
Abstract
Background
When there are no strand-specific biases in mutation and selection rates (that is, in the substitution rates) between the two strands of DNA, the average nucleotide composition is theoretically expected to be A = T and G = C within each strand. Deviations from these equalities are therefore evidence for an asymmetry in selection and/or mutation between the two strands. By focusing on weakly selected regions that could be oriented with respect to replication in 43 out of 51 completely sequenced bacterial chromosomes, we have been able to detect asymmetric directional mutation pressures.
Results
Most of the 43 chromosomes were found to be relatively enriched in G over C and T over A, and slightly depleted in G+C, in their weakly selected positions (intergenic regions and third codon positions) in the leading strand compared with the lagging strand. Deviations from A = T and G = C were highly correlated between third codon positions and intergenic regions, with a lower degree of deviation in intergenic regions, and were not correlated with overall genomic G+C content.
Conclusions
During the course of bacterial chromosome evolution, the effects of asymmetric directional mutation pressures are commonly observed in weakly selected positions. The degree of deviation from equality is highly variable among species, and within species is higher in third codon positions than in intergenic regions. The orientation of these effects is almost universal and is compatible in most cases with the hypothesis of an excess of cytosine deamination in the single-stranded state during DNA replication. However, the variation in G+C content between species is influenced by factors other than asymmetric mutation pressure.
Similar content being viewed by others


Background
The G+C content of bacterial DNA ranges from around 25% to around 75% [1,2]. In contrast to the nuclear genomes of some vertebrates, the intragenomic heterogeneity of G+C content is generally small in bacteria [3,4], although there are some exceptions [5,6,7]. The wide interspecies variation and narrow intraspecies heterogeneity of the DNA G+C content was interpreted to be the result of differences in the bidirectional mutation rates between AT and GC pairs [8]. This kind of mutation pressure is said to be the G+C pressure. From Watson-Crick base-pairing rules [9], the G+C content is exactly the same for the two complementary strands in a DNA duplex, so the G+C pressure effects are symmetric with respect to the two strands. The effects of G+C pressure are dramatically increased in regions where selective pressure is weak: for instance, in the third codon positions in protein-coding sequences, where most substitutions do not alter the encoded amino acid, the average G+C content ranges from at least 7% to 95% between bacteria of different species [7,10,11]. It should be stressed that symmetric directional mutation effects are not restricted to purely neutral regions, and strongly influence amino-acid frequencies in proteins [12,13,14,15,16].
The two strands of a DNA duplex must have the same G+C content, but the different bases can still vary in frequency. For instance, one strand may have more G than C, say G/(G+C) = 0.8, and by complementarity the other one will have more C than G, G/(G+C) = 0.2 in this case. The strand-specific asymmetries in DNA have been the subject of considerable research [17,18,19,20,21,22,23]. Briefly, the theoretical predictions are as follows: when mutation and selection are assumed to be symmetric with respect to the two strands of DNA, parity rule 1 (PR1) holds where the following pairs of substitution rates are equal: rAT = rTA, rGC = rCG, rAG = rTC, rGA = rCT, rAC = rTG, rCA = rGT [20]. Here, rAT, and so on, are substitution rates of A → T, and so on, in a specific strand. Under the PR1 hypothesis, the intrastrand base composition at equilibrium is expected to be always in a particular state, the parity rule 2 (PR2) state, such that A = T and G = C within each strand [20,21,24]. This PR2 state is more than just an equilibrium point, because convergence towards PR2 continues even if the substitution rates are modified: there is no way to escape from the PR2 state under the PR1 hypothesis [25]. However, a bias in mutation or selection between the two strands of DNA can generate deviations from PR2 (that is, A ≠ T, or G ≠ C, or both). In this situation, the underlying cause, regardless of its mutational or selective nature, is asymmetric between the two strands.
Note that there is a serious confusion in the literature about substitution matrices that are 'symmetric' in the sense that the two strands evolve with the same pattern of substitutions (PR1 hypothesis), and 'symmetric' in the sense that the terms above and below the diagonal are the same (the SYM model, where rij = rji). The SYM model adopted previously by others, especially in the field of molecular phylogeny [26], assumes the equality of intrastrand mutation rates between any two bases: in contrast to the PR1 model, the SYM model does not incorporate DNA base-pairing rules. The SYM model has long been recognized as unrealistic because it predicts A = T = G = C at equilibrium without biases. The PR1 model is not a special case of the SYM model; rejection of the SYM model does not lead to rejection of the PR1 model.
The effect of asymmetric mutation pressure on the amino-acid content of proteins has been reported [27,28,29]. In terms of amino-acid composition, the diversifying selection on integral membrane proteins (enriched in hydrophobic amino acids) and on cytoplasmic proteins (enriched in hydrophilic amino acids) is generally the most important factor in within-proteome variability [30]. In Borrelia burgdorferi, however, the effects of this selective pressure are quantitatively less important than those resulting from asymmetric mutation pressure [31], stressing again that mutation pressure is far from being a negligible phenomenon.
Deviations from PR2 have been studied using many different approaches, including correspondence discriminant analysis [32], linear discriminant analysis with rotating windows [28], correspondence analysis applied to relative synonymous codon usage [27,33,34], analysis of variance [35], a comparative genomics approach [22,36,37,38], simple DNA walks [39], detrended DNA walks split by codon positions [40,41,42], skewed oligomer distributions [43], and moving windows plots with direct [44,45] or cumulated [46,47] or codon position-dependent [48] skew indices. Here we have used a simple graphical representation, very close to the raw data, that provides a straightforward visualization and interpretation of results.
Consider a complete annotated bacterial chromosome in which origin and terminus of replication have been located so that we know whether a subsequence is on the leading strand or the lagging strand at replication (Figure 1). Let A3, C3, G3, and T3 denote the base counts in the sense strand of a given coding sequence when only the weakly selected third codon positions are considered. Deviations from PR2 in third codon positions are characterized by G3/(G3+C3) and A3/(A3+T3) to draw PR2-plots [7].

Representation of the strands used for analysis in this paper. The leading strand for replication is black and the lagging one is shaded gray; note the switch at the origin of replication. The sense strand of coding sequences is represented by white arrows. The sense sequence of a coding region that is transcribed in the same direction as the motion of the replication fork is in the leading strand, whereas a sense sequence that is transcribed in the opposite direction is in the lagging strand. The strand used for large intergenic spaces, represented by large boxes, is always the published strand from 5' to 3'. The strand used for potentially transcribed untranslated spaces, that is, small intergenic regions among co-oriented genes, is represented by small boxes.
