
Abstract
This review of vasopressin in septic shock differs from previous reviews by providing more information on the physiology and pathophysiology of vasopressin and vasopressin receptors, particularly because of recent interest in more specific AVPR1a agonists and new information from the Vasopressin and Septic Shock Trial (VASST), a randomized trial of vasopressin versus norepinephrine in septic shock. Relevant literature regarding vasopressin and other AVPR1a agonists was reviewed and synthesized. Vasopressin, a key stress hormone in response to hypotension, stimulates a family of receptors: AVPR1a, AVPR1b, AVPR2, oxytocin receptors and purinergic receptors. Rationales for use of vasopressin in septic shock are as follows: first, a deficiency of vasopressin in septic shock; second, low-dose vasopressin infusion improves blood pressure, decreases requirements for norepinephrine and improves renal function; and third, a recent randomized, controlled, concealed trial of vasopressin versus norepinephrine (VASST) suggests low-dose vasopressin may decrease mortality of less severe septic shock. Previous clinical studies of vasopressin in septic shock were small or not controlled. There was no difference in 28-day mortality between vasopressin-treated versus norepinephrine-treated patients (35% versus 39%, respectively) in VASST. There was potential benefit in the prospectively defined stratum of patients with less severe septic shock (5 to 14 μg/minute norepinephrine at randomization): vasopressin may have lowered mortality compared with norepinephrine (26% versus 36%, respectively, P = 0.04 within stratum). The result was robust: vasopressin also decreased mortality (compared with norepinephrine) if less severe septic shock was defined by the lowest quartile of arterial lactate or by use of one (versus more than one) vasopressor at baseline. Other investigators found greater hemodynamic effects of higher dose of vasopressin (0.06 units/minute) but also unique adverse effects (elevated liver enzymes and serum bilirubin). Use of higher dose vasopressin requires further evaluation of efficacy and safety. There are very few studies of interactions of therapies in critical care - or septic shock - and effects on mortality. Therefore, the interaction of vasopressin infusion, corticosteroid treatment and mortality of septic shock was evaluated in VASST. Low-dose vasopressin infusion plus corticosteroids significantly decreased 28-day mortality compared with corticosteroids plus norepinephrine (44% versus 35%, respectively, P = 0.03; P = 0.008 interaction statistic). Prospective randomized controlled trials would be necessary to confirm this interesting interaction. In conclusion, low-dose vasopressin may be effective in patients who have less severe septic shock already receiving norepinephrine (such as patients with modest norepinephrine infusion (5 to 15 μg/minute) or low serum lactate levels). The interaction of vasopressin infusion and corticosteroid treatment in septic shock requires further study.
Similar content being viewed by others

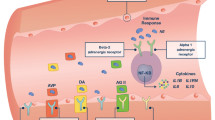

Vasopressin receptors
Vasopressin stimulates a family of receptors: AVPR1a (also known as V1 receptor, mainly vascular), AVPR1b (V3 receptor, mainly central), AVPR2 (V2 receptor, mainly renal), oxytocin receptors and purinergic receptors. AVPR1a, a G-protein coupled receptor, is responsible for vasoconstriction associated with vasopressin and is expressed on vascular smooth muscle, hepatocytes and platelets (Figure 1). G proteins stimulate a phosphatidyl-inositol-calcium signaling pathway, causing smooth muscle contraction [1–3]. Stimulation of the AVPR1a receptor also induces production of the potent vasodilator nitric oxide in coronary vessels [4] and pulmonary vessels [5–7]. Genetic variants of AVPR1a have been associated with essential hypertension [2], autism [8], and generosity [9]. The effects of sepsis on AVPR1a are complex and include downregulation of the receptor [10, 11], yet there can be increased sensitivity to vasopressin in sepsis (compared with normal controls who are hemodynamically stable) [12].

Vasopressin occupies the AVPR1a receptor. Vasopressin (line and beads) occupies the AVPR1a receptor.
AVPR1b (or V3 receptor) is expressed in the anterior pituitary gland and hippocampus. Stimulation of AVPR1b by vasopressin releases adrenocorticotropic hormone (ACTH) because vasopressin flows from the posterior pituitary through pituitary portal capillaries to bind to the AVPR1b on corticotrophic cells of the anterior pituitary. Vasopressin thus interacts with the corticosteroid axis in response to stresses such as hypotension [13, 14]. Vasopressin and corticotrophin-releasing hormone stimulate different signaling systems and have synergistic effects on release of ACTH. AVPR1b knockout mice have an impaired stress response because of blunted ACTH response [15]. In contrast, overexpression of AVPR1b has been associated with pituitary adenomas and ectopic ACTH syndrome [16].
AVPR2 (V2 receptor) is expressed in the renal collecting duct. Vasopressin is a trophic factor of the ascending limb of Henle and thereby creates the gradient that mediates vasopressin's antidiuretic effect. AVPR2 stimulation increases retention of water (antidiuretic activity) by increasing cyclic AMP, which causes movement of aquaporin-2 water channels from cytoplasm to the apical membrane of collecting duct cells. If there is vasopressin deficiency, aquaporin-2 channels internalize from the apical membrane to subapical vesicles so that there is no active water reabsorption. Absence of vasopressin also results in decreased synthesis of aquaporin-2, further impairing water reabsorption. The V2 receptor also mediates vasodilation by stimulating the nitric oxide pathway [17, 18].
Over 150 mutations of the AVPR2 receptor have been reported associated with nephrogenic diabetes insipidus. Stimulation of the AVPR2 receptor increases release of von Willebrand factor, von Willebrand factor multimers, and risk of clotting. Stimulation of AVPR2 is therefore the mechanism for increased coagulation in response to desmopressin. Interestingly, von Willebrand factor levels are increased in sepsis [19, 20].
