from these PNSP clusters. Additional raw sequences were extracted from the July 31, 2002 NCBI GenBank dbEST build with the Entrez Search and Retrieval System (Table 1) [47]. Meloidogyne incognita and M. javanica datasets from NCBI (NMi and NMj respectively) contain the individual ESTs generated by the PNSP, and from which the clusters for the WMi and WMj datasets were generated. In addition, the NMi and NMj datasets included some sequences from sources other than the PNSP. M. hapla sequences (NMh) were also retrieved from NCBI. Entrez was used to extract all available nuclear sequences for D. melanogaster, C. elegans and bacterial sequences from the GenBank non-redundant (nr) database (May 1, 2002 build).
Candidate search algorithm
Analyses of the WMi and WMj data were performed via a local installation of WU-BLAST 2.0 [48]. Each sequence in WMi and WMj was extracted into individual FASTA format files using Perl scripts and submitted for three six-phase translated WU-BLASTX searches, once each against the C. elegans, Drosophila and bacterial protein databases. WU-BLASTX parameters were E = 10, W = 3, T = 12. E-values were extracted for the best match for each query sequence in each of the three searches.
Meloidogyne sequences from NCBI were analyzed using the Tera-BLAST Hardware Accelerated BLAST algorithm (TimeLogic, Crystal Bay, NV). Single FASTA files were submitted for three six-phase translated Tera-TBLASTX queries against six-phase translated C. elegans and Drosophila genomic databases. Tera-TBLASTX parameters were Open Penalty = 8, Extend Penalty = 2, Word Size = 4, Query Increment = 3 and Neighborhood Threshold = 18. Perl scripts were employed to parse the query name and associated best e-value from each of the nine analyses (three each for NMi, NMj and NMh).
As a first round of phylogenetic filtering, automated comparison of e-values for each sequence allowed us to eliminate sequences with a best match to either C. elegans or Drosophila from further analysis. The remaining sequences, those with a best match to bacteria of order 1.0e-10 or better, provided a preliminary pool of candidates for each dataset. A BLASTX search was carried out for each candidate against the nr database, using the above parameters. The results from this second filter were examined and any sequence with a significant match to a metazoan other than a closely related plant-parasitic nematode was removed from further analysis. An e-value of 1.0e-10 was the threshold used to declare a match. The remaining sequences provided our final set of candidates for horizontally transferred genes (Tables 1, 3).
Codon usage analysis
The protein alignment of the M. incognita and R. leguminosarum nodL sequences was trimmed such that only identical amino acids remained, and the sequences back-translated, retaining the correct codon usage. Ten thousand pairs of simulated sequences were generated by independently permuting the homologous codon pairs in the actual data. In other words, the probability that the ith codon in the first simulated sequence was assigned the ith codon from the actual M. incognita sequence and the ith codon in the second simulated sequence was assigned the ith codon from the actual R. leguminosarum sequence was set to 0.5 and the probability that the ith codon assignments in the simulated sequences were reversed was also set to 0.5. Codon adaptation indices were computed for each simulated sequence using the EMBOSS suite of sequence analysis tools [49].
Phylogenetic analysis of candidates
For each candidate, the protein sequences for the top 15 matches with an e-value of 1.0e-10 or less were extracted from the BLASTX search against the nr database. If there were not 15 matches with an e-value meeting this criterion, all sequences with e-values lower than 1.0e-10 were selected. Alignments of these sequences with the translated candidate sequence were constructed with CLUSTALX [50]; improvements to the CLUSTALX alignments were performed manually. Sequences from the same species with more than 95% identity after alignment were considered possible paralogs and deemed redundant information for this analysis. Only one sequence from each of these sets was used in further analysis. Poorly aligned sequences were also discarded.
Distances between aligned proteins were estimated with the Dayhoff amino-acid replacement model [51]. Tree topologies were then inferred from these distances via neighbor-joining [52] and 1,000 non-parametric bootstrap replicates were used to estimate clade support [53]. Maximum likelihood analysis produced topologies consistent with the neighbor-joining analysis. All phylogenetic reconstructions were performed with the PHYLIP and PAML software packages [54, 55].
