
Abstract
The modification of chromatin structure is important for a number of nuclear functions, exemplified by the regulation of transcription. This review discusses recent studies of covalent histone modifications and the enzymatic machines that generate them.
Similar content being viewed by others

In eukaryotes, genomic DNA is packaged with histone proteins into chromatin, compacting DNA some 10,000-fold. Such condensation of DNA provides a considerable obstacle to the nuclear machinery that drives processes such as replication, transcription or DNA repair. Importantly, the structure of chromatin dynamically changes, permitting localized decondensation and remodeling that facilitates the progress of nuclear machinery. An emerging theme in the field of chromatin research has been the significant role that posttranslational modifications of histones play in regulating nuclear function. Over the past few years, considerable progress has been made into the identification of the enzymatic machines that modify these proteins, and this review is devoted to our current understanding of the array of core histone modifications and the factors that regulate them.
The basic repeating unit of chromatin is the nucleosome, typically composed of an octamer of the four core histones H2A, H2B, H3 and H4 and 146 basepairs of DNA wrapped around the histones [1]. Each core histone is composed of a structured domain and an unstructured amino-terminal 'tail' of 25-40 residues. This unstructured tail extends through the DNA gyres and into the space surrounding the nucleosomes. Histone tails provide sites for a variety of posttranslational modifications, including acetylation, phosphorylation and methylation. It is becoming increasingly apparent that such modifications of histone tails determine the interactions of histones with other proteins, which may in turn also regulate chromatin structure [2]. Identifying the multitude of histone modifications, the enzymes that generate them and the nuclear response to any given pattern of alterations poses a fascinating challenge.
Histone acetylation
The acetylation and deacetylation of the ε-amino groups of conserved lysine residues present in histone tails has long been linked to transcriptional activity [3] and has been the most intensively studied histone modification. Acetylated histones are usually associated with transcriptionally active chromatin and deacetylated histones with inactive chromatin. In addition to the relationship between histone acetylation and the transcriptional capacity of chromatin, acetylation is also involved in processes such as replication and nucleosome assembly, higher-order chromatin packing and interactions of non-histone proteins with nucleosomes [4]. In particular, the highly conserved histone H3 lysines at amino-terminal amino-acid positions 9, 14, 18 and 23, and H4 lysines 5, 8,12 and 16, are frequently targeted for modification [5]. The neutralization of the basic charge of the histone tails by acetylation is thought to reduce their affinity for DNA and to alter histone:histone interactions between adjacent nucleosomes as well as the interactions of histones with other regulatory proteins [4,5]. These changes contribute to a chromatin environment believed to be permissive for transcription. Acetylation of lysine 12 in histone H4, however, is linked to transcriptional silencing in yeast and Drosophila [6,7], and some acetyltransferases, such as Sas3 and Hat1, have been linked to silencing in yeast [8,9].
Strong molecular evidence for a direct link between acetylation and transcription was provided when the conserved transcriptional regulator Gcn5 was found to have histone acetyltransferase (HAT) activity [10]. Yeast and human Gcn5 and the orthologous human protein, PCAF, were subsequently found to be associated with other transcriptional adaptors or coactivators in multisubunit complexes that regulate Gcn5 specificity and recruitment to target promoters [2,5]. Gcn5 and PCAF commonly modify lysine 14 of histone H3. This modification was initially suggested to represent a transcription-linked acetylation mark [11]. As part of native complexes, however, these enzymes can modify an expanded repertoire of lysines [12,13]. One extensively studied native Gcn5-containing complex is the yeast 'SAGA' complex, a highly conserved complex containing a large number of transcriptional coactivators from the Spt, Ada, TAF and Tra1 families of proteins, which preferentially modifies nucleosomal histones H3 and H2B [5]. A localized focus of acetylation [2] is created when this activity is specifically recruited to promoters by acidic activators [14] or following the action of the ATP-dependent SWI/SNF chromatin-remodeling complex [15,16]. Chromatin-remodeling complexes are thought to 'loosen' nucleosomes to allow transcription factors to gain access to their targets within chromatin.