Figure 2a shows diagrammatically what would be expected under the null hypothesis PR1: the PR2 state should be observed. Although there will statistical fluctuations, spreading genes across the space indicated by each circle, the circles are centered at the midpoint of the plot (0.5, 0.5) which corresponds to the PR2 state. The range of dispersion is greater for the leading-strand group, because we have assumed here (for clarity, to avoid overlap in the figures), that there is an excess of coding sequences in the leading strand, as is usually observed in bacteria [48]. Figure 2b shows diagrammatically what would be expected when replication-associated mutation pressure enriches the leading strand in G and T. The leading-strand coding sequences, whose sense strand is, by definition, the leading strand for replication (Figure 1) are therefore also enriched in G and T. Thus, x1, their average G3/(G3+C3) value, is greater than 0.5, and y1, their average A3/(A3+T3) value, is less than 0.5. From DNA duplex base-pairing rules, if the leading strand for replication is enriched in G and T, its complementary lagging strand is conversely enriched in C and A. Therefore, we have a perfectly symmetric situation for the lagging-strand coding sequences, whose sense strand is, by definition, the lagging strand for replication (Figure 1), with x2 = 1 - x1 and y2 = 1 - y1. Note that the overall average is biased towards the leading-strand group because of the excess of leading-strand coding sequences, but that the midpoint (xc = (x1 + x2)/2, yc = (y1 + y2)/2) is not affected by this uneven distribution. Figure 2c shows diagrammatically what would be expected with asymmetric forces, such as codon usage that optimizes translation efficiency [49,50] or transcription-induced mutation pressure [51]. These affect the sense strand of both the leading and lagging coding sequences in the same direction, because the analyzed strand is always the sense strand. Note that the deviation from the null hypothesis could be higher [22] for the leading group because a greater number of highly expressed genes are found in this group [48], so that both transcription-induced mutation and translation-induced selection [52,53] could be higher in these genes. However, this effect was found to be negligible [35], and never produces a symmetric distribution around the center of the two groups, as in previous case (Figure 2b), because the two groups are expected to be moved in the same direction for transcription- and translation-induced biases, instead of in opposite directions for replication-induced biases. Figure 2d shows diagrammatically what would be expected when all phenomena are combined. The distance between the averages of the two groups,

Expected deviations in PR2 plots in coding sequences and small intergenic regions among co-oriented genes. (a) The null hypothesis: mutation and selection are symmetric with respect to the two DNA strands; (b) replication-induced mutation pressure is asymmetric; (c) transcription- or translation-associated mutation or selection pressures are asymmetric; (d) all forces in (b) and (c) combined are asymmetric.

is the contribution of replication-associated effects to PR2 deviations. The distance from the center of the plot to the midpoint of B I (xc; yc),

is the overall contribution of transcription- and translation-associated effects to PR2 deviations, where B I is the replication-associated bias.
Consider now a small transcribed untranslated intergenic region between two co-oriented coding sequences. Let A, C, G, T denote its base counts in the cis-sense, that is in the same strand as the sense strand of the two flanking coding sequences (Figure 1). As previously described, deviations from PR2 are characterized by using G/(G+C) and A/(A+T) values to draw PR2-plots. Expected effects in the cis-sense strand are the same as in the sense strand (Figure 2) except for translation-induced selection, which is absent in untranslated regions. Therefore, a difference in B II when compared with the analysis of coding sequences is evidence of translation-associated effects on PR2 deviations in coding sequences. Last, consider a large untranscribed intergenic region and note its base counts (A, C, G, T) in the published strand of the chromosome (Figure 1).
Figure 3a shows what would be expected in spacers under replication-induced effects alone when the leading strand is enriched in G and T. This is essentially the same as the situation for third codon position when there is only replication-induced mutation pressure (Figure 2b), because all bases are involved, not just those in protein-coding genes. The sole difference is that the leading and lagging group are now of similar size, because when working with the published strand, there are approximately the same number of intergenic regions in the two groups (Figure 1). Therefore, when B I is similarly oriented in the two analyses, this is an indication of a common underlying replication-induced mutation pressure, the difference in intensity being controlled by the difference in selective pressure between the two kinds of region.

Expected deviations in PR2 plots in large intergenic spaces (a) when replication-induced mutation pressure is asymmetric and (b) in an extreme case of asymmetric transcription-induced forces(see text for details).
In practice, however, it is impossible to verify from DNA database annotations whether large intergenic regions are completely free from transcription. Consider an extreme theoretical case in which all large intergenic regions are transcribed (Figure 3b). For simplicity, the whole intergenic region is assumed to be transcribed from the same strand: the whole intergenic region is either the template strand or the anti-template strand, but not a mixture of both. (The anti-template strand has the same nucleotide sequence as the transcribed RNA because the complementary strand is the template strand for transcription.) This simplification does not alter the reasoning, because a proper partition of intergenic regions would remove the mixed cases. In the leading-strand group, positioned to the right of the origin (Figure 1), there is an excess of anti-template strand for transcription because of the selection against head-on collisions between RNA and DNA polymerase complexes [54,55,56]. Therefore, the anti-template strand subgroup is bigger than the template one in the leading group, and by a symmetrical reasoning, on the left to the origin, the template strand subgroup is bigger than the anti-template one in the lagging-strand group. Figure 3b shows what is expected if transcription leads to an excess of G and T bases in the anti-template strand. Note that in this extreme case transcription effects yield B I ≠ 0, but the general pattern (Figure 3b) is completely different from replication-induced mutation pressure effects (Figure 2b), making it easy to discriminate between the two effects.
Results
Distribution of G+C content
Thanks to the availability of an increasing number of complete bacterial genomes with a high G+C content, there was a representative distribution of G+C content in the dataset (Table 1), and the values of G+C content in third codon positions (P3, as previously defined in [11]; see also Materials and methods), in first and second codon positions averaged (P12), and in intergenic spaces (GC IGR ), were found to be consistent with previously known results [10,11] both in terms of their range of distribution and the correlation between them (Figure 4). Intragenomic P3 distributions of all species examined (all histograms are given in the tables in Additional data files under the 'GCorder' flag) are unimodal and narrow for a major class of genes comprising more than 90% of the total genes, although average P3 values differ widely between species. Some species show scattering of P3 distribution. In particular, a previous study of 254 genes in Pseudomonas [7] revealed a sizeable minor class of genes (28%) forming a trail toward lower G+C content (genes with P3 < 0.75). In our present study of 5,255 genes, the minor class of genes (with P3 < 0.75) is only 8% of the total, indicating that the previous sample was biased towards the minor class. Moreover, the width of the distribution (standard deviation) of the majority of genes is narrower in species with both extreme ranges of P3 than in those with middle ranges. These features of the P3 distribution are expected from the theory of bidirectional mutation rates and their equilibrium [8]. In this theory, mutation rates, u (GC → AT) and v (AT → GC), will lead to a G+C content (P3) at equilibrium of v/(u+v). The expected standard deviation is

Distribution of G+C content in the dataset. The G+C content of the 51 bacterial chromosomes under analysis was highly variable from 25.5% in the Ureaplasma urealyticum chromosome to 67.9% in Halobacterium sp., with a larger distribution in third codon positions (x-axis P3 from 11.2% to 88.0%) than in intergenic spaces (y-axis GCIGR from 15.5% to 63.4%) than in first and second position (y-axis P12 from 33.4% to 62.2%) as expected [10,11]. Regression slopes (or ε values) and their standard deviations for P12 and GCIGR were 0.343 ± 0.021 and 0.586 ± 0.024, respectively.