Sepsis may downregulate vasopressin receptors. Expression of AVPR1a is downregulated in studies of models of sepsis, thereby limiting smooth muscle contraction and increase of blood pressure [10, 11, 21], AVPR2 receptors [22] and AVPR3 receptors [23]. Downregulation AVPR1a in the lung, liver, kidney and heart is probably caused by increased TNFα, IL-1β, IL-6 and IFNγ [10, 21]. The magnitude of downregulation of AVPR1a may be large because continuous infusion of lipopolysaccharide decreased AVPR1A expression by 43% [11]. Models of sepsis also downregulate AVPR1b and AVPR2. Lipopoly-saccharide decreases mRNA levels of the AVPR1b receptor in the pituitary [23] and downregulates AVPR2 expression in rat renal medulla [22].
Vasopressin also simulates oxytocin receptors to cause vasodilation [24, 25]. Stimulation of oxytocin receptors increases nitric oxide and thus causes vasodilation [26]. Stimulation of oxytocin receptors also increases release of atrial natriuretic peptide [27, 28]. The oxytocin receptor is downregulated in the heart in response to ischemia- reperfusion injury; this downregulation of the receptor may explain the potentially detrimental effects of vasopressin in myocardial ischemia-reperfusion, including increased mortality and decreased myocardial contractility [26].
Purinergic receptors are also stimulated by vasopressin [27]. At low doses, vasopressin acts on purinergic receptors that mediate endothelial vasodilation; this effect is reversed when high-dose vasopressin stimulates the AVPR1a receptor to mediate vasoconstriction.
The vasopressin neurons of the hypothalamus and neurohypophysis are innervated by a variety of pathways that carry information regarding, firstly, blood volume/blood pressure and, secondly, osmolality. Purinergic receptors are also important components of the stimulation of release of vasopressin. ATP is a neurotransmitter that signals through the purinergic P2 receptors (P2X2 [28], P2X3 [29, 30] and P2Y1 [31, 32]) to increase cytosolic free calcium [33], which then stimulates release of vasopressin from the neurohypophysis [34–37]. The P2X5 receptor is expressed and co-localized with vasopressin on paraventricular and supraoptic nuclei of the hypo-thalamus, suggesting that P2X5 is also important in the stimulation of vasopressin release [38]. The stimulatory effects of ATP on vasopressin release are synergized (that is, an increased amount and duration of vasopressin release) by co-stimulation with norepinephrine [39] acting as neurotransmitters via P2X [36, 39, 40] and P2Y1 [41] receptors. This synergistic action of ATP and nor-epinephrine in the neurohypophysis may be important for the sustained release of vasopressin during hypotension [36]. The stimulation of vasopressin release by ATP is terminated by adenosine, a metabolite of ATP, and is thus a natural negative feedback mechanism of vasopressin release [42].
ATP also stimulates P2Y1 and P2Y2 receptors of the convoluted tubule of the nephron to modulate vasopressin antidiuretic effects [43, 44]. Indeed, P2Y2 knock-out mice have salt-resistant hypertension [45]. Further-more, the cardiac effects of vasopressin are mediated at least in part through P2X and P2Y receptors [46].
Synthesis, release and clearance of vasopressin
Vasopressin is synthesized and released into the systemic circulation from the posterior pituitary gland (Figure 2). Vasopressin, like many hormones, is synthesized as a prohormone and then is cleaved to form the mature active hormone. Serum levels of vasopressin - a nonapeptide - represent the interactions of the synthesis, release, and metabolism of vasopressin. Synthesis of preprovasopressin occurs in neurophypophyseal neurons (also known as magnocellular neurons) of paraventricular and supraoptic nuclei of the hypothalamus. Provasopressin is packaged in neurosecretory granules and transported along the suprahypophyseal tract to the posterior pituitary. Subsequently, there is conversion to provasopressin followed by conversion of provasopressin by subtilisin-like proprotein convertase (SPC3) to vasopressin.

The hypothalamic pituitary axis and vasopressin. Provasopressin is synthesized in neurohypophyseal neurons of the paraventricular nuclei (PVN) and supraoptic nuclei (SON) of the hypothalamus. Provasopressin is transported along the supraoptic neurohypophyseal tract in neurosecretory granules to the posterior pituitary gland. Provasopressin is cleaved by enzymes to vasopressin during transport to the posterior pituitary gland. Hypotension and hyperosmolality stimulate release of stored vasopressin (immediate) and synthesis (delayed) of vasopressin. In response to hypotension, vasopressin is released into the systemic circulation and also into the pituitary portal capillaries, flows to the anterior pituitary, binds to the AVPR1b receptor of corticotrophs, and stimulates release of adrenocorticotropic hormone. MNPO, median pre-optic nucleus.
Vasopressin is metabolized by several promiscuous vasopressinases such as LNPEP and IRAP. LNPEP metabolizes vasopressin and also clears oxytocin (a vasodilator), GLUT4 (which modulates cellular uptake of glucose) and angiotensin-converting enzyme 4 (which converts angiotensinogen to the potent vasoconstrictor angiotensin), indicating that variations in levels and function of LNPEP could have complex effects on hemodynamics and blood pressure in sepsis.
Sepsis initially increases vasopressin levels by 20-fold to 200-fold to supraphysiologic levels by several mechanisms. Early studies by Wilson and colleagues in endotoxin-induced shock and Escherichia coli-induced shock found that vasopressin levels were >500 pg/ml in dogs and >300 pg/ml in baboons within 15 minutes of initiation of sepsis [47]. This increase of vasopressin levels occurred when cardiac output was decreased but before hypotension, suggesting that stimuli other than hypotension - such as increased levels of mediators of inflammation and decreased cardiac output - stimulated the increase of vasopressin levels. Furthermore, endotoxin stimulates the release of vasopressin into hypophyseal portal blood. Hypotension and hypernatremia stimulate release of stored vasopressin (immediate) and synthesis of vasopressin (delayed). Vasopressin stores are thus ample and available for instantaneous release from the posterior pituitary in response to sepsis, septic shock and hypotension.