HATs
Since the identification of Gcn5 as an archetypal nuclear HAT, numerous other coactivator proteins have been found to possess HAT activity and have frequently been identified as components of high-molecular-weight complexes composed of proteins with homology (or identity) to transcriptional regulators [4]. There are now several reported families of acetyltransferases, comprising over twenty enzymes, which generate specific patterns of free and/or nucleosome-associated histone acetylation. The first family of HATs is the GNAT superfamily (Gcn5-related N-acetytransferases), which includes proteins involved with, or linked to, transcriptional initiation (Gcn5 and PCAF), elongation (Elp3), histone deposition and telomeric silencing (Hat1). The p300/CBP HAT family is comprised of the highly related p300 and CBP proteins, which share sequence homology with GNATs [5]. Members of the p300/CBP family have been extensively described as coactivators for multiple transcription factors. The HAT activity of p300 and CBP is required for their role in transactivation, and these enzymes have been found to associate with other acetyltransferases, indicating that multiple HAT enzymes may be recruited to act cooperatively during gene activation [4].
The MYST family of HATs is named after the founding members MOZ, Ybf2/Sas3, Sas2 and Tip60. Notable members of this family include the human oncogene MOZ. In one type of chromosomal translocation associated with acute myeloid leukemias, MOZ is fused in-frame to the CBP acetyltransferase; leukemogenesis follows presumably as a result of aberrant chromatin acetylation [17]. The yeast homolog of MOZ is Sas3, the catalytic subunit of the nucleosomal H3-specific HAT complex, NuA3 [18]. This complex is predicted to function in transcriptional elongation and replication [18] and in silencing of the yeast HM mating-type loci [8]. Unlike SAGA, the NuA3 complex is apparently not recruited to yeast promoters by acidic activators, however [14]. Interestingly, nucleosomes retard transcription elongation by RNA polymerase, and nucleosomal acetylation by the Elp3-dependent elongator [19] or Sas3-dependent NuA3 complexes may overcome this barrier.
The essential yeast Esa1 acetyltransferase is a MYST family member that predominantly modifies histones H4 and H2A within the native NuA4 HAT complex [20]. Esa1 and Gcn5 both contribute to the regulation of the PHO5 gene [21,22], which encodes an acid phosphatase, in a manner that generates widespread acetylation of a 4.25 kb region around the PHO5 gene [23], which in turn allows opening of the chromatin at the promoter. Although this acetylation is antagonized by a pattern of widespread deacetylation, the targeting of these HATs to the promoter allows a transient and localized increase of acetyl groups sufficient to stimulate transcription. Esa1 is homologous to the Drosophila MOF protein, which catalyzes acetylation of H4 lysine 16 on the male X chromosome and a subsequent two-fold increase in transcription, a phenomenon known as dosage compensation (reviewed in [24]). The NuA4 complex has a number of components also found in the human Tip60 complex, which is thought to function in DNA-damage responses and the regulation of apoptosis [25] suggesting a conserved mechanism for signaling the existence of DNA damage through histone acetylation. Finally, the human MYST family member HBO1 is the first HAT shown to associate with the protein complex that binds DNA at the origin of replication [26], underscoring a role for acetylation in multiple aspects of chromatin metabolism.
Other acetyltransferases are grouped as basal transcription factors, such as the TAFII250 component of the TFIID complex, or as nuclear hormone-receptor cofactors, such as ACTR and SRC1. These HATs and their substrate specificities have been reviewed elsewhere [4,5]. It is notable that many HATs have also been reported to modify other transcription factors, such as the p53 tumor suppressor and the basal transcription factors TFIIE and TFIIF.