where b is an average of the number of third codon positions per gene [8].
Difference in G+C content between leading and lagging strands
Statistically highly significant differences (p < 0.01) in P3 between coding sequences located on the leading and lagging strands were found in 23 out of 43 chromosomes. In most cases (21 out of the 23) genes on the leading strand were found to have a lower P3 content than those on the lagging one. (Two exceptions were Mycoplasma genitalium, which is known to have a peculiar behavior with respect to G+C content [5,6], and Caulobacter crescentus.) As far as we know, this is the first time that such a systematic difference has been reported, but note that the differences were always very small. The maximum was only 0.035, for the difference in P3 content of leading and lagging strands of Vibrio cholerae chromosome 2, as compared to the 0.768 average P3 difference between Halobacterium sp. and Ureaplasma urealyticum, or the 0.050 accepted range of genomic G+C content polymorphism [57]. There was no significant correlation between the average P3 and the difference in P3 between the two groups of genes. A highly significant difference in G+C content between the leading and the lagging sequences was found in only five chromosomes for large intergenic regions, and three chromosomes for small intergenic spaces.
PR2-biases in coding sequences
Out of 43 chromosomes, the differences in PR2-biases between the leading and lagging groups were highly significant (p < 0.01) in 39 chromosomes for G3/(G3+C3) and in 34 for A3/(A3+T3) and in 32 chromosomes for simultaneously G3/(G3+C3) and A3/(A3+T3) (see Additional data files). In the PR2-bias-plot analysis, we found a general pattern of x1 >x2 and y1 <y2, as in Figure 2b, meaning that coding sequences in the leading strand had higher G3/(G3+C3) values and lower A3/(A3+T3) values than those on the lagging strand: leading coding sequences were enriched in keto-bases (G and T) in the third codon position compared to lagging coding sequences. There were, however, exceptions to this general trend: out of the 39 chromosomes that were highly significant for G3/(G3+C3), all had higher values in the leading strand, but out of the 34 chromosomes that were highly significant for A3/(A3+T3), three chromosomes did not follow the general trend (Lactococcus lactis and Staphylococcus aureus strains Mu50 and N315). Leading and lagging coding sequences are separated in PR2 plots as expected under replication-associated effects, and the leading group is almost always down right of the lagging group. However, the extent to which the groups are separated differs between species. The two most extreme species are B. burgdorferi, where the two groups of genes are completely resolved, and M. genitalium, where the two groups are almost completely overlapping (Figure 5). Other species with a spectacular difference in PR2 plots were Chlamydia muridarum, Chlamydia trachomatis and Treponema pallidum. Out of 43 chromosomes, the residual bias B II was in most cases oriented as in Figure 2c, with xc < 0.5 (all except Pyrococcus abyssi and T. pallidum) and yc < 0.5 (all except Thermotoga maritima). The relative contribution of replication-coupled bias (B I ) to the total bias (B I plus B II ) ranged from 6% (Mycoplasma pulmonis) to 86% (B. burgdorferi) with an average value of 52%.

Example of detailed results for two extreme cases with B. burgdorferi (left) and M. genitalium (right). (a) P3-P12 plot for coding sequences; (b) PR2-plot in third codon position; (c) PR2 plot in large intergenic spaces. Leading-group sequences are represented by black circles, and lagging ones by white circles.
PR2-biases in large intergenic regions
Out of 43 chromosomes, the differences in PR2-biases between the leading and lagging groups were highly significant (p < 0.01) in 36 chromosomes for G/(G+C) and in 34 for A/(A+T) and in 32 chromosomes for simultaneously G/(G+C) and A/(A+T) (see table in Additional data files). All the 36 chromosomes highly significant for G/(G+C) had higher values in the leading strand (that is, x1 >x2 as in coding sequences). Out of the 34 chromosomes highly significant for A/(A+T), 28 had lower values in the leading strand (that is, y1 <y2 as in coding sequences). Exceptions were Bacillus subtilis, Bacillus halodurans, L. lactis, S. aureus strains Mu50 and N315, and Streptococcus pyogenes. The distribution of intergenic regions in a PR2-plot was as expected under replication-associated effects (Figure 3a), and is particularly visible in the case of B. burgdorferi (Figure 5c). Unlike the third codon position, the leading and lagging groups clustered symmetrically around the center point of the PR2-bias plot. In intergenic regions, both xc and yc were always near 0.5, so that the residual bias B II was always close to zero. The relative contribution of replication-coupled bias (B I ) to the total bias (B I plus B II ) was therefore very high, on average 90%, and ranged from 88% (Helicobacter pylori) to 99.6% (B. subtilis) in the intergenic regions of species with significant B I values.
PR2-biases in potentially transcribed untranslated regions
Out of 43 chromosomes, the differences in PR2-biases between the leading and lagging small intergenic spaces among co-oriented genes were highly significant (p < 0.01) in 26 chromosomes for G/(G+C) and in 7 chromosomes for A/(A+T) and in 7 chromosomes for both G/(G+C) and A/(A+T) together (see table in Additional data files). All the 26 chromosomes with highly significant difference in G/(G+C) showed the same pattern as in third codon positions in coding sequences with x1 >x2. Of the seven chromosomes highly significant for A/(A+T), all showed the same pattern as in third codon positions in coding sequences with y1 <y2, except for B. subtilis and S. pyogenes. Out of 43 chromosomes, the orientation of the residual bias B II showed no clear tendency (only 31 chromosomes had xc > 0.5 and 23 yc > 0.5; the trend, if any, would be in the opposite direction, as in third codon positions). The relative contribution of replication-coupled bias (B I ) to the total bias (B I plus B II ) ranged from 10% (H. pylori strain 26695) to 93% (Thermoplasma acidophilum) with an average value of 58%.
Comparison of third codon positions and intergenic spaces
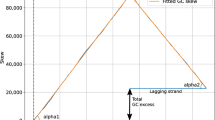
The differences between PR2-biases in intergenic regions between the leading and lagging strands (B I ) were significantly and highly correlated with the PR2-biases in the third codon position as is evident from the regression coefficient of approximately 0.6 and correlation coefficient (r2 = 0.77) among 43 chromosomes (Figure 6). The correlation was still significant when the extreme B. burgdorferi was removed from analysis (r2 = 0.68). The 95% confidence interval for the value of the slope of the regression line [0.50-0.71] is less than 1, meaning that replication-associated biases were significantly smaller in intergenic regions than in third codon positions. This slope value was very close to the one obtained for the regression line between G+C content in intergenic regions versus third codon positions (Figure 4).