Additional analyses of the putative nodL gene were conducted with Version 3.0b4 of the MrBayes software [56]. For these analyses, the Jones-Taylor-Thornton model of amino-acid replacement [57] was adopted and variation of replacement rates among sites was incorporated by a discretized gamma distribution with four rate categories [58]. Each Markov chain Monte Carlo analysis used four heated chains and employed a burn-in period of 10,000 cycles, followed by 990,000 additional cycles. Convergence of the Markov chain was diagnosed by performing two different runs from different initial parameter states. Prior distributions for all parameters were the default distributions incorporated in the MrBayes software.
Additional data files
The following files are available with the online version of this article: the e-values of the best matches for the initial BLASTX searches against bacteria, C. elegans and Drosophila for the WMi dataset (Additional data file 1), together with a mapping file (Additional data file 2) that gives the MI contig number associated with each filename; the best match and e-value for the BLASTX search of the WMj dataset against bacteria (Additional data file 3), C. elegans (Additional data file 4) and Drosophila (Additional data file 5); the e-value for the best match to bacteria, C. elegans and Drosophila resulting from a TBLASTX search for the NMi dataset (Additional data file 6), the NMj dataset (Additional data file 7), the NMh dataset (Additional data file 8), where a value of 100 indicates no match found; a text file with details of the data given in each of these dataset files (Additional data file 9); the alignment in Phylip format used to calculate the NodL tree (Additional data file 10); the alignment in Phylip format of the original sequences, before any manual adjustments were made (Additional data file 11); and a key giving the gi number listed in the alignment for each species (Additional data file 12).
References
Boucher G, Lambshead PJD: Ecological biodiversity of marine nematodes in samples from temperate, tropical, and deep-sea regions. Conserv Biol. 1994, 9: 1594-1604. 10.1046/j.1523-1739.1995.09061594.x.
Koenning SR, Overstreet C, Noling JW, Donald PA, Becker JO, Fortnum BA: Survey of crop losses in response to phytoparasitic nematodes in the United States for 1994. J Nematol. 1999, 31: 587-618.
Bird DMcK, Koltai H: Plant parasitic nematodes: habitats, hormones, and horizontally-acquired genes. J Plant Growth Regul. 2000, 19: 183-194.
Blaxter ML, DeLey P, Garey J, Liu LX, Scheldeman P, Vierstraete A, Vanfletern J, Mackey LY, Dorris M, Frisse LM, et al: A molecular evolutionary framework for the phylum Nematoda. Nature. 1998, 392: 71-75. 10.1038/32160.
Jain R, Rivera MC, Lake JA: Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci USA. 1999, 96: 3801-3806. 10.1073/pnas.96.7.3801.
Lawrence JG: Gene transfer, speciation, and the evolution of bacterial genomes. Curr Opin Microbiol. 1999, 2: 519-523. 10.1016/S1369-5274(99)00010-7.
Ochman H, Lawrence JG, Groisman EA: Lateral gene transfer and the nature of bacterial innovation. Nature. 2000, 405: 299-304. 10.1038/35012500.
Dehal P, Satou Y, Campbell RK, Chapman J, Degnan B, De Tomaso A, Davidson B, Di Gregorio A, Gelpke M, Goodstein DM, et al: The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science. 2002, 298: 2157-2167. 10.1126/science.1080049.
Andersson JO, Sjögren ÅM, Davis LAM, Embley TM, Roger AJ: Phylogenetic analyses of diplomonad genes reveal frequent lateral gene transfers affecting eukaryotes. Curr Biol. 2003, 13: 94-104. 10.1016/S0960-9822(03)00003-4.
Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q: Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998, 282: 754-759. 10.1126/science.282.5389.754.