Vasopressin levels increase early in septic shock because hypotension is the most potent stimulus for release and synthesis of vasopressin. Indeed, if an animal is challenged with conflicting signals to vasopressin release (such as hypotension and hyponatremia), the animal will increase vasopressin. After the initial release of vasopressin, which increases vasopressin levels in septic shock, vasopressin levels then decline rapidly - because of depletion of stored vasopressin - to levels that are inappropriately low (compared with similarly hypotensive patients who have cardiogenic shock [48, 49]). The decrease in vasopressin levels is initially due to the depletion of vasopressin in the post-hypophyseal centers, but vasopressin levels remain extremely, inappropriately low [50, 51], suggesting that there is also a sustained impairment of synthesis and release of vasopressin. Indeed, consistent with these earlier studies, vasopressin levels remained extremely low for up to 7 days after onset of septic shock in the Vasopressin and Septic Shock Trial (VASST) (Figure 3) [52]. Few endocrine systems are so rapidly activated (to increase serum levels) and then are so rapidly exhausted (such that serum levels decrease) as the vasopressin axis in sepsis.

Plasma vasopressin levels in the Vasopressin and Septic Shock Trial. Plasma vasopressin levels in patients in the Vasopressin and Septic Shock Trial who were infused with vasopressin compared with norepinephrine. Plasma vasopressin levels over time in patients receiving a vasopressin infusion (n = 54, black squares), patients in the vasopressin group once vasopressin infusions had stopped (open squares), and patients in the norepinephrine group (n = 53, grey circles). Values are median and interquartile range (IQR).
Ironically, even though vasopressin levels fall to very low levels in septic shock, some evidence suggests that low plasma levels of vasopressin may not be associated with increased mortality. A recent study found that plasma levels of provasopressin were significantly higher in nonsurvivors of septic shock (Table 1) [53]. Therefore, it is intriguing to speculate that this lack of association of vasopressin levels and mortality could be interpreted to indicate that the decrease of vasopressin levels could be adaptive. The studies of levels of vasopressin and outcome must also be interpreted with an understanding of when vasopressin levels were measured during the course of illness, because changing levels of vasopressin (increased early and decreased later) could be linked to outcomes.
The discussion of appropriate levels of vasopressin in septic shock leads logically to the following fundamental question regarding the design of vasopressin intervention studies. Should vasopressin be infused as very-low-dose replacement therapy (opotherapy), at higher doses for vasopressin's vasoconstrictive properties, or at supra-physiologic levels? At low plasma levels of vasopressin (<10 pmol/l), vasopressin's V2 receptor antidiuretic actions predominate. At higher levels, the vasoconstrictive properties of vasopressin are initiated and become progressively predominant. There is also, however, vasodilation caused by vasopressin (caused by vasopressin's stimulation of AVPR1a, AVPR1b, and oxytocin receptors).
The early increase in vasopressin release in septic shock is modulated by inducible nitric oxide synthase-induced increase of nitric oxide [54, 55]. Heme-oxygenase activation in sepsis attenuates the increase of AVP in septic shock [56].
Copeptin, a 39-amino-acid glycopeptide, is the C terminal of provasopressin (similar to the C peptide of insulin [6]) and is stable in plasma, and is therefore easier to measure than vasopressin. Some studies have therefore suggested that copeptin could be used as a surrogate for measurement of vasopressin [57–60]. Landry and colleagues first discovered that vasopressin levels are inappropriately low in septic shock [45, 49] (Table 1). Other workers found that vasopressin levels were lower in emergency patients who progressed to septic shock (3.6 pg/ml) compared with patients who had sepsis (10.6 pg/ml) and severe sepsis (21.8 pg/ml) [61].
Critically ill patients have higher copeptin levels than normal volunteers [57, 59, 60, 62, 63]. Morgenthaler and colleagues found increased copeptin levels in human sepsis and septic shock (means: normal volunteers, 4 pM; critically ill patients, no sepsis, 27 pM; sepsis patients, 50 pM; severe sepsis patients, 74 pM; septic shock patients, 171 pM) [57]. Furthermore, patients who had hemodynamic dysfunction had higher vasopressin levels (and copeptin levels) than patients without hemodynamic dysfunction (AVP, 14.1 pg/ml versus 8.7 pg/ml). Patients who are post cardiovascular surgery usually have higher vasopressin and copeptin levels than patients with septic shock [57, 59, 60]. Copeptin was higher in nonsurvivors (versus survivors) in at least two studies of adult septic shock (171 pM versus 87 pM [57] and 70 pM versus 24 pM [58]) and in one study of pediatric septic shock [64]. Lodha and colleagues [64] found that serum vasopressin levels in pediatric septic shock are similar to the results of Jochberger and colleagues [53]. Vasopressin levels were increased in septic shock (116 pg/ml) and severe sepsis (106 pg/ml), and vasopressin levels did not change over 96 hours.
Do serum levels of copeptin and vasopressin correlate tightly, such that copeptin is a surrogate for vasopressin? I suggest copeptin levels should not be used as a surrogate for vasopressin levels at this time without further evaluation.
There are fascinating interactions of vasopressin with the baroreceptor reflex and neural control of the cardiovascular system. It is well known that vasopressin infusion causes bradycardia in normal animals and humans because of stimulation of the baroreceptor reflex by vasopressin. In addition, infused vasopressin also enters the brain and modulates the sensitivity of the baroreceptor reflex. Vasopressin is also an important neurotransmitter in the brain that regulates the brain-stem control of the cardiovascular system. Vasopressin is also a key regulator of the behavioral response to stress.
Effects of infused vasopressin in septic shock
Vasopressin infusion increases vasopressin levels rapidly [48, 49]. The effects of vasopressin infusion on copeptin levels are not known. Responses to vasopressin and norepinephrine, however, are modified in sepsis. One fundamental concept shown repeatedly in animal studies of sepsis is that the effects of vasopressin (both beneficial and adverse) in models of sepsis depend critically on the volume status of the animal [65]. For example, volume loading in models of sepsis is critical to minimizing and ameliorating the adverse mesenteric vasoconstriction that can produce intestinal ischemia [12]. These observations highlight the central importance of adequate volume resuscitation of patients before infusion of vasopressin is considered.