Comparison of the absolute contribution of replication-associated bias B I between intergenic and third codon positions.
Correlation between P3and replication-associated biases
If differences in the G+C content between species were dictated by asymmetric replication-associated mutation pressure, one would expect a correlation between the extent of PR2-bias and G+C content; however, no significant correlation between B I in coding sequences and P3 were found (Figure 7).

Correlation between GC content and the extent of strand biases in third codon positions.
Discussion
Asymmetric mutation pressure
A general feature observed in the present study is that, whenever there is a difference in base composition between the leading and the lagging strand, the G and T bases are enriched in the leading strand, and the G+C content is somewhat lower in the leading strand (see Results). The direction of observed biases are universal in bacteria [28], although not every species is biased [58]. This pattern is not restricted to the bacterial world, as GT-enriched leading strands have been documented in mitochondria [59,60,61], in the chloroplast genome of Euglena gracilis [62], in viruses [45,46,47], and in the telomeric ends of the Saccharomyces cerevisiae genome [63]. Not all sequences are biased, as shown by the well-documented β-globin region in the Homo sapiens genome [37]. Remarkably, absolutely no exceptions to the relative enrichment in G of the leading strand were found here, either in third codon positions or intergenic positions. However, the sole general rule in biology is that there are no general rules, and during the time of revision of this manuscript the first exception was documented in the chromosome of Streptomyces coelicolor [64]. The relative enrichment in T of the leading strand suffers from some exceptions that, interestingly, are all found here in the low-G+C group of Gram-positive bacteria (namely B. subtilis, B. halodurans, L. lactis, S. aureus, and S. pyogenes).
Because of the strong correlation in direction and intensity of PR2 deviation between third codon positions and intergenic spaces (Figure 6), the most likely explanation for the GT enrichment of the leading strand is a replication-induced asymmetric mutation pressure between the leading and lagging strand.
Potential underlying causes
The universal excess of G and T in the leading strand is common to distantly related species, suggesting that asymmetric directional mutation pressure has a chemical basis. This does not mean that the mechanism is simple: there are 12 substitution rates under asymmetric molecular evolution, but only four base frequencies; many different substitution scenarios can explain the observed biases. Here we discuss only simple hypotheses in which only one of the 12 substitution rates is assumed to differ between the two strands: for example, only an excess of A → G or of C → T in the leading strand is able to produce a GT-rich leading strand. Because the leading strand spends more time in the single-stranded state than the lagging strand during DNA replication [65], it is natural to look for mutations that are increased in the single-stranded state as possible explanations.
Oxidations
We are not aware of any studies comparing the rate of base oxidation between the single-stranded and double-stranded state in DNA. However, in a study of peroxyl radicals [66], which have a longer biological half-life than other O2-derived oxidants, all products formed were from the major groove of the bases, implying that steric hindrance from Watson-Crick base pairing in double-stranded DNA is not a factor. Consequently, similar yields of products in both single- and double-stranded DNA were found. Additionally, 8-hydroxyguanine, a major product of oxidative damage in DNA, induces mainly G → T and A → C mutations [67,68]. In Escherichia coli, it has also been reported that 2-hydroxy-dATP induces G → T mutations [69]. These results do not explain GT excess in the leading strand and therefore are not good candidates for a single mechanism of asymmetric mutation pressures.
Deaminations
Within this group, only cytosine deamination to uracil (or 5-methylcytosine deamination directly to thymine) yields a C → T mutation, and only adenine deamination to hypoxanthine yields an A → G mutation. Interestingly, there is experimental evidence in vitro that the rate of these deaminations is increased in the single-stranded state. Deamination of cytosine occurs over 100 times faster in single-stranded than in double-stranded DNA [70,71]. This is also most likely true for adenine, but its deamination rate is one-fortieth of the rate of cytosine [72], so that it is not possible to determine its rate in double-stranded DNA. The rate-determining step in cytosine deamination involves attack by water at the C-4 of the cytosine to give a tetrahedral intermediate, difficult to obtain in the rigid structure of double-stranded DNA because of Watson-Crick base pairing with guanine [73]. Single-stranded-DNA-binding proteins do not offer specific protection from this type of mutation during DNA replication [74] but may differ between species in their ability to protect from solvent attack. The single-stranded state during transcription induces C → T mutations [75], and this effect increases with transcription level [76] or when the duration of the single-stranded state is increased using a slower, mutant, RNA polymerase [77]. Also consistent is the report [78] that GT enrichment increases with the time spent in the single-stranded state during mitochondrial DNA replication. In a study of the mutation pattern estimated from pseudogene data in B. burgdorferi, it was found that C → T mutation was the most frequent in the leading strand [79]. The small depletion in G+C content observed here in the leading strand is also consistent with an excess of C → T substitutions in the leading strand because C → T substitutions decrease the G+C content. This effect is also expected to be a second-order one because of the centripetal action of symmetric directional mutation pressure on the G+C content of both strands. Therefore, we found the cytosine deamination theory compatible in most cases with the data, but a more definitive answer may come from biochemical studies on an extreme case such as B. burgdorferi.
G+C content variation between species is not controlled by a unique asymmetric mutation pressure
From inspection of our data, we can reject the simple model in which a unique asymmetric mutation pressure causes variation in G+C content between species. For instance, if only C → T mutations were the underlying phenomenon, we would expect low G+C in species with the highest B I . Figure 7 shows that this is clearly not the case. Moreover, if asymmetric directional mutation pressures were the major origin of variation in G+C content between species, we would expect a much more important difference in term of G+C content between the genes on the leading strand and those on the lagging strand.
Intergenic regions could be under greater selective constraints than expected
If we exclude as an explanation that many unannotated genes remain to be discovered, or that the boundaries of annotated genes should be greatly extended, so that some intergenic regions under analysis are not actually intergenic, then, there are two non-exclusive interpretations of the fact that B I is smaller in intergenic regions than in third codon positions. On the one hand, one may postulate that intergenic regions are under weaker selection pressure against inversions, so that deviations from PR2 are cancelled. On the other hand, one may assume that some positions are not free to deviate from PR2 because of selective pressure for some function. For instance, there are 100 copies per enterobacterial genome of palindromic sequences in intergenic spaces [80], which may be under selective pressure to preserve their palindromic character and therefore follow PR2 (as pure palindromic sequences are effectively base-paired). This interpretation is compatible with the fact that interspecific G+C content variation is higher at the third codon position than in intergenic spaces [10,11], as shown here in Figure 4. Moreover, the surprisingly low level of polymorphism in large intergenic regions among natural strains of E. coli [81] is in favor of selective constraints.