Wolf YI, Aravind L, Koonin EV: Rickettsiae and Chlamydiae: evidence of horizontal gene transfer and gene exchange. Trends Genet. 1999, 15: 173-175. 10.1016/S0168-9525(99)01704-7.
Royo J, Gimez E, Hueros G: CMP-KDO synthetase: A plant gene borrowed from Gram-negative eubacteria. Trends Genet. 2000, 16: 432-433. 10.1016/S0168-9525(00)02102-8.
Lange BM, Rujan T, Martin W, Croteau R: Isoprenoid biosynthesis: the evolution of two ancient and distinct pathways across genomes. Proc Natl Acad Sci USA. 2000, 97: 13172-13177. 10.1073/pnas.240454797.
International Human Genome Sequencing Consortium: Initial sequencing and analysis of the human genome. Nature. 2001, 409: 860-921. 10.1038/35057062.
Brinkman FSL, Blanchard JL, Cherkasov A, Av-Gay Y, Brunham RC, Fernandez RC, Finlay BB, Otto SP, Ouellette BFF, Keeling PJ, et al: Evidence that plant-like genes in Chlamydia species reflect an ancestral relationship between Chlamydiaceae, cyanobacteria, and the choloroplast. Genome Res. 2002, 12: 1159-1167. 10.1101/gr.341802.
Stanhope MJ, Lupas A, Italia MJ, Koretke KK, Volker C, Brown JR: Phylogenetic analyses do not support horizontal gene transfers from bacteria to vertebrates. Nature. 2001, 411: 940-944. 10.1038/35082058.
Popeijus H, Overmars H, Jones J, Blok V, Goverse A, Helder J, Schots A, Bakker J, Smant G: Degradation of plant cell walls by a nematode. Nature. 2000, 406: 36-37. 10.1038/35017641.
Smant G, Stokkermans JPWG, Yan Y, De Boer JM, Baum TJ, Wang X, Hussey RS, Gommers FJ, Henrissat B, et al: Endogenous cellulases in animals: Isolation of β-1,4-endoglucanase genes from two species of plant-parasitic cyst nematodes. Proc Natl Acad Sci USA. 1998, 95: 4906-4911. 10.1073/pnas.95.9.4906.
Yan Y, Smant G, Stokkermans J, Qin L, Helder J, Baum T, Schots A, Davis E: Genomic organization of four β-1,4-endoglucanase genes in plant-parasitic cyst nematodes and its evolutionary implications. Gene. 1998, 220: 61-70. 10.1016/S0378-1119(98)00413-2.
Lambert KN, Allen KD, Sussex IM: Cloning and characterization of an esophageal-gland-specific chorismate mutase from the phytoparasitic nematode Meloidogyne javanica. Mol Plant Microbe Interact. 1999, 12: 328-336.
Jaubert S, Laffaire J-B, Abad P, Rosso M-N: A polygalacturonase of animal origin isolated from the root-knot nematode Meloidogyne incognita. FEBS Lett. 2002, 522: 109-112. 10.1016/S0014-5793(02)02906-X.
McCarter JP, Abad P, Jones J, Bird DMcK: Rapid gene discovery in plant parasitic nematodes via expressed sequence tags. Nematology. 2000, 2: 719-731. 10.1163/156854100509574.
Veronico P, Jones J, Di Vito M, De Giorgi C: Horizontal transfer of a bacterial gene involved in polyglutamate biosynthesis to the plant-parasitic nematode Meloidogyne artiellia. FEBS Lett. 2001, 508: 470-474. 10.1016/S0014-5793(01)03132-5.
Aguinaldo AM, Turbeville JM, Linford LS, Rivera MC, Garey JR, Raff RA, Lake JA: Evidence for a clade of nematodes, arthropods and other moulting animals. Nature. 1997, 387: 489-493. 10.1038/387489a0.
Mallatt J, Winchell CJ: Testing the new animal phylogeny: first use of combined large-subunit and small-subunit rRNA gene sequences to classify the protostomes. Mol Biol Evol. 2002, 19: 289-301.