Fecal peritonitis was used to evaluate the vascular effects on mesenteric artery vasoconstriction in response to norepinephrine, vasopressin, and a selective AVPR1a agonist (F180) [12]. The vasoconstrictor responses to vasopressin and the AVPR1a agonist were enhanced whereas responses to norepinephrine were severely impaired. There thus appeared to be vasopressin hyper-sensitivity in the mesenteric circulation.
Nakajima and colleagues evaluated vasopressin, nor-epinephrine, and L-arginine effects on intestinal circulation in a septic shock model [66]. Lipopolysaccharide decreased perfused intestinal villi flow; and vasopressors increased the mean arterial pressure (MAP) and prevented further falls in perfused intestinal villi. Knotzer and colleagues studied vasopressin in an acute endotoxin model of septic shock and found decreased jejunal oxygenation [67]. There was no effect (that is, no further worsening) from vasopressin.
In contrast, other studies have found decreased vascular responsiveness to vasopressin in models of septic shock [68]. There was a synergistic effect of vasopressin and norepinephrine on vascular responsiveness.
The effects of vasopressin differed in several vascular beds in sepsis [69, 70]. Renal blood flow was lower with norepinephrine than vasopressin; hepatic blood flow was lower with vasopressin than norepinephrine, while coronary blood flow was similar with vasopressin and norepinephrine.
Patients who have septic shock have decreased forearm vascular resistance, as a component of decreased systemic vascular resistance. The forearm vasoconstrictor response to phenylephrine (α-agonist) and angiotensin II was impaired whereas the response to vasopressin was enhanced in septic shock compared with normal [71].
There have been many small studies of the effects of vasopressin infusion reviewed recently [72]. Vasopressin infusion decreases the dose requirement of norepinephrine, increases urine output and creatinine clearance, and sometimes decreases cardiac output [48, 49, 62, 63, 73–78]. These studies, however, were not powered for the 28-day mortality rate or organ dysfunction.
The side effects of vasopressin studied in animal models of septic shock are conflicting because of differences in models (acute, type of septic insult, chronic), doses of vasopressin, and duration of studies (hours versus days). For example, vasopressin infusion (0.06 units/kg/hour) increased the MAP but also decreased mesenteric blood flow by one-half and decreased microcirculatory flow to gastric mucosa (by 23%) and to jejunal mucosa (by 27%) in porcine fecal peritonitis [79].
Adverse effects of vasopressin reported in studies of humans include mesenteric ischemia, myocardial ischemia, digital ischemia, skin necrosis, and hyponatremia, to name a few [62, 63, 67, 76, 78, 80]. These studies were small, nonblinded, and often uncontrolled, however, so the true risks of low-dose vasopressin infusion in septic shock are unknown. Vasopressin has complex effects on the immune and inflammatory responses. Ironically, commonly used catecholamines (epinephrine and, to a lesser extent, norepinephrine) induce an inflammatory response in hepatocytes [81] similar to lipopolysaccharide. Effects such as these could be harmful directly or indirectly in patients who have septic shock.
The effects of infused vasopressin on endogenous vasopressin synthesis and release are not known. Early withdrawal of vasopressin infusion in septic shock can lead to a rapid drop in vasopressin levels (Figure 3), sometimes to such low levels of vasopressin that hemodynamic instability could recur. The risk of vasopressin deficiency after withdrawal of vasopressin infusion may be increased in patients who are also on corticosteroids because corticosteroids inhibit vasopressin secretion.
The Vasopressin and Septic Shock Trial
VASST, a randomized controlled trial of vasopressin versus norepinephrine in patients with septic shock, was performed to address uncertainties regarding vasopressin in septic shock [52]. The primary hypothesis was that vasopressin, compared with norepinephrine infusion, would decrease 28-day mortality from 60% to 50%. An important secondary hypothesis was that the effects of vasopressin, compared with norepinephrine, would be more pronounced in patients with more severe septic shock. We therefore stratified patients into less and more severe septic shock.
We studied adults with septic shock on norepinephrine treatment. Septic shock was defined by infection, two or more of the systemic inflammatory response syndrome criteria [82], at least one new dysfunction, and vasopressor support (5 μg/minute norepinephrine (or equivalent) for 6 hours). Exclusion criteria are listed in the online supplement [52]. It should be noted that the MAP of patients at baseline in VASST was 72 mmHg, suggesting to some that the patients may not have been in septic shock. All patients were on at least 5 μg/minute norepinephrine (or equivalent), however, which satisfies the commonly used definitions of septic shock [83–86].
The study drugs were vasopressin or norepinephrine; stratification variables were the center and severity of shock. Less severe septic shock was defined as infusion of 5 to 14 μg/minute norepinephrine (or equivalent) while more severe septic shock was defined as ≥15 μg/minute norepinephrine (or equivalent) in the hour before randomization. Blinded vasopressin infusion was titrated from 0.01 U/minute to 0.03 U/minute, while blinded norepinephrine infusion was titrated from 5 μg/minute to 15 μg/minute. Clinicians titrated open-label vasopressors to maintain a MAP of 65 to 75 mmHg. Weaning of open-label vasopressors occurred when the goal MAP had been achieved during study drug infusion. The study drug was weaned when MAP was maintained for 8 hours without open-label vasopressors. Infusion of the study drug was continued until the patient died, had a serious adverse event, or stabilized so that open-label vasopressors were no longer needed.
Patients enrolled in VASST were severely ill (for example, high Acute Physiology and Chronic Health Evaluation II scores, high fraction with new organ dysfunction, high norepinephrine infusion rates). The MAP was similar in the two treatment groups (Figure 4a). The heart rate was significantly lower in the vasopressin group than in the norepinephrine group (first 4 days) (Figure 4b). As in previous human studies, vasopressin infusion allowed a rapid decrease of the norepinephrine dose (over the first 4 days) (Figure 5).