Transcription-coupled PR2-biases
Francino and Ochman [22,81] pointed out that transcription is likely to cause more mutations in the sense strand than in the anti-sense strand because of its transient single-stranded state during transcription. We found no evidence for this in intergenic spaces (> 100 bp), which could be interpreted either as an absence of transcription-induced mutation pressure or by a low proportion of transcribed fragments in these regions. Third codon positions are known to be transcribed, so that we expect an effect of transcription-induced mutation pressure on the residual deviation from PR2 (B II ). However, selection for translation optimization interferes, so that we cannot interpret unambiguously the almost universal orientation of B II (down left in PR2-plot, as in Figure 2c) as the result of transcription-induced mutation pressure. If we interpret results this way, we would have to postulate a different mechanism for replication-induced and transcription-induced mutation pressures, as B I and B II have different orientations. Moreover, in small intergenic spaces, which are potentially transcribed untranslated regions, no general pattern is evident for the residual deviation from PR2, so that no universal transcription-induced mutation pressure was found here as a strong contributor to the deviation from the PR2 state.
Asymmetric mutation pressure and chirochore structure
A chirochore is a contiguous segment of the chromosome homogeneous with respect to its deviation from PR2 [44]. A strong asymmetric mutation pressure induces a chirochore structure, as observed in B. burgdorferi [82], but the converse need not to be true: a weak asymmetric mutation pressure does not imply an absence of chirochore. For example in M. genitalium, replication-associated biases, B I /(B I + B II ), are the weakest among species under study (B I = 12%), but there is still a strong chirochore structure [83] because of a large excess (80%) of coding sequences in the leading strand. The deviation from PR2 in the first and second codon positions, because of selection at the amino-acid level (for example [84]), is then a major factor. It is not known whether chirochores could be adaptative, but recent results suggest a functional polarization of the E. coli chromosome [85,86] and may be relevant to this problem.
PR2-plot analysis
A prerequisite for using the simple graphical representations and quantifications used here is that the coding sequences and intergenic spaces can be divided into two groups: the leading and the lagging group. This is not necessarily always possible when, for instance, all coding sequences are in the leading group (this situation has never been encountered in bacteria but could be problematic in small genomes such as those of mitochondria). Another limitation is encountered when there are genes in both groups but we are unable to assign sequences to the correct group because of limited information about the location of origin and terminus of replication used during the course of evolution. Although an estimation of B I is not possible in this latter case, an oblong distribution of points in PR2-plots may suggest a non-zero value for B I . When more than one strain was sequenced for a species (that is, Chlamydophila pneumoniae, E. coli, H. pylori, Mycobacterium tuberculosis, Neisseria meningitidis, and S. aureus) B I and B II values in third codon position are extremely stable within a species (see table in the Additional data files), but the maximum number of three strains available here for one species does not allow us to estimate their within-species variability, and therefore their taxonomic utility.
Materials and methods
Source of data
Data on 51 complete bacterial chromosomes were retrieved from the EMGLib server [87,88] and used to extract annotated coding sequences and intergenic spaces (Figure 1). These 51 chromosomes were from 49 different strains (there are two chromosomes in Deinococcus radiodurans and Vibrio cholerae), from 41 different species (there were three strains of C. pneumoniae, E. coli; two strains of H. pylori, M. tuberculosis, N. meningitidis, S. aureus) as detailed in Table 1.
Leading and lagging group assignment
To classify sequences into the leading and the lagging groups (Figure 1), we must be able to locate them relative to the origin and terminus of replication. Out of 51 bacterial chromosomes, the locations of the origin and the terminus were known from experimental evidence or were estimated from chirochore boundaries [44] in 43 chromosomes with the program Oriloc [89,90]. Details of assignments are given in EMGLib database under a '/strand=' qualifier. To discriminate between the origin and the terminus of replication, we used the assumption that organisms tend to avoid head-on collisions between the DNA replication apparatus and the RNA polymerase transcription complex [54,55,56]. We also assumed that coding sequences were more abundant on the leading than on the lagging strand. As far as we know, there is no exception to this rule, and the report that the trend is exacerbated in highly expressed genes supports this assumption [48]. Moreover, the comparison of the maps of closely related bacterial chromosomes shows that observed chromosome rearrangements tend to preserve an excess of genes in the leading strand [91,92,93,94,95,96,97,98]. Our figures show sequences on the leading strand as filled circles, and those on the lagging strand as open circles.
Coding-sequence analysis
The nucleotide composition of coding sequences was always calculated from the sense strand (Figure 1) for each codon position. The sense strand (or anti-template strand) is the DNA strand opposite to the template strand for transcription. The average G+C content of the third codon position (P3) and that of the first and second codon positions (P12) were computed to draw neutrality plots relative to P3 as previously described [11]: stop codons and codons from odd-numbered sets ATG (Met), TGG (Trp) and ATA (Ile) were excluded from the calculations of P-values below to avoid an extra cause of potential deviation from PR2. The coding sequences after position 1,950 kb on D. radiodurans chromosome 1 were discarded because of a frameshift error in the annotations. Some of the apparent coding sequences identified in genome sequences could be artifacts that do not correspond to actually expressible genes [99]. To overcome this problem, coding sequences of less than 300 bp were removed from the analysis (less than 10% of sequences were discarded). As the probability of obtaining an open reading frame with more than 300 bp by chance is low, most (> 99%) sequences analyzed should correspond to actual coding sequences [100].
Intergenic-sequence analysis
GCIGR denotes the G+C content, and deviations from PR2 were characterized by G/(G+C) and A/(A+T) to draw PR2-plots in a way similar to coding-sequence analyses. Intergenic sequences were split into two non-overlapping groups (Figure 1). The first group contained small intergenic sequences among co-oriented genes that are potentially transcribed untranslated spacers. The strand used to compute nucleotide composition - chosen to allow direct comparisons with coding-sequence analyses - is the same as the anti-template strand for transcription of the two flanking genes (CDS, tRNA, rRNA). Therefore, when the two flanking genes are coding sequences, the strand is the same as the strand used for the analysis of coding sequences, that is the sense strand (Figure 1). Their minimum size was set to 50 bp, a trade-off between the likelihood of being transcribed and the avoidance of too much scattering in PR2-plots. Their maximum size was set to 100 bp to avoid data overlap with large intergenic regions. The second group contained large intergenic regions that are potentially untranscribed untranslated regions. Their minimum size was set to 100 bp, which is an amount of information equivalent to the third codon position of 100 codons, the minium size required in coding-sequence analyses. The analyzed strand cannot be defined unambiguously with respect to the flanking genes when they are convergently or divergently transcribed. The nucleotide composition was therefore always calculated from the published strand (Figure 1).