Hedges SB: The origin and evolution of model organisms. Nat Rev Genet. 2002, 3: 838-849. 10.1038/nrg929.
Blair JE, Ikeo K, Gojobori T, Hedges SB: The evolutionary position of nematodes. BMC Evol Biol. 2002, 2: 7-10.1186/1471-2148-2-7.
McCarter JP, Mitreva MD, Martin J, Dante M, Wylie T, Rao U, Pape D, Bowers Y, Theising B, Murphy C, et al: Analysis and functional classification of transcripts from the root-knot nematode Meloidogyne incognita. Genome Biol. 2003, 4: R26-10.1186/gb-2003-4-4-r26.
McCarter JP, Clifton SW, Bird DMcK, Waterston R: Nematode gene sequences; Update for June 2002. J Nematol. 2002, 34: 71-74.
Nematode.net. [http://www.nematode.net]
Koltai H, Dhandaydham M, Opperman C, Thomas J, Bird DMcK: Overlapping plant signal transduction pathways induced by a parasitic-nematode and a rhizobial endosymbiont. Mol Plant Microbe Interact. 2001, 14: 1168-1177.
Ochman H, Moran NA: Genes lost and genes found: evolution of bacterial pathogensis and symbiosis. Science. 2001, 292: 1096-1098. 10.1126/science.1058543.
Wood DW, Setubal JC, Kaul R, Monks DE, Kitajima JP, Okura VK, Zhou Y, Chen L, Wood GE, Almedia Jr NF, et al: The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science. 2001, 294: 2317-2323. 10.1126/science.1066804.
Ludwig RA: Physiological roles of glutamine synthetase I and II in ammonium assimilation in Rhizobium sp. 32H1. J Bacteriol. 1980, 141: 1209-1216.
Turner SL, Young JPW: The glutamine synthetases of rhizobia: phylogenetics and evolutionary implications. Mol Biol Evol. 2000, 17: 309-319.
Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer ELL: The Pfam protein families database. Nucleic Acids Res. 2002, 30: 276-280. 10.1093/nar/30.1.276.
Downie JA, Young JPW: Genome sequencing: the ABC of symbiosis. Nature. 2001, 412: 597-598. 10.1038/35088167.
Van Rhijn P, Vanderleyden J: The Rhizobium-plant symbiosis. Microbiol Rev. 1995, 59: 124-142.
Göttfert M: Regulation and function of rhizobial nodulation genes. FEMS Microbiol Rev. 1993, 10: 39-64.
Ditt RF, Nester EW, Comai L: Plant gene expression response to Agrobacterium tumefaciens. Proc Natl Acad Sci USA. 2001, 98: 10954-10959. 10.1073/pnas.191383498.
Ray C, Abbott AG, Hussey RS: Trans-splicing of a Meloidogyne incognita mRNA encoding a putative esophageal gland protein. Mol Biochem Parasitol. 1994, 68: 93-101. 10.1016/0166-6851(94)00153-7.
Lawrence JG, Ochman H: Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol. 1997, 44: 383-397.
Capela D, Barloy-Hubler F, Gouzy J, Bothe G, Ampe F, Batut J, Boistard P, Becker A, Boutry M, Cadieu E, et al: Analysis of the chromosome sequence of the legume symbiont Sinorhizobium meliloti strain 1021. Proc Natl Acad Sci USA. 2001, 98: 9877-9882. 10.1073/pnas.161294398.
Sharp PM, Li W-H: The codon adaptation index - a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15: 1281-1295.
Koski LB, Morton RA, Golding GB: Codon bias and base composition are poor indicators of horizontally transferred genes. Mol Biol Evol. 2001, 18: 404-412.
Sullivan JT, Ronson CW: Evolution of rhizobia by acquisition of a 500-kb symbiosis island that integrates into a phe-tRNA gene. Proc Natl Acad Sci USA. 1998, 95: 5145-5149. 10.1073/pnas.95.9.5145.