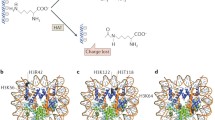
Modification of histone tails by acetylation is known to increase the access of transcription factors to DNA through structural changes in nucleosomes or nucleosomal arrays. Acetylated histones are also specifically recognized by other proteins. The bromodomain, found in transcription factors and HATs such as TAFII250, PCAF and GCN5 is a protein domain that allows for the preferential recognition of histone tails when they are acetylated at specific lysine residues [27,28,29] (Figure 1). Similarly, other histone modifications are binding sites for other specialized protein domains (see below), consistent with the hypothesis that patterns of covalent histone modifications form a platform recognized by other proteins and used to transduce downstream events [30]. Three recent reports [31,32,33] have suggested that histone acetylation may precede the recruitment of ATP-dependent chromatin-remodeling activities during transcriptional activation. Gcn5 can participate in stabilizing SWI/SNF binding to a promoter, and this interaction seems to be mediated through the Gcn5 bromodomain [31]. Furthermore, Gcn5 recruitment and histone acetylation precedes recruitment of the SWI/SNF complex during activation of the interferon-β promoter [32], and transactivation by RAR/RXR nuclear receptors involves histone acetylation prior to the action of human SWI/SNF [33].

A model for the generation of transcriptionally active and inactive chromatin domains by post-translational histone modification. (a) Histone acetyltransferases (HATs) generate patterns of acetylation (Ac) recognized by other transcriptional regulators, such as the bromodomain-containing (Bromo) factors TAFII250, PCAF and GCN5, leading to chromatin 'opening' and gene activation. (b) Conversely, the conserved histone methyltransferase (HMT) SUV39H1 methylates (Me) histone H3, which is then bound by the heterochromatin-associated chromodomain (Chromo) protein HP1/Swi6, establishing a silent chromatin domain.
HDACs
A large number of histone deacetylases (HDACs) have now been identified, many of which act as corepressors of transcription. The yeast HDACs Rpd3 and Hda1 are known to be recruited by repressor proteins to promoters, causing a localized deacteylation of chromatin [2]. Specialized regions of chromatin have been described, such as telomeres, centromeres and silent yeast mating-type loci, which are transcriptionally inactive and form hypoacetylated heterochromatin-like domains. Inhibition of HDAC activity and inactivation of sites for hypoacetylation in histone H4 are known to disrupt the formation of such highly condensed (heterochromatic) regions [34]. In yeast, heterochromatin formation is mediated by the Sir2, Sir3 and Sir4 silencing proteins. Recently, Sir2 has been found to have NAD-dependent HDAC activity [35,36,37] and, in one report, NAD-dependent histone-ribosylation activity [38]. The deacetylation of lysine 16 of H4 seems important for the interaction of Sir3 and Sir4 proteins and subsequent spreading of heterochromatin.
Histone phosphorylation
In contrast to the relative wealth of information about the large number of acetyltransferases and deacetylases, relatively little is known about the enzymes that generate other histone modifications. Important progress has been made, however, towards understanding the role of histone phosphorylation in processes such as transcription, DNA repair, apoptosis and chromosome condensation [39].
Phosphorylation of serine 10 in histone H3 has been shown to correlate with gene activation in mammalian cells [40] and with the induction of transcription during heat-shock response in Drosophila [41]. Quiescent fibroblasts treated with epidermal growth factor undergo rapid serine 10 phosphorylation, coincident with the induction of early response genes such as c-fos. This phosphorylation is catalyzed by the Rsk-2 kinase, and notably cells derived from Rsk-2-deficient Coffin-Lowry Syndrome patients do not undergo serine 10 phosphorylation or c-fos induction in response to the epidermal growth factor (EGF) [42].
The mechanism by which phosphorylation contributes to transcriptional activation is not well understood. The addition of negatively charged phosphate groups to histone tails neutralizes their basic charge and is thought to reduce their affinity for DNA. Furthermore, it has been found that several acetyltransferases have increased HAT activity on serine 10-phosphorylated substrates, and that mutation of serine 10 decreases activation of Gcn5-regulated genes [43,44]. Thus, phosphorylation may contribute to transcriptional activation through the stimulation of HAT activity on the same histone tail. Indeed, phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes has been demonstrated upon gene activation [45].
Phosphorylation of H2A has also long been correlated with mitotic chromosome condensation, and again serine 10 appears to play a key role [39]. For example, mutation of serine 10 in Tetrahymena histones causes abnormal chromosomal condensation and defective chromosome separation during anaphase. Recently, the Ipl1/aurora kinase in yeast and nematodes, and the NIMA kinase in Aspergillus nidulans, have been reported to regulate H3 serine 10 phosphorylation [46,47]. The normal expression of both enzymes correlates with mitosis and they can directly modify H3 serine 10. Disregulation of Ipl1 or NIMA leads to subsequent disruption of the normal process of chromosome condensation or segregation. It has been suggested that the protein phosphatase Glc7/PP1 dephosphorylates H3 after mitosis.
Phosphorylation of histone H3 is also known to occur after activation of DNA-damage signaling pathways. For example, a conserved motif (ASQE, in the single-letter amino-acid code) found in the carboxyl terminus of yeast H2A and the mammalian H2A variant H2A.X is rapidly phosphorylated upon exposure to DNA-damaging agents [48,49]. Serine 139 has been identified as the site for this modification, and its phosphorylation in response to damage is dependent on the phosphatidylinositol-3-OH kinase Mec1 in yeast. Mec1-dependent serine 139 phosphorylation is apparently required for efficient non-homologous end-joining repair of DNA. This suggests that phosphorylation mediates an alteration of chromatin structure, which in turn facilitates repair.
Histone methylation
Methylation of histones was first described in 1964 [50]. Direct evidence linking methylation and transcription was only found some 35 years later, when the histone H3 arginine-specific histone methyltransferase (HMT) CARM1 was shown to interact and cooperate with the steroid-hormone-receptor coactivator GRIP-1 in transcriptional activation [51]. Initial studies of histone modifications have indicated that histones H3 (lysines 4, 9 and 27) and H4 (lysine 20) are frequently preferentially methylated [52]. Modified lysines have the ability to be mono-, di- or tri- methylated, adding a further potential complexity to the posttranslational status of H3 and H4 tails.
Recently, the heterochromatin-enriched human SUV39H1, murine Suv39h1 and fission yeast Clr4 proteins were found to methylate lysine 9 of histone H3 selectively [53,54]. Overexpression of SUV39H1 induces ectopic heterochromatin [55], the formation of heterochromatin in a normally euchromatic location, suggesting that methylation of H3 lysine 9 may generate a chromatin architecture preferred for binding by other heterochromatin-specific proteins. SUV39H1 family members are homologs of the heterochromatin-associated Drosophila SU(VAR)3-9 and Schizosaccharomyces pombe Clr4 proteins, both of which have been identified genetically as suppressors of position-effect variegation, a phenomenon in which the relocation (by rearrangement or transposition) of a locus to heterochromatin results in abnormal silencing of the locus in a proportion of the cells that would normally express it. All three proteins are members of the SET-domain family of proteins. The heterochromatin protein HP1 colocalizes with SUV39H1 to heterochromatin, where it mediates gene silencing. Three new studies reveal that HP1 and the S. pombe homolog Swi6 specifically bind with high affinity to histone H3 that has been methylated at lysine 9 by the SUV39H family of HMTs [54,56,57] (Figure 1). The specificity of this recognition is underscored by the fact that methylated H3 lysine 4 is apparently not bound by HP1. The evolutionarily conserved chromodomain regions of HP1 mediates the interaction with methyl lysine 9. In S. pombe, methylase activity of Clr4 is required for the recruitment of Swi6 to silenced heterochromatin and for transcriptional silencing [54]. Notably, methylation of lysine 9 interferes with phosphorylation of the adjacent serine 10 by the Ipl1/aurora kinase, but is also inhibited by prior acetylation of lysine 9 [53]. Thus, unmodified tails such as those available following replication-coupled histone deposition may favor SUV39H1-mediated methylation events and subsequent spreading of heterochromatin.
A potentially vast number of other histone modifications within the unstructured tails and structured carboxyl termini of histones may await discovery or further investigation. These include ADP-ribosylation and ubiquination events. For example, the carboxyl terminus of histone H2B is ubiquitinated in yeast in a Rad6-dependent fashion. Loss of this ubiquination site leads to defects in mitosis and meiosis [58]. The transcription factor TAFII250, mentioned above as a histone acetyltransferase, also shows histone ubiquitination activity. Drosophila TAFII250 modifies the linker histone H1 by monoubiquitination, and this modification is required for full activation of the Dorsal transcription factor [59]. In summary, recent discoveries concerning histone modifications and the enzymes that mediate them have revealed significant interplay between different covalent marks and the proteins that recognize them. Mapping the full repertoire of histone modifications, the players involved and the structural and functional responses to them will inevitably continue to be a major focus of chromatin research in years to come.
References
Luger K, Mader AW, Richmond R, Sargent DF, Richmond TJ: Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997, 389: 251-260. 10.1038/38444.
Wu J, Grunstein M: 25 years after the nucleosome model: chromatin modifications. Trends Biochem Sci. 2000, 25: 619-623. 10.1016/S0968-0004(00)01718-7.
Allfrey VG, Faulkner R, Mirsky AE: Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. 1964, 51: 786-794.
Grant PA, Berger SL: Histone acetyltransferase complexes. Semin Cell Dev Biol. 1999, 10: 169-177. 10.1006/scdb.1999.0298.
Roth SY, Denu JM, Allis CD: Histone acetyltransferases. Annu Rev Biochem. 2001, 70: 81-120. 10.1146/annurev.biochem.70.1.81.
Turner BM, Birley AJ, Lavender J: Histone H4 isoforms acetylated at specific lysine residues define individual chromosomes and chromatin domains in Drosophila polytene nuclei. Cell. 1992, 69: 375-384. 10.1016/0092-8674(92)90417-B.
Braunstein M, Sobel RE, Allis CD, Turner BM, Broach JR: Efficient transcriptional silencing in Saccharomyces cerevisiae requires a heterochromatin histone acetylation pattern. Mol Cell Biol. 1996, 16: 4349-4356.
Reifsnyder C, Lowell J, Clarke A, Pillus L: Yeast SAS silencing genes and human genes associated with AML and HIV-1 Tat interactions are homologous with acetyltransferases. Nat Genet. 1996, 14: 42-49. 10.1038/ng0996-42.
Kelly TJ, Qin S, Gottschling DE, Parthun MR: Type B histone acetyltransferase Hat1p participates in telomeric silencing. Mol Cell Biol. 2000, 20: 7051-7058. 10.1128/MCB.20.19.7051-7058.2000.
Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, Allis CD: Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996, 84: 843-851. 10.1016/S0092-8674(00)81063-6.
Kuo M-H, Brownell JE, Sobel RE, Ranalli TA, Cook RG, Edmondson DG, Roth SY, Allis CD: Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature. 1996, 383: 269-271. 10.1038/383269a0.
Grant PA, Eberharter A, John S, Cook RG, Turner BM, Workman JL: Expanded lysine acetylation specificity of Gcn5 in native complexes. J Biol Chem. 1999, 274: 5895-5900. 10.1074/jbc.274.9.5895.
Zhang W, Bone JR, Edmondson DG, Turner BM, Roth SY: Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. EMBO J. 1998, 17: 3155-3167. 10.1093/emboj/17.11.3155.
Utley RT, Ikeda K, Grant PA, Cote J, Steger DJ, Eberharter A, John S, Workman JL: Transcriptional activators target histone acetyltransferase complexes to nucleosomes. Nature. 1998, 394: 498-502. 10.1038/28886.
Krebs JE, Fry CJ, Samuels ML, Peterson CL: Global role for chromatin remodeling enzymes in mitotic gene expression. Cell. 2000, 102: 587-598. 10.1016/S0092-8674(00)00081-7.
Cosma MP, Tanaka T, Nasmyth K: Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell. 1999, 97: 299-311. 10.1016/S0092-8674(00)80740-0.
Borrow J, Stanton VP, Andresen JM, Becher R, Behm FG, Chaganti RS, Civin CI, Disteche C, Dube I, Frischauf AM, et al: The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet. 1996, 14: 33-41. 10.1038/ng0996-33.
John S, Howe L, Tafrov ST, Grant PA, Sternglanz R, Workman JL: The something about silencing protein, Sas3, is the catalytic subunit of NuA3, a yTAF(II)30-containing HAT complex that interacts with the Spt16 subunit of the yeast CP (Cdc68/Pob3)-FACT complex. Genes Dev. 2000, 14: 1196-1208.
Wittschieben BO, Otero G, de Bizemont T, Fellows J, Erdjument-Bromage H, Ohba R, Li Y, Allis CD, Tempst P, Svejstrup JQ: A novel histone acetyltransferase is an integral subunit of elongating RNA polymerase II holoenzyme. Mol Cell. 1999, 4: 123-128. 10.1016/S1097-2765(00)80194-X.
Allard S, Utley RT, Savard J, Clarke A, Grant P, Brandl CJ, Pillus L, Workman JL, Cote J: NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J. 1999, 18: 5108-5119. 10.1093/emboj/18.18.5108.
Gregory PD, Schmid A, Zavari M, Lui L, Berger SL, Horz W: Absence of Gcn5 HAT activity defines a novel state in the opening of chromatin at the PHO5 promoter in yeast. Mol Cell. 1998, 1: 495-505. 10.1016/S1097-2765(00)80050-7.
Eisen A, Utley RT, Nourani A, Allard S, Schmidt P, Lane WS, Lucchesi JC, Cote J: The yeast NuA4 and Drosophila MSL complexes contain homologous subunits important for transcriptional regulation. J Biol Chem. 2001, 276: 3484-3491. 10.1074/jbc.M008159200.
Vogelauer M, Wu J, Suka N, Grunstein M: Global histone acetylation and deacetylation in yeast. Nature. 2000, 408: 495-498. 10.1038/35044127.
Pannuti A, Lucchesi JC: Recycling to remodel: evolution of dosage-compensation complexes. Curr Opin Genet Dev. 2000, 10: 644-650. 10.1016/S0959-437X(00)00136-2.
Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y: Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000, 102: 463-473. 10.1016/S0092-8674(00)00051-9.
Iizuka M, Stillman B: Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. J Biol Chem. 1999, 274: 23027-23034. 10.1074/jbc.274.33.23027.
Owen DJ, Ornaghi P, Yang JC, Lowe N, Evans PR, Ballario P, Neuhaus D, Filetici P, Travers AA: The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000, 19: 6141-6149. 10.1093/emboj/19.22.6141.
Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM: Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999, 399: 491-496. 10.1038/20974.
Jacobson RH, Ladurner AG, King DS, Tjian R: Structure and function of a human TAFII250 double bromodomain module. Science. 2000, 288: 1422-1425. 10.1126/science.288.5470.1422.
Strahl BD, Allis CD: The language of covalent histone modifications. Nature. 2000, 403: 41-45. 10.1038/47412.
Syntichaki P, Topalidou I, Thireos G: The Gcn5 bromodomain co-ordinates nucleosome remodelling. Nature. 2000, 404: 414-417. 10.1038/35006136.
Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D: Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell. 2000, 103: 667-678. 10.1016/S0092-8674(00)00169-0.
Dilworth FJ, Fromental-Ramain C, Yamamoto K, Chambon P: ATP-driven chromatin remodeling activity and histone acetyltransferases act sequentially during transactivation by RAR/RXR in vitro. Mol Cell. 2000, 6: 1049-1058. 10.1016/S1097-2765(00)00103-9.
Ekwall K, Olsson T, Turner BM, Cranston G, Allshire RC: Transient inhibition of histone deacetylation alters the structural and functional imprint at fission yeast centromeres. Cell. 1997, 91: 1021-1032. 10.1016/S0092-8674(00)80492-4.
Imai S, Armstrong CM, Kaeberlein M, Guarente L: Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000, 403: 795-800. 10.1038/35001622.
Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R: The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci USA. 2000, 97: 5807-5811. 10.1073/pnas.110148297.
Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, et al: A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci USA. 2000, 97: 6658-6663. 10.1073/pnas.97.12.6658.
Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D: An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell. 1999, 99: 735-745. 10.1016/S0092-8674(00)81671-2.
Cheung P, Allis CD, Sassone-Corsi P: Signaling to chromatin through histone modifications. Cell. 2000, 103: 263-271. 10.1016/S0092-8674(00)00118-5.
Thomson S, Mahadevan LC, Clayton AL: MAP kinase-mediated signalling to nucleosomes and immediate-early gene induction. Semin Cell Dev Biol. 1999, 10: 205-214. 10.1006/scdb.1999.0302.
Nowak SJ, Corces VG: Phosphorylation of histone H3 correlates with transcriptionally active loci. Genes Dev. 2000, 14: 3003-3013. 10.1101/gad.848800.
Sassone-Corsi P, Mizzen CA, Cheung P, Crosio C, Monaco L, Jacquot S, Hanauer A, Allis CD: Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science. 1999, 285: 886-891. 10.1126/science.285.5429.886.
Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM, Allis CD: Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol Cell. 2000, 5: 905-915. 10.1016/S1097-2765(00)80256-7.
Lo WS, Trievel RC, Rojas JR, Duggan L, Hsu JY, Allis CD, Marmorstein R, Berger SL: Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol Cell. 2000, 5: 917-926. 10.1016/S1097-2765(00)80257-9.
Clayton AL, Rose S, Barratt MJ, Mahadevan LC: Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J. 2000, 19: 3714-3726. 10.1093/emboj/19.14.3714.
De Souza CP, Osmani AH, Wu LP, Spotts JL, Osmani SA: Mitotic histone H3 phosphorylation by the NIMA kinase in Aspergillus nidulans. Cell. 2000, 102: 293-302. 10.1016/S0092-8674(00)00035-0.
Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, Grushcow JM, Brame CJ, Caldwell JA, Hunt DF, et al: Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000, 102: 279-291. 10.1016/S0092-8674(00)00034-9.
Downs JA, Lowndes NF, Jackson SP: A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 2000, 408: 1001-1004. 10.1038/35050000.
Rogakou EP, Boon C, Redon C, Bonner WM: Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999, 146: 905-916. 10.1083/jcb.146.5.905.
Murray K: The occurence of ε-N-methyl lysine in histones. Biochemistry. 1964, 3: 10-15. 10.1021/bi00889a003.
Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR: Regulation of transcription by a protein methyltransferase. Science. 1999, 284: 2174-2177. 10.1126/science.284.5423.2174.
Strahl BD, Ohba R, Cook RG, Allis CD: Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc Natl Acad Sci USA. 1999, 96: 14967-14972. 10.1073/pnas.96.26.14967.
Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, et al: Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000, 406: 593-599. 10.1038/35020506.
Nakayama JI, Rice JC, Strahl BD, Allis CD, Grewal SIS: Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001, epub ahead of print:
Melcher M, Schmid M, Aagaard L, Selenko P, Laible G, Jenuwein T: Structure-function analysis of SUV39H1 reveals a dominant role in heterochromatin organization, chromosome segregation, and mitotic progression. Mol Cell Biol. 2000, 20: 3728-3741. 10.1128/MCB.20.10.3728-3741.2000.
Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T: Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001, 410: 116-120. 10.1038/35065132.
Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T: Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001, 410: 120-124. 10.1038/35065138.
Robzyk K, Recht J, Osley MA: Rad6-dependent ubiquitination of histone H2B in yeast. Science. 2000, 287: 501-504. 10.1126/science.287.5452.501.
Pham AD, Sauer F: Ubiquitin-activating/conjugating activity of TAFII250, a mediator of activation of gene expression in Drosophila. Science. 2000, 289: 2357-2360. 10.1126/science.289.5488.2357.
Acknowledgements
I am grateful to David Allis, Thomas Jenuwein and Tony Kouzarides for the communication of results prior to publication and to David Allis for critical reading of this review. P.G. is the recipient of a Burroughs Wellcome Fund Career Award in the Biomedical Sciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Grant, P.A. A tale of histone modifications. Genome Biol 2, reviews0003.1 (2001). https://doi.org/10.1186/gb-2001-2-4-reviews0003
Published:
DOI: https://doi.org/10.1186/gb-2001-2-4-reviews0003