Statistical tests and their interpretation
The significance of the analyses was assessed using unpaired t-test implemented in Statview 5.0 (SAS Institute Inc., North Carolina, USA) to compare the mean between two groups - here the leading and the lagging group. The null hypothesis is the equality of the mean value between the two groups. The critical value p is the probability of taking the wrong decision of rejecting the null hypothesis when the null hypothesis is true. Two notes of caution should be introduced about the interpretation of results in the present context. First, we made runs of 43 comparisons so that the odds of getting at least one wrong significant result, 1 - (1 - p)43, is 0.89, 0.35, 0.04 for p = 0.05, p = 0.01, p = 0.001 respectively. We selected the highly significant critical level (p < 0.01) so that odds of getting one, two, or three wrong decisions were 0.28, 0.06, and 0.01, respectively, for each run. Therefore, a simple figure to keep in mind when looking at the results is that the odds of getting by chance three or more highly significant differences in a run were less than 1 in 100. The second note of caution is that we were working with large data sets, typically one thousand values within each group, so that the power of the test, that is its ability to reject the null hypothesis, was high. As a matter of consequence, even a small difference between the mean of the two groups was considered, correctly from the statistical test point of view, as significant. However, statistically significant does not mean automatically biologically significant because a second-order or a side effect could easily be evidenced this way. To help interpretation, the simple graphical representations used here, very close to raw data, were connected through hyperlinks to the tabulated test results. By this means, readers can quickly form their own opinions by viewing the original data.
Additional data files
Tables of the intragenomic P3 distributions of all species examined, and the PR2-biases between leading and lagging groups for coding sequences, and small and large intergenic regions, together with associated histograms are available as additional data files in pdf format, with the tables also in text format (Tables 1,2,3,4).
References
Lee KY, Wahl R, Barbu E: Contenu en bases puriques et pyrimidiques des acides désoxyribonucléiques des bactéries. Ann Inst Pasteur. 1956, 91: 212-224.
Belozersky AN, Spirin AS: A correlation between the compositions of deoxyribonucleic and ribonucleic acids. Nature. 1958, 182: 111-112.
Sueoka N, Marmur J, Doty P: Heterogeneity in deoxyriboneucleic acids. II. Dependence of the density of deoxyribonucleic acids on guanine-cytosine. Nature. 1959, 183: 1427-1431.
Rolfe R, Meselson M: The relative homogeneity of microbial DNA. Proc Natl Acad Sci USA. 1959, 45: 1039-1043.
Kerr ARW, Peden JF, Sharp PM: Systematic base composition variation around the genome of Mycoplasma genitalium, but not Mycoplasma pneumoniae. Mol Microbiol. 1997, 25: 1177-1179. 10.1046/j.1365-2958.1997.5461902.x.
McInerney JO: Prokaryotic genome evolution as assessed by multivariate analysis of codon usage patterns. Microb Comp Genomics. 1997, 2: 1-10.
Sueoka N: Two aspects of DNA base composition: G+C content and translation-coupled deviation from intra-strand rule of A = T and G = C. J Mol Evol. 1999, 49: 49-62.
Sueoka N: On the genetic basis of variation and heterogeneity of DNA base compositon. Proc Natl Acad Sci USA. 1962, 48: 582-592.
Watson JD, Crick FHC: A structure for deoxyribose nucleic acid. Nature. 1953, 171: 737-738.
Muto A, Osawa S: The guanine and cytosine content of genomic DNA and bacterial evolution. Proc Natl Acad Sci USA. 1987, 84: 166-169.
Sueoka N: Directional mutation pressure and neutral molecular evolution. Proc Natl Acad Sci USA. 1988, 85: 2653-2657.
Sueoka N: Correlation between base composition of deoxyribonucleic acid and amino acid composition of protein. Proc Natl Acad Sci USA. 1961, 47: 1141-1149.
Lobry JR: Influence of genomic G+C content on average amino-acid composition of proteins from 59 bacterial species. Gene. 1997, 205: 309-316. 10.1016/S0378-1119(97)00403-4.
Gu X, Hewett-Emmet D, Li W-H: Directional mutational pressure affects the amino acid composition and hydrophobicity of proteins in bacteria. Genetica. 1998, 102/103: 383-391. 10.1023/A:1017028102013.
Singer GAC, Hickey DA: Nucleotide bias causes a genomewide bias in the amino acid composition of proteins. Mol Biol Evol. 2000, 17: 1581-1588.
Knight RD, Freeland SJ, Landweber L: A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2001, 2: research0010.1-research0010.13. 10.1186/gb-2001-2-4-research0010.
Wu C-I, Maeda N: Inequality in mutation rates of the two strands of DNA. Nature. 1987, 327: 169-170. 10.1038/327169a0.
Wu C-I: DNA strand asymmetry. Nature. 1991, 352: 114-114. 10.1038/352114b0.
Furusawa M, Doi H: Promotion of evolution: disparity in the frequency of strand-specific misreading between the lagging and leading DNA strands enhances disproportionate accumulation of mutations. J Theor Biol. 1992, 157: 127-133.
Sueoka N: Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J Mol Evol. 1995, 40: 318-325. Erratum 42:323.
Lobry JR: Properties of a general model of DNA evolution under no-strand-bias conditions. J Mol Evol. 1995, 40: 326-330. Erratum 41:680.
Francino MP, Ochman H: Strand asymmetries in DNA evolution. Trends Genet. 1997, 13: 240-245. 10.1016/S0168-9525(97)01118-9.
Frank AC, Lobry JR: Asymmetric substitution patterns: a review of possible underlying mutational or selective mechanisms. Gene. 1999, 238: 65-77. 10.1016/S0378-1119(99)00297-8.
Valenzuela CY: Non random DNA evolution. Biol Res. 1997, 30: 117-123.
Lobry JR, Lobry C: Evolution of DNA base composition under no-strand-bias conditions when the substitution rates are not constant. Mol Biol Evol. 1999, 16: 719-723.
Zharkikh A: Estimation of evolutionary distances between nucleotide sequences. J Mol Evol. 1994, 39: 315-329.
Lafay B, Lloyd AT, McLean MJ, Devine KM, Sharp PM, Wolfe KH: Proteome composition and codon usage in spirochaete: species-specific and DNA strand-specific mutational biases. Nucleic Acids Res. 1999, 27: 1642-1649. 10.1093/nar/27.7.1642.
Rocha EPC, Danchin A, Viari A: Universal replication biases in bacteria. Mol Microbiol. 1999, 32: 11-16. 10.1046/j.1365-2958.1999.01334.x.
Mackiewicz P, Gierlik A, Kowalczuk M, Dudek MR, Cebrat S: How does replication-associated mutational pressure influence amino acid composition of proteins?. Genome Res. 1999, 9: 409-416.
Lobry JR, Gautier C: Hydrophobicity and aromaticity are the major trends of amino-acid usage in 999 Escherichia coli chromosome-encoded genes. Nucleic Acids Res. 1994, 22: 3174-3180.
Lobry JR: The black hole of symmetric molecular evolution. Mémoire d'habilitation à diriger des recherches 34-2000: Université Claude Bernard - Lyon I, France. 2000, [http://pbil.univ-lyon1.fr/members/lobry/hdr/HDR_ENG.pdf]
Perrière G, Lobry JR, Thioulouse J: Correspondence discriminant analysis: a multivariate method for comparing classes of protein and nucleic acid sequences. Comput Appl Biosci. 1996, 12: 519-524.