Wheeler DL, Church DM, Lash AE, Leipe DD, Madden TL, Pontius JU, Schuler GD, Schriml LM, Tatusova TA, Wagner L, Rapp BA: Database resources of the National Center for Biotechnology Information: 2002 update. Nucleic Acids Res. 2002, 30: 13-16. 10.1093/nar/30.1.13.
WU-BLAST. [http://blast.wustl.edu]
Rice P, Longden I, Bleasby A: EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16: 276-277. 10.1016/S0168-9525(00)02024-2.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The Clustal_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25: 4876-4882. 10.1093/nar/25.24.4876.
Dayhoff MO: Atlas of Protein Sequence and Structure. Volume 5 Supplement 3. 1978, Washington, DC: National Biomedical Research Foundation
Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987, 4: 406-425.
Felsenstein J: Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985, 39: 783-791.
Felsenstein J: PHYLIP (Phylogeny Inference Package) version 3.6a2. 1993, Seattle, WA: Department of Genetics, University of Washington
Yang Z: PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997, 13: 555-556.
Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogeny. Bioinformatics. 2001, 17: 754-755. 10.1093/bioinformatics/17.8.754.
Jones DT, Taylor WR, Thornton JM: The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 1992, 8: 275-282.
Yang Z: Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol. 1994, 39: 306-314.
Tandingan-De Ley I, De Ley P, Vierstraete A, Karssen G, Moens M, Vanfleteren J: Phylogenetic analyses of Meloidogyne small subunit rDNA. J Nematol. 2002, 34: 319-327.
Acknowledgements
We thank M. Burke for his technological support and advice, H. Kishino for his helpful comments and insights, and M. Dante and J. Martin for NemaGene clusters. This research was supported by NSF grant DBI-0077503 to D.B. J.P.M. was supported by a Helen Hay Whitney/Merck Postdoctoral Fellowship. E.H.S. and J.L.T. were supported by NSF grant INT-990934, and J.L.T. was further supported by BIRD of Japan Science and Technology Corporation. D.B. and J.M. are equity holders of Divergence Inc.; none of this research was funded by Divergence Inc.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
13059_2003_549_MOESM1_ESM.tab
Additional data file 1: The e-values of the best matches for the initial BLASTX searches against bacteria, C. elegans and Drosophila for the WMi dataset (TAB 22 KB)
13059_2003_549_MOESM3_ESM.tab
Additional data file 3: The best match and e-value for the BLASTX search of the WMj dataset against bacteria (TAB 131 KB)
13059_2003_549_MOESM4_ESM.tab
Additional data file 4: The best match and e-value for the BLASTX search of the WMj dataset against C. elegans (TAB 219 KB)
13059_2003_549_MOESM5_ESM.tab
Additional data file 5: The best match and e-value for the BLASTX search of the WMj dataset against Drosophila (TAB 208 KB)
13059_2003_549_MOESM6_ESM.tab
Additional data file 6: The e-value for the best match to bacteria, C. elegans and Drosophila resulting from a TBLASTX search for the NMi dataset (TAB 2 MB)
13059_2003_549_MOESM7_ESM.tab
Additional data file 7: The e-value for the best match to bacteria, C. elegans and Drosophila resulting from a TBLASTX search for the NMj dataset (TAB 856 KB)
13059_2003_549_MOESM8_ESM.tab
Additional data file 8: The e-value for the best match to bacteria, C. elegans and Drosophila resulting from a TBLASTX search for the NMh dataset (TAB 81 KB)
13059_2003_549_MOESM11_ESM.phy
Additional data file 11: The alignment in Phylip format of the original sequences, before any manual adjustments were made (PHY 4 KB)
Rights and permissions
About this article
Cite this article
Scholl, E.H., Thorne, J.L., McCarter, J.P. et al. Horizontally transferred genes in plant-parasitic nematodes: a high-throughput genomic approach. Genome Biol 4, R39 (2003). https://doi.org/10.1186/gb-2003-4-6-r39
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/gb-2003-4-6-r39