Mean arterial pressure and heart rate in the Vasopressin and Septic Shock Trial. Comparison of (a) mean arterial pressure (MAP) and (b) heart rate in the norepinephrine group (grey circles) and in the vasopressin group (black squares) of the Vasopressin and Septic Shock Trial. Values are mean ± standard deviation (SD). Heart rate was significantly lower in the vasopressin group than in the norepinephrine group over the first 4 days (P < 0.001). There were no statistically significant differences between the norepinephrine and vasopressin groups in MAP.

Doses of norepinephrine infusions in the Vasopressin and Septic Shock Trial. Rates of total norepinephrine infusion (open-label and study drug) in the Vasopressin and Septic Shock Trial for the vasopressin-treated group (black squares) and for the norepinephrine-treated group (grey circles) amongst all patients who were treated with open-label norepinephrine at baseline. The rates of norepinephrine infusion were significantly lower in the vasopressin group than in the norepinephrine group over the first 4 days (P < 0.001). Values are median and interquartile range (IQR).
Plasma vasopressin was extremely low at baseline (median 3.2 pmol/l) and did not change in the norepinephrine group. Low-dose vasopressin infusion increased vasopressin levels to 73.6 pmol/l (median at 6 hours) and 98.0 pmol/l (24 hours) (Figure 3). These levels of vasopressin may be supraphysiological because those levels of vasopressin are rarely observed in the very acute phase of septic shock. Some studies found that vasopressin levels increased 200-fold, however, suggesting the circulating levels of vasopressin in VASST were not supraphysiologic.
There was no significant difference between vasopressin-treated and norepinephrine-treated groups in 28-day mortality (35.4% versus 39.3%, P = 0.26; absolute risk reduction (ARR) = -2.9 to 10.7%), in 90-day mortality (43.9% versus 49.6%, respectively, P = 0.11; ARR = -1.3 to 12.8%), or in organ dysfunction rates (Figure 6). VASST was prospectively powered to detect an absolute difference in mortality of 10% from an expected 60%. Interestingly, the overall mortality rate was considerably lower (37%) in VASST than previously reported.

Survival curve for Vasopressin and Septic Shock Trial vasopressin-infused patients compared with norepinephrine-infused patients. Ninety-day Kaplan-Meier survival curves for all randomized and infused patients (P = 0.27 at day 28; P = 0.10 at day 90). Solid line, vasopressin-infused group; dotted line, norepinephrine-infused group. P values were calculated using the log-rank statistic.
Vasopressin may have decreased mortality in patients who had less severe septic shock in VASST. At baseline, patients who had less severe septic shock had lower Acute Physiology and Chronic Health Evaluation II score, higher arterial pressure, lower lactate, lower norepinephrine dose, and a lower proportion presenting organ dysfunction than patients who had more severe septic shock. There were trends suggesting an association of vasopressin infusion with decreased 28-day (and 90-day) mortality in less severe septic shock. Post hoc analyses stratified by indicators of severity of shock suggested a possible advantage of vasopressin. Stratification by lactate quartile and by number of vasopressors at baseline was significant. In distinct contrast, there were no differences in mortality in more severe septic shock between vasopressin and norepinephrine. The test for the interaction of treatment group by severity of shock subgroup, however, was not significant (P = 0.10).
The potential benefits of vasopressin (compared with norepinephrine) in patients who had less severe septic shock were somewhat delayed so might not have been directly related to rapid reversal of septic shock. Kaplan-Meier curves show that the vasopressin-treated patients separated from the norepinephrine-treated patients at about day 10 (Figure 7). This delayed separation of the Kaplan-Meier curves could be due to vasopressin-induced delayed benefit of improved shock, delayed benefits of the decreased dose of norepinephrine, or effects of vasopressin on inflammation and immunity.

Survival curve for patients with more severe and less severe septic shock. Ninety-day Kaplan-Meier survival curves for patients with more severe and less severe septic shock in the Vasopressin and Septic Shock Trial comparing vasopressin-infused patients with norepinephrine-infused patients within each stratum. (a) Patients in the more severe stratum (P = 0.77 at day 28 and P = 0.92 at day 90). (b) Patients in the less severe stratum (P = 0.05 at day 28 and P = 0.03 at day 90). Solid black line, vasopressin-treated group; dotted line, norepinephrine-treated group; vertical line, day 28. P values were calculated using the log-rank statistic.
Serious adverse event rates were similar in the vasopressin and norepinephrine groups (10.3% versus 10.5%). Vasopressin infusion may increase the risk of cardiac arrest, yet in VASST there were more cardiac arrests in the norepinephrine group than in the vasopressin group (2.1% versus 0.8%, P = 0.14). Other adverse effects of vasopressin and norepinephrine are decreased cardiac output, mesenteric ischemia, hyponatremia (vasopressin), skin necrosis and digital ischemia. In VASST, there was a trend to more digital ischemia in the vasopressin group versus the norepinephrine group (2.0% versus 0.5%, P = 0.11).
VASST studied the effects of vasopressin on patients who were already receiving norepinephrine infusion. Determining whether it is the temporal influence of the combination of vasopressin plus norepinephrine or of vasopressin alone that was potentially beneficial in VASST is therefore difficult. Studies of vasopressin as the first and only agent versus vasopressin added to - and allowing tapering of - norepinephrine would resolve such controversy.
Some would argue that it may have been unrealistic to design a randomized controlled trial to detect an ARR of10% in septic shock. Indeed, if the sample size was larger, VASST could have detected a statistically significant ARR of perhaps 5% - which some would argue would be clinically significant and lead to changes in practice. For example, a sample size of 2,286 per group would have been required to have 80% power to detect a statistically significant (P <0.05) decrease in mortality in VASST from 39% (norepinephrine group) to 35% (vasopressin group). Similarly, a sample size of 73 per group would detect a decrease of mortality from 39% to 32%.
Strengths of VASST were the multicenter design, the large sample size powered for mortality, blinding, the use of low-dose vasopressin, blinded evaluation of serious adverse events, well-defined inclusion and exclusion criteria, and assessment of pharmacokinetics for several days of vasopressin infusion. Some limitations, however, do apply to VASST. First, the vasopressin infusion rate was predetermined for a low dose. Second, we could not measure plasma vasopressin levels to guide the dose or duration of drug infusion. Third, the MAP at baseline in VASST was about 72 to 73 mmHg, so VASST evaluated low- dose vasopressin as a catecholamine-sparing drug. Finally, the mean time from meeting the VASST inclusion criteria to study drug infusion was 12 hours - Rivers and colleagues identified benefits of early goal-directed therapy when applied in the first 6 hours [87], so delays greater than these 6 hours may be important. There is some evidence from VASST that early treatment with vasopressin may optimize its benefits. There was a trend to decreased mortality with vasopressin (33.2% versus norepinephrine 40.5%, P = 0.12) in patients treated within 12 hours. Early vasopressin infusion may optimize benefits similar to early goal-directed therapy [87].
Vasopressin as the initial agent in septic shock
Some would ask whether vasopressin infusion could be used as the first agent in patients who have not responded to adequate volume resuscitation, especially since vasopressin was most effective in patients who had less severe septic shock in VASST. VASST did not evaluate vasopressin as the initial agent in septic shock.
A retrospective cohort study of patients who had septic shock who received either norepinephrine (n = 49), vasopressin (n = 50), or dopamine (n = 51) as the first vasoactive agent in septic shock found no difference in mortality (65%, 52%, and 60%, respectively) [88]. Furthermore, there are no studies of the safety of vasopressin as the first agent in septic shock. We therefore do not believe there is yet adequate evidence to recommend vasopressin as the initial agent in septic shock.
Interactions of vasopressin infusion and corticosteroid treatment in septic shock
There have been remarkably few large studies of interactions of the commonly used therapies in critical care and the effects on mortality. Many of the patients in VASST were treated with corticosteroids because patients had septic shock. We therefore evaluated the interaction of vasopressin infusion, corticosteroid treatment, and mortality of septic shock in VASST [89]. Low-dose vasopressin infusion plus corticosteroids significantly decreased 28-day mortality compared with corticosteroid plus norepinephrine treatment (44.7 to 35.9%, P = 0.03; P = 0.008 interaction statistic). In contrast, if patients were not treated with corticosteroid, then vasopressin may have increased mortality compared with nor-epinephrine (33.7 versus 21.3% respectively, P = 0.06). In patients who received vasopressin infusion, corticosteroids increased plasma vasopressin levels significantly to 33% at 6 hours (P = 0.001) and to 67% at 24 hours (P = 0.032) versus patients who did not receive corticosteroids.
To the knowledge of the VASST investigators, this substudy of VASST is the first clinical study of interactions of vasopressin and corticosteroids on mortality and organ dysfunction in human septic shock. Many patients probably receive the combination of vasopressin and corticosteroids in clinical practice. Despite this, there are few studies of the interaction of vasopressin and corticosteroids in septic shock. The effects of corticosteroids on vasopressin levels and efficacy are controversial. Studies find that corticosteroids increase vasopressin mRNA and restore responsiveness to vasopressin, have no effect on vasopressin levels, and even suppress vasopressin gene expression [90–93].
The effects of vasopressin on the corticosteroid axis are clearer [90, 94, 95]. When vasopressin binds to AVR1b (V3 receptor), vasopressin increases corticotroph responsiveness to corticotropin-releasing factor, thus increasing ACTH even in conditions of stress when corticosteroid levels are increased [73, 75, 90, 94, 95]. The vasopressin-induced increase of ACTH (unlike effects of corticotropin-releasing factor on ACTH) is resistant to corticosteroid-negative feedback because binding to AVR1b is coupled to phospholipase C and is not regulated by corticosteroid levels. Vasopressin infusion did not change serum ACTH or cortisol levels in a cohort of septic shock [73, 75].
It is not known how vasopressin plus corticosteroids (versus norepinephrine plus corticosteroids) could have altered mortality rates. Potential mechanisms could be that corticosteroids increased vasopressin levels or enhanced responsiveness to vasopressin [91], or the combination of vasopressin and corticosteroids altered inflammation and immunity beneficially. Annane suggested that interactions with nitric oxide could explain this interaction [96]. Furthermore, vasopressinases such as LNPEP catalyze the metabolism of vasopressin in the circulation. Interestingly, LNPEP and other vasopressinases are corticosteroid responsive [96].
The effects of corticosteroids on vasopressin receptors have not been extensively studied. Dexamethasone attenuates the downregulation of AVPR1a receptors in sepsis by inhibition of NFκB.
Use of higher doses of vasopressin
Several investigators have explored the use of higher doses of vasopressin in septic shock [62, 63, 73–75], and indeed have argued that this is one reason why vasopressin might not have decreased mortality in VASST [97]. Cohort studies, case-control studies, and small randomized controlled trials have used doses of vasopressin of about 0.06 units/minute (compared with 0.03 units/minute in VASST), and found that vasopressin significantly decreased use of norepinephrine and increased blood pressure [62, 63, 73, 75]. Some of these studies, however, also detected potentially adverse side effects of vasopressin such as decreased intestinal mucosal perfusion [98, 99], increased bilirubin [62, 63], increased serum transaminases [62, 63, 73, 75], and decreased platelet counts [62, 73]. Note that some of these studies used much higher doses than recommended in human septic shock [98–100]. We have agreed with the suggestion [97] that a large randomized trial of these higher doses of vasopressin (0.06 units/minute) to address safety and efficacy questions in septic shock is meritorious [97].
Other AVPR1A agonists: terlipressin and others
There are a number of alternatives to vasopressin in septic shock (including ornipressin, terlipressin, specific AVPR1a agonists, and others), of which terlipressin has been most studied [65, 101–112]. Terlipressin is a 12-amino-acid synthetic analog of lysine vasopressin, a precursor of vasopressin, and has a longer elimination half-life than vasopressin (50 minutes for terlipressin compared with 6 minutes for vasopressin) [113–116]. Terlipressin also has greater selectivity for the AVPR1a receptor than does vasopressin [110, 113]. Terlipressin is metabolized by exopeptidases to yield small sustained levels of lysine vasopressin in the circulation, accounting for its longer biological half-life compared with native vasopressin. Similar to vasopressin, terlipressin increases blood pressure, decreases dose requirements for norepinephrine in septic shock and increases renal function (urine output and creatinine clearance) [65, 101, 104–107]. Terlipressin may be effective for transporting patients with septic shock who are on norepinephrine because of the relatively long half-life of terlipressin [117]. Terlipressin improves renal function and outcomes of hepatorenal syndrome [118–120] and so, like vasopressin [121], could also improve renal function in septic shock. Terlipressin may also be given as a continuous infusion [122–125], which then increases blood pressure, decreases dose requirements of norepinephrine, and may have less rebound hypotension when discontinued compared with vasopressin [122].
Like vasopressin [73], terlipressin may decrease cardiac output in septic shock [101], especially if there is myocardial depression or pre-existing myocardial dysfunction (such as pre-existing congestive heart failure, ongoing myocardial ischemia) [126]. The decrease of cardiac output due to terlipressin is prevented in animals [126] and humans by co-administration of dobutamine [111], which increases the left ventricular stroke work index [111]. Furthermore, terlipressin may increase coronary vasoconstriction [127] and induce myocardial ischemia because both vasopressin and terlipressin may potentiate adrenergic vasoconstriction in coronary arteries [128] (as well as mesenteric arteries [129]). Terlipressin may also have direct coronary vasoconstrictive properties because of binding to AVPR1a [127]. Terlipressin has been associated with ventricular arrhythmias [130], with decreased platelet counts [101, 122] and - similar to vasopressin [66, 79, 107] - with mesenteric ischemia, in part because both agents may potentiate adrenergic vasoconstriction in the mesenteric circulation [129].
Conundrums and one view to consensus
There are conundrums in the vasopressin literature. Large clinical trials such as VASST suggest that vasopressin may be more effective in patients who have less severe septic shock and in combination with corticosteroids. Yet experimental studies suggest that AVPR1a sensitivity and the prevalence of adrenal dysfunction increase with severity of shock. The conflicting evidence about AVPR1a sensitivity (some studies suggest increased AVPR1a sensitivity while others found less sensitivity in models of sepsis) suggests this is an ongoing controversy. Regarding the interaction of vasopressin and corticosteroids, perhaps the beneficial interaction is not related to the presence of adrenal dysfunction but rather is related to corticosteroid-induced increase of vasopressin levels [52, 89]. Vasopressin levels were higher in patients who had preserved adrenal function - and higher corticosteroid levels - compared with patients who had impaired adrenal function (Table 1).
Another puzzle is the observation that the efficacy of corticosteroids is only moderate in moderate septic shock (CORTICUS) [86] and is greater in more severe septic shock. We have recently shown that corticosteroids appear to increase plasma vasopressin levels in both less severe and more severe septic shock, but the effect was statistically significant only in less severe septic shock. The beneficial interaction of vasopressin infusion and corticosteroid treatment may therefore span the spectrum of severity of shock.
Another question is the relationship of vasopressin to dopamine, to dobutamine plus norepinephrine, and to epinephrine - considering that VASST focused on patients of whom the majority were already on norepinephrine infusion. In VASST, some patients were on dopamine or epinephrine instead of norepinephrine; however, the numbers of these patients were too small to assess accurately the effects of vasopressin on patients who were receiving these vasoactive agents. It is likely that the findings of studies of vasopressin effects on norepinephrine dose also apply to dopamine when dopamine is used as a vasopressor (that is, doses >5 μg/kg/minute). It is quite likely that vasopressin can be used effectively with dobutamine because several animal and human studies have shown that the combination of vasopressin and dobutamine is beneficial in limiting any decrease of cardiac output, oxygen delivery, and mixed venous oxygen saturation caused by vasopressin. It is also likely that vasopressin may also be used in patients who are already on epinephrine infusion. The combination of dopexamine and vasopressin attenuated decreases of the cardiac index and oxygen delivery - compared with vasopressin alone - but dopexamine also decreased blood pressure compared with vasopressin; this suggests that dopexamine may not be a good agent to attenuate cardiovascular adverse effects of vasopressin.
Clearly there is a common, profound deficiency of vasopressin in septic shock. However, it is not known when and how quickly this vasopressin deficiency recovers. Siami and colleagues addressed this important question by measuring plasma vasopressin levels of normonatremic patients who had septic shock for more than 72 hours who were given a stimulus: osmotic stimulation using 500 ml hypertonic saline [131]. Interestingly, 52% (17 of 33 patients) of patients were nonresponders to this stimulation. In responders, plasma vasopressin increased from 4.8 to 14.4 pg/ml, a rate of increase of 6.2 pg/ml/hour. In contrast, nonresponders' plasma vasopressin did not increase significantly (from 2.8 to 4 pg/ml, a rate of increase of 0.7 pg/ml/hour). Nonresponders were more frequently bacteremic and had hepatic dys-function compared with responders. Hypertonic saline lead to comparable changes in central venous pressure, MAP and systolic arterial pressure in responders and nonresponders; thus, it is unlikely that the differences in response of plasma vasopressin levels were due to differences in baroreceptor reflex stimulation. Osmoregulation by vasopressin is therefore impaired in about one-half of this small sample of patients who had sustained septic shock. It is likely that vasopressin regulation of blood pressure is similarly impaired for at least this duration.
What is the status of the vasopressin axis shortly after patients are weaned off vasopressin infusion? Bauer and colleagues found that patients dropped their blood pressure more often when vasopressin infusion was stopped before stopping infusion of norepinephrine [132]. Their study suggests there can be ongoing inadequate vasopressin axis reserve. This hypothesis is not directly known, but the vasopressin axis is probably only partially recovered for some time after discontinuation of vasopressin infusion and a secondary insult (for example, new nosocomial infection) might lead to septic shock more readily because of continued impaired vasopressin axis responses.
Newer alternative strategies to support ventricular function in patients whose ventricular function declines on vasopressin
Vasopressin infusion has been reported to decrease cardiac output in some patients who have septic shock. Dobutamine infusion is often added to increase cardiac output [133]. Levosimendan is a novel inotropic agent that acts by sensitizing calcium channels leading to vasodilatation, and often (but not always) leading to increases in cardiac output. Rehberg and colleagues showed that levosimendan plus vasopressin improved aspects of hemodynamic parameters (such as the left ventricular stroke work index) and net fluid balance in sheep that had fecal peritonitis compared with vasopressin therapy alone [134]. The combination of vasopressin plus levosimendan also increased global perfusion (as assessed by mixed venous oxygen saturation) and decreased pulmonary artery pressure. This novel combination also appeared to improve the net fluid balance and prevented decreases in colloid osmotic pressure compared with vasopressin alone. Levosimendan is not yet available for clinical use in North America, but its study is intriguing and bears further clinical evaluation.
If patients suffer a decrease of cardiac output and tissue perfusion in response to vasopressin, therefore, one should re-evaluate the volume status - volume resuscitate if deficient, and if volume replete the current evidence supports adding dobutamine infusion to augment cardiac output.
Genomics and vasopressin
Recent studies suggest that there could be notable genomic variation in vasopressinases (also known as LNPEP), the enzyme that metabolizes vasopressin (as well as oxytocin and GLUT4). Furthermore, Nakada and colleagues found that patients who had the genotype TT of the single nucleotide polymorphism rs4869317 of the LNPEP gene had increased mortality (compared with patients who had the AT/AA genotypes) in a discovery cohort of patients who had septic shock, and that this finding was replicated in an independent cohort of patients who had septic shock [135]. This marker (rs4860317 TT) was also associated with increased cardiovascular dysfunction. Furthermore, these authors discovered that patients who were on a vasopressin infusion who had the rs4869317 TT genotype had lower plasma vasopressin concentrations because of increased vasopressin clearance [135]. To summarize, the TT genotype of rs4869317 was associated with increased mortality and increased vasopressin clearance in patients who had septic shock.
Patients who have the TT genotype of rs4869317 would also be expected to have altered AVPR1a and AVPR2 responses to vasopressin because of altered vasopressin levels available to these different vasopressin receptors. Indeed, the plasma sodium concentration was higher in the first 48 hours of cardiovascular surgery in patients who had the rs4869317 TT genotype compared with patients who had the AT/AA genotypes. This finding aligns with the diminished vasopressin levels and increased mortality rates of patients with the TT genotype of rs4869317 who had septic shock. The genomic variation of the LNPEP gene of patients could therefore alter the response to vasopressin (and other vasopressin analogues) in septic shock, postcardiovascular surgery and other conditions such as hepatorenal syndrome.
The role for the use of genetic testing to provide more patient-centered care is exciting, requires more investigation and could be of benefit in the selection of patients for treatment with vasopressin or vasopressin analogs.
New conditions in which there is a vasopressin deficiency
Patients with end-stage liver disease characteristically have a vasodilated circulation that can mimic sepsis and septic shock. Therefore it is reasonable to hypothesize that there could be a vasopressin deficiency in such patients. Wagener and colleagues evaluated a cohort of such patients by measuring plasma vasopressin levels and responses to infusion of vasopressin [136]. Plasma vasopressin levels were inappropriately low in patients who had end- stage hepatic disease undergoing liver transplantation compared with control surgical patients. Vasopressin increased systemic vascular resistance and arterial pressure. Patients who have end-stage hepatic disease thus appear to have vasopressin deficiency and may benefit from vasopressin infusion (once fluid resuscitation is adequate).
These observations of potential benefits of vasopressin in end-stage liver disease are congruent with the studies showing benefits of terlipressin in patients who have severe liver disease and renal dysfunction - hepatorenal syndrome.
More selective V1a agonism in septic shock: another approach
As discussed, there could be advantages to more selective V1a agonism treatment in septic shock such as purer vasoconstriction, limited antidiuretic effects, lack of stimulation of thrombosis (because of lower increase of von Willebrand multimers) and perhaps better protection from increased permeability. Rehberg and colleagues designed a clever study in which they gave a V2 receptor antagonist (propionyl-1-d-(Tyr(Et)2-Val4-Abu6-Arg8,9)-vasopressin) with or without a V1a agonist (arginine vasopressin) or placebo to chronically instrumented sheep who had early septic shock [137]. This V2 receptor antagonist with arginine vasopressin maintained hemodynamics (cardiac index and MAP) and increased the left ventricular stroke work index. Most notably, the combination of the V2 receptor antagonist with arginine vasopressin (compared with arginine vasopressin alone) improved markers of tissue perfusion (for example, pH and arterial lactate), while also attenuating liver and renal dysfunction. Markers of lung injury (hemeoxygenase and 3-nitrotyrosine concentrations) were also more favorably protected in the animals that received this combination of V2 receptor antagonist with arginine vasopressin compared with arginine vasopressin alone. Also intriguing is the observation that the administration of arginine vasopressin alone increased plasma vasopressin levels, whereas the combination of this V2 receptor antagonist with arginine vasopressin increased plasma vasopressin levels to a lesser degree.
This innovative animal model study provides further evidence to support further clinical investigation into the role of selective V1a agonism in human septic shock.