McInerney JO: Replicational and transcriptional selection on codon usage in Borrelia burgdorferi. Proc Natl Acad Sci USA. 1998, 95: 10698-10703. 10.1073/pnas.95.18.10698.
Romero H, Zavala A, Musto H: Codon usage in Chlamydia trachomatis is the result of strand-specific mutational biases and a complex pattern of selective forces. Nucleic Acids Res. 2000, 28: 2084-2090. 10.1093/nar/28.10.2084.
Tillier ERM, Collins RA: The contributions of replication orientation, gene direction, and signal sequences to base-composition asymmetries in bacterial genomes. J Mol Evol. 2000, 50: 249-257.
Francino MP, Ochman H: A comparative genomics approach to DNA asymmetry. Ann New York Acad Sci. 1999, 870: 428-431.
Francino MP, Ochman H: Strand symmetry around the beta-globin origin of replication in primates. Mol Biol Evol. 2000, 17: 416-422.
Rocha EPC, Danchin A: Ongoing evolution of strand composition in bacterial genomes. Mol Biol Evol. 2001, 18: 1789-1799.
Lobry JR: A simple vectorial representation of DNA sequences for the detection of replication origins in bacteria. Biochimie. 1996, 78: 323-326. 10.1016/0300-9084(96)84764-X.
Cebrat S, Dudek MR: The effect of DNA phase structure on DNA walks. Eur Phys J B. 1998, 3: 271-276. 10.1007/s100510050313.
Mackiewicz P, Gierlik A, Kowalczuk M, Dudek MR, Cebrat S: Asymmetry of nucleotide composition of prokaryotic chromosomes. J Appl Genet. 1999, 40: 1-14.
Mackiewicz P, Gierlik A, Kowalczuk M, Szczepanik D, Dudek MR, Cebrat S: Mechanisms generating long-range correlation in nucleotide composition of the Borrelia burgdorferi genome. Physica A. 1999, 273: 103-115. 10.1016/S0378-4371(99)00345-3.
Salzberg SL, Salzberg AJ, Kerlavage AR, Tomb J-F: Skewed oligomers and origins of replication. Gene. 1998, 217: 57-67. 10.1016/S0378-1119(98)00374-6.
Lobry JR: Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 1996, 13: 660-665.
Mrázek J, Karlin S: Strand compositional asymmetry in bacterial and large viral genomes. Proc Natl Acad Sci USA. 1998, 95: 3720-3725. 10.1073/pnas.95.7.3720.
Grigoriev A: Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 1998, 26: 2286-2290. 10.1093/nar/26.10.2286.
Grigoriev A: Strand-specific compositional asymmetries in double-stranded DNA viruses. Virus Res. 1999, 60: 1-19. 10.1016/S0168-1702(98)00139-7.
Mclean MJ, Wolfe KH, Devine KM: Base composition skews, replication orientation and gene orientation in 12 prokaryote genomes. J Mol Evol. 1998, 47: 691-696.
Ikemura T: Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J Mol Biol. 1981, 146: 1-21.
Sharp PM, Li W-H: An evolutionary perspective on synonymous codon usage in unicellular organisms. J Mol Evol. 1986, 24: 28-38.
Francino MP, Chao L, Riley MA, Ochman H: Asymmetries generated by transcription-coupled repair in enterobacterial genes. Science. 1996, 272: 107-109.
Gouy M, Gautier C: Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 1982, 10: 7055-7075.
Kanaya S, Yamada Y, Kudo Y, Ikemura T: Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene. 1999, 238: 143-155. 10.1016/S0378-1119(99)00225-5.
Brewer BJ: When polymerases collide: replication and the transciptional organization of the E. coli chromosome. Cell. 1988, 53: 679-686.
French S: Consequences of replication fork movement through transcription units in vivo. Science. 1992, 258: 1362-1365.
Liu B, Alberts BM: Head-on collision between a DNA replication apparatus and RNA polymerase transcription complex. Science. 1995, 267: 1131-1137.
Brenner DJ, Fanning GR, Skerman FJ, Falkow S: Polynucleotide sequence divergence among strains of Escherichia coli and closely related organisms. J Bacteriol. 1972, 109: 953-965.
Rocha EPC, Danchin A, Viari A: Bacterial DNA strand compositional asymmetry: Response. Trends Microbiol. 1999, 7: 308-308. 10.1016/S0966-842X(99)01561-9.
Andersson SGE, Kurland CG: An extreme codon preference strategy: codon reassignment. Mol Biol Evol. 1991, 8: 530-544.
Asakawa S, Kumazawa Y, Araki T, Himeno H, Miura K, Watanabe K: Strand-specific nucleotide composition bias in echninoderm and vertebrate mitochondrial genomes. J Mol Evol. 1991, 32: 511-520.
Tanaka M, Ozawa T: Strand asymmetry in human mitochondrial DNA mutations. Genomics. 1994, 22: 327-335. 10.1006/geno.1994.1391.
Morton BR: Strand asymmetry and codon usage bias in the chloroplast genome of Euglena gracilis. Proc Natl Acad Sci USA. 1999, 96: 5123-5128. 10.1073/pnas.96.9.5123.
Gierlik A, Kowalczuk M, Mackiewicz P, Dudek MR, Cebrat S: Is there replication-associated mutational pressure in the Saccharomyces cerevisiae genome?. J Theor Biol. 2000, 202: 305-314. 10.1006/jtbi.1999.1062.
Bentley SD, Chater KF, Cerdeño-Tárraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, et al: Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature. 2002, 417: 141-147. 10.1038/417141a.
Okazaki R, Okazaki T, Sakabe K, Sugimoto K, Sugino A: Mechanism of DNA chain growth. I. Possible discontinuity and unusual secondary structure of newly synthesized chains. Proc Natl Acad Sci USA. 1968, 59: 598-605.
Simandan T, Sun J, Dix TA: Oxidation of DNA bases, deoxyribonucleosides and homopolymers by peroxyl radicals. Biochem J. 1998, 335: 233-240.
Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA: 8-hydroxyguanine, an abundant form of oxidative DNA damage, causes G → T and A → C substitutions. J Biol Chem. 1992, 267: 166-172.
Mo J-Y, Maki H, Sekiguchi M: Hydrolytic elimination of a mutagenic nucleotide, 8-oxodGTP, by human 18-kilodalton protein: sanitization of nucleotide pool. Proc Natl Acad Sci USA. 1992, 89: 11021-11025.
Kamiya H, Maki H, Kasai H: Two DNA polymerases of Escherichia coli display distinct mis-insertion specificities for 2-hydroxy-dATP during DNA synthesis. Biochemistry. 2000, 39: 9508-9513. 10.1021/bi000683v.
