
Abstract
Metal ions are essential nutrients, yet they can also be toxic if they over-accumulate. Homeostatic mechanisms and detoxification systems therefore precisely control their intracellular levels and distribution. The tools of functional genomics are rapidly accelerating understanding in this field, particularly in the yeast Saccharomyces cerevisiae.
Similar content being viewed by others


Metals such as iron, copper, and zinc are essential nutrients for all organisms and play important roles in many biochemical processes. For example, iron and copper readily donate and accept electrons and are important cofactors in electron transport and in many redox-active metalloenzymes. Zinc, a strong Lewis acid (an electron-pair acceptor), is a catalytic component of over 300 enzymes and plays critical structural roles in many proteins, for example in zinc fingers. On the other hand, these same metal ions, as well as non-nutrient metals such as cadmium and mercury, can be extremely toxic to cells if they overaccumulate. Organisms have therefore evolved precise homeostatic regulatory systems to control the uptake, distribution, storage, and detoxification of metal ions. While researchers using the traditional approaches of genetics and biochemistry have made tremendous strides in understanding these systems, the application of functional genomics is greatly accelerating the pace of this research. Some of the best examples of the impact of functional genomics come from studies of the yeast Saccharomyces cerevisiae; this organism will thus be the focus of this article.
Functional genomics has its foundation in the large-scale projects for collecting expressed sequence tags (ESTs) and for genome sequencing, begun in the 1980s. Expression profiling is the direct offspring of these sequencing projects. With genome or EST sequences in hand, microarrays or macroarrays can be constructed to analyze gene-expression profiles at the genome-wide level over a range of conditions. These techniques have been especially useful in identifying genes that are directly regulated in response to changes in metal-ion status.
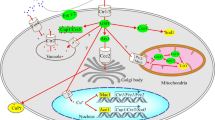
For example, our group has examined the genes regulated by the Zap1p transcription factor in S. cerevisiae using DNA microarrays and promoter motif analysis [1] (Figure 1). Zap1p activates the transcription of its target genes in zinc-limited but not in zinc-replete yeast cells. This study identified 46 genes that are likely to be direct targets of Zap1p, including the genes encoding Zrt3p, which is responsible for mobilizing stored zinc out of the vacuole [2], and more surprisingly, Zrc1p, a zinc transporter responsible for sequestering excess zinc in the vacuole. Zrc1p could protect cells from the zinc overload that occurs when a zinc-limited cell is resupplied with even small quantities of zinc (C.W. MacDiarmid and D.E., unpublished observation). It is notable that both zrt3 and zrc1 mutants grow normally under zinc-replete or zinc-limiting conditions and, thus, would have been very difficult to identify using classical genetic approaches.

Identification of the Zap1p regulon. Zap1p target genes were identified using the predictions that they would: be expressed at higher levels in zinc-limited wild-type cells than in zinc-replete cells; expressed at higher levels in zinc-limited wild-type cells than in zinc-limited zap1 mutants; and contain one or more potential Zap1p-binding sites (zinc-responsive elements, ZREs) in their promoters. This gives 46 candidate Zap1p target genes; a further 65 may be indirect targets or lack the consensus ZRE. Adapted from [1].
Similar studies have also identified the target genes of the Aft1p and Mac1p transcription factors in yeast [3,4,5]. Aft1p regulates genes involved in iron metabolism, and Mac1p regulates genes involved in copper homeostasis. Like Zap1p, these factors activate the expression of target genes in metal-limiting conditions, to facilitate uptake and distribution of the metal ion. Although many Aft1p-regulated genes were already known, microarray analysis identified four additional target genes, ARN1-ARN4, which encode homologous members of the major facilitator superfamily of transporters [3]. Further studies have shown that these proteins transport iron bound to different siderophore compounds (siderophores are iron-binding agents produced and secreted by other microbes in order to obtain iron) [6,7].
In a complementary study, Foury and Talibi [4] identified genes upregulated in a yfh1 mutant strain of yeast. YFH1 encodes the yeast homolog of the human gene involved in Friedreich ataxia, a neurodegenerative disease. Deletion of the YFH1 gene causes accumulation of iron in mitochondria, which depletes cytosolic iron, thereby activating Aft1p. This study identified 70 genes that were upregulated in yfh1 mutants and that are thus likely to be Aft1p targets. Notable among these are the ARN genes, several novel cell-wall proteins that are perhaps involved in iron acquisition, a heme oxygenase possibly involved in the recycling of heme iron, and TIS11, a homolog of mammalian tristetraproline protein implicated in controlling the stability of mRNAs involved in iron homeostasis [4].
Robertson et al. [8] have extended our understanding of iron homeostasis by examining the roles of protein kinase A catalytic subunits. The yeast genome encodes three such subunits, Tpk1p, Tpk2p, and Tpk3p. These kinases phosphorylate target proteins when cAMP levels are high, for example during growth on fermentable carbon sources. Using DNA microarrays and expression profiling of wild-type and tpk mutant strains, they found that Tpk2p activity represses many of the Aft1p target genes. A simple hypothesis that explains this result is that Tpk2p negatively regulates Aft1p activity by direct phosphorylation. This circuitry would increase iron uptake during the switch to respiratory growth, when additional iron would be required for heme synthesis.
The Mac1p transcription factor activates genes in response to copper limitation, and its target genes, as determined by expression profiling, include four genes directly involved in copper uptake [5]. Perhaps one of the most intriguing applications of expression profiling was reported by Moler et al. [9]. Using the data from several previously published microarray experiments not directly dealing with metals, these investigators identified potential Aft1p- and Mac1p-regulated genes from the clustering of their expression profiles with previously known targets (using a naive Bayes model to cluster the genes). The prior studies upon which this analysis was based addressed cell-cycle control, responses to heat shock and cold shock, and the shift from fermentable to non-fermentable carbon sources. Evidently, expression levels of Aft1p and Mac1p targets happen to change in concert under these different growth conditions, allowing clustering of their expression profiles. Thus, an unexpected attribute of expression profiling is its ability to identify genes involved in processes that are not even the direct focus of the experiment. Careful re-analysis of all microarray data will surely reveal abundant information about regulatory relationships.
Obviously, expression profiling of mRNAs will not detect changes in protein levels resulting from post-transcriptional effects. Proteome profiling can therefore provide additional information. A recent study of the response to cadmium stress in yeast provides an excellent example of the application of proteomics to the biology of metals [10]. Untreated cells and cells treated with 1 mM cadmium for 1 hour were pulse-labeled with 35S-methionine and the products analyzed by comparative two-dimensional gel electrophoresis. More than 50 proteins were expressed at increased levels after cadmium treatment, and expression of about 40 others was reduced. One striking result is the induction of nine enzymes involved in sulfur amino-acid biosynthesis, and the concomitant increase in glutathione levels (Figure 2). These data are consistent with the previous observation that cadmium-glutathione conjugates are transported into the vacuole to detoxify the metal ion [11]. Two other proteins induced by cadmium treatment are thioredoxin and thioredoxin reductase. The importance of these proteins in cadmium resistance was demonstrated by the pronounced cadmium hypersensitivity of mutants defective in these genes. Thus, this study has demonstrated that the two cellular thiol redox systems, glutathione and thioredoxin, are critical components of the cell's defense against cadmium.

Effect of cadmium treatment on accumulation of glutathione biosynthetic enzymes. Proteins whose levels are increased in cadmium-treated cells are boxed; the fold increase in expression level is given in brackets. Adapted from [10].
Significant studies of metals using functional genomics have also been performed using other organisms. Among prokaryotes, expression profiling studies addressing metal biology have used Bacillus subtilis [12] and Escherichia coli [13]. For mammals, expression profiling was used by Schaffner and colleagues to identify potential targets of the zinc-responsive transcription factor MTF-1 [14]. Moreover, recent studies have used microarrays for 'toxicogenomics' - the use of functional genomics to understand the mechanisms underlying the toxicity of environmental agents [15]. For example, studies have addressed changes in transcription resulting from nickel exposure to lung tissue [16] and from lead exposure in astrocytes [17]. The latter study showed that expression of vascular endothelial growth factor (VEGF) is induced in lead-treated astrocytes via a pathway that includes protein kinase C and the transcription factor AP-1, providing the first evidence that lead increases expression of a growth factor that may contribute to its neurotoxicity.
In summary, during its short history, functional genomics has made major contributions in the field of metals in biology. These advances have primarily addressed the critical mechanisms of metal-ion homeostasis and detoxification. With continued use of these techniques, and the application of new approaches (such as protein microarrays) [18], functional genomics will continue to accelerate research in this area.
References
Lyons TJ, Gasch AP, Gaither LA, Botstein D, Brown PO, Eide DJ: Genome-wide characterization of the Zap1p zinc-responsive regulon in yeast. Proc Natl Acad Sci USA. 2000, 97: 7957-7962. 10.1073/pnas.97.14.7957.
MacDiarmid CW, Gaither LA, Eide D: Zinc transporters that regulate vacuolar zinc storage in Saccharomyces cerevisiae. EMBO J. 2000, 19: 2845-2855. 10.1093/emboj/19.12.2845.
Yun CW, Ferea T, Rashford J, Ardon O, Brown PO, Botstein D, Kaplan J, Philpott CC: Desferrioxamine-mediated iron uptake in Saccharomyces cerevisiae. Evidence for two pathways of iron uptake. J Biol Chem. 2000, 275: 10709-10715. 10.1074/jbc.275.14.10709.
Foury F, Talibi D: Mitochondrial control of iron homeostasis. J Biol Chem. 2001, 276: 7762-7768. 10.1074/jbc.M005804200.
Gross C, Kelleher M, Iyer V, Brown PO, Winge DR: Identification of the copper regulon in Saccharomyces cerevisiae by DNA microarrays. J Biol Chem. 2000, 275: 32310-32316. 10.1074/jbc.M005946200.
Yun CW, Tiedeman JS, Moore RE, Philpott CC: Siderophoreiron uptake in Saccharomyces cerevisiae. Identification of ferrichrome and fusarinine transporters. J Biol Chem. 2000, 275: 16354-16359. 10.1074/jbc.M001456200.
Heymann P, Ernst JF, Winkelmann G: Identification and substrate specificity of a ferrichrome-type siderophore transporter (Arn1p) in Saccharomyces cerevisiae. FEMS Microbiol Lett. 2000, 186: 221-227. 10.1016/S0378-1097(00)00152-X.
Robertson LS, Causton HC, Young RA, Fink GR: The yeast A kinases differentially regulate iron uptake and respiratory function. Proc Natl Acad Sci USA. 2000, 97: 5984-5988. 10.1073/pnas.100113397.
Moler EJ, Radisky DC, Mian IS: Integrating naive Bayes models and external knowledge to examine copper and iron homeostasis in S. cerevisiae. Physiol Genomics. 2000, 4: 127-135.
Vido K, Spector D, Lagniel G, Lopez S, Toledano MB, Labarre J: A proteome analysis of the cadmium response in Saccharomyces cerevisiae. J Biol Chem. 2001, 276: 8469-8474. 10.1074/jbc.M008708200.
Li ZS, Lu YP, Zhen RG, Szczypka M, Thiele DJ, Rea PA: A new pathway for vacuolar cadmium sequestration in Saccharomyces cerevisiae: YCF1-catalyzed transport of bis(glutathionato)cadmium. Proc Natl Acad Sci USA. 1997, 94: 42-47. 10.1073/pnas.94.1.42.
Ye RW, Tao W, Bedzyk L, Young T, Chen M, Li L: Global gene expression profiles of Bacillus subtilis grown under anaerobic conditions. J Bacteriol. 2000, 182: 4458-4465. 10.1128/JB.182.16.4458-4465.2000.
Brocklehurst KR, Morby AP: Metal-ion tolerance in Escherichia coli: analysis of transcriptional profiles by gene array technology. Microbiology. 2000, 146: 2277-2282.
Lichtlen P, Wang Y, Belser T, Georgiev O, Certa U, Sack R, Schaffner W: Target gene search for the metal-responsive transcription factor MTF-1. Nucleic Acids Res. 2001, 29: 1514-1523. 10.1093/nar/29.7.1514.
Iannaccone PM: Toxicogenomics: "The call of the wild chip". Environ Health Perspect. 2001, 109: A8-A11.
McDowell SA, Gammon K, Bachurski CJ, Wiest JS, Leikauf JE, Prows DR, Leikauf GD: Differential gene expression in the initiation and progression of nickel-induced acute lung injury. Am J Respir Cell Mol Biol. 2000, 23: 466-474.
Hossain MA, Bouton CML, Pevsner J, Laterra J: Induction of vascular endothelial growth factor in human astrocytes by lead. J Biol Chem. 2000, 275: 27874-27882.
Kumar A, Snyder M: Emerging technologies in yeast genomics. Nature Rev Genet. 2001, 2: 302-312. 10.1038/35066084.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eide, D.J. Functional genomics and metal metabolism. Genome Biol 2, reviews1028.1 (2001). https://doi.org/10.1186/gb-2001-2-10-reviews1028
Published:
DOI: https://doi.org/10.1186/gb-2001-2-10-reviews1028