Lindahl T, Nyberg B: Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry. 1974, 13: 3405-3410.
Frederico LA, Kunkel TA, Shaw BR: A sensitive genetic assay for the detection of cytosine deamination: determination of rate constants and the activation energy. Biochemistry. 1990, 29: 2532-2537.
Karran P, Lindahl T: Hypoxanthine in deoxyribonucleic acid: generation by heat-induced hydrolysis of adenine residues and release in free form by a deoxyribonucleic acid glycosylase from calf thymus. Biochemistry. 1980, 19: 6005-6011.
Frederico LA, Kunkel TA, Shaw BR: Cytosine deamination in mismatched base pairs. Biochemistry. 1993, 32: 6523-6530.
Raghunathan S, Kozlov AG, Lohman TM, Waksman G: Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nature Struct Biol. 2000, 8: 648-652. 10.1038/77943.
Beletskii A, Bhagwat AS: Transcription-induced mutations: increase in C to T mutations in the nontranscribed strand during transcription in Escherichia coli. Proc Natl Acad Sci USA. 1996, 93: 13919-13924. 10.1073/pnas.93.24.13919.
Beletskii A, Bhagwat AS: Correlation between transcription and C to T mutations in the non-transcribed DNA strand. Biol Chem. 1998, 379: 549-551.
Beletskii A, Grigoriev A, Joyce S, Bhagwat AS: Mutations induced by bacteriophage T7 RNA polymerase and their effects on the composition of the T7 genome. J Mol Biol. 2000, 300: 1057-1065. 10.1006/jmbi.2000.3944.
Reyes A, Gissi C, Pesole G, Saccone C: Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol. 1998, 15: 957-966.
Kowalczuk M, Mackiewicz P, Mackiewicz D, Nowicka A, Dudkiewicz M, Dudek RM, Cebrat S: High correlation between the turnover of nucleotides under mutational pressure and the DNA composition. BMC Evol Biol. 2001, 1: 13-10.1186/1471-2148-1-13.
Bachellier S, Clement JM, Hofnung M: Short palindromic repetitive DNA elements in enterobacteria: a survey. Res Microbiol. 1999, 150: 627-639. 10.1016/S0923-2508(99)00128-X.
Francino MP, Ochman H: Deamination as the basis of strand-asymmetric evolution in transcribed Escherichia coli sequences. Mol Biol Evol. 2001, 18: 1147-1150.
Picardeau M, Lobry JR, Hinnebusch BJ: Physical mapping of an origin of bidirectional replication at the center of the Borrelia burgdoferi linear chromosome. Mol Microbiol. 1999, 32: 437-445. 10.1046/j.1365-2958.1999.01368.x.
Lobry JR: Origin of replication of Mycoplasma genitalium. Science. 1996, 272: 745-746.
Kano-Sueoka T, Lobry JR, Sueoka N: Intra-strand biases in bacteriophage T4 genome. Gene. 1999, 238: 59-64. 10.1016/S0378-1119(99)00296-6.
Perals K, Cornet F, Merlet Y, Delon I, Louarn JM: Functional polarization of the Escherichia coli chromosome terminus: the dif site acts in chromosome dimer resolution only when located between long stretches of opposite polarity. Mol Microbiol. 2000, 36: 33-43. 10.1046/j.1365-2958.2000.01847.x.
Capitaux H, Cornet F, Corre J, Guijo M-I, Pérals K, Rebollo JE, Louarn J-M: Polarization of the Escherichia coli chromosome. A view from the terminus. Biochimie. 2001, 83: 161-170. 10.1016/S0300-9084(00)01202-5.
Perrière G, Bessieres P, Labedan B: EMGLib: the enhanced microbial genomes library. Nucleic Acids Res. 2000, 28: 68-71. 10.1093/nar/28.1.68.
The enhanced microbial genomes library. [http://pbil.univ-lyon1.fr/emglib/emglib.html]
Frank AC, Lobry JR: Oriloc: prediction of replication boundaries in unannotated bacterial chromosomes. Bioinformatics. 2000, 16: 560-561. 10.1093/bioinformatics/16.6.560.
Tillier ERM, Collins RA: Genome rearrangement by replication-directed translocation. Nature Genet. 2000, 26: 195-197. 10.1038/79918.
Takami H, Nakasone K, Takaki Y, Maeno G, Sasaki R, Masui N, Fuji F, Hirama C, Nakamura Y, Ogasawara N, et al: Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucl Acids Res. 2000, 28: 4317-4331. 10.1093/nar/28.21.4317.
Read TD, Brunham RC, Shen C, Gill SR, Heidelberg JF, White O, Hickey EK, Peterson J, Utterback T, Berry K, et al: Genome sequence of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucl Acids Res. 2000, 28: 1397-1406. 10.1093/nar/28.6.1397.
Eisen JA, Heidelberg JF, White O, Salzberg SL: Evidence for symmetric chromosomal inversions around the replication origin in bacteria. Genome Biol. 2000, 1: research0011.1-0011.9. 10.1186/gb-2000-1-6-research0011.
Mackiewicz P, Mackiewicz D, Kowalczuk M, Cebrat S: Flip-flop around the origin and terminus of replication in prokaryotic genomes. Genome Biol. 2001, 2: interactions1004.1-1004.4. 10.1186/gb-2001-2-12-interactions1004.
Mackiewicz P, Mackiewicz D, Gierlik A, Kowalczuk M, Nowicka A, Dudkiewicz M, Dudek MR, Cebrat S: The differential killing of genes by inversions in prokaryotic genomes. J Mol Evol. 2001, 53: 615-621. 10.1007/s002390010248.
Moran NA, Mira A: The process of genome shrinkage in the obligate symbiont Buchnera aphidicola. Genome Biol. 2001, 2: research0054.1-0054.12. 10.1186/gb-2001-2-12-research0054.
Suyama M, Bork P: Evolution of prokaryotic gene order: genome rearrangements in closely related species. Trends Genet. 2001, 17: 10-13. 10.1016/S0168-9525(00)02159-4.
Fickett JW: ORFs and genes: how strong a connection?. J Comp Biol. 1995, 2: 117-123.
Oliver JL, Marin A: A relationship between GC content and coding-sequence length. J Mol Evol. 1996, 43: 216-223.
Acknowledgements
We thank the anonymous referees for their helpful comments, and we thank Rob Knight for his kind help in improving this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
13059_2001_395_MOESM1_ESM.pdf
Additional data file 1: Tables of the intragenomic P3 distributions of all species examined, and the PR2-biases between leading and lagging groups for coding sequences, and small and large intergenic regions, together with associated histograms (PDF 10 MB)
Rights and permissions
About this article
Cite this article
Lobry, J.R., Sueoka, N. Asymmetric directional mutation pressures in bacteria. Genome Biol 3, research0058.1 (2002). https://doi.org/10.1186/gb-2002-3-10-research0058
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/gb-2002-3-10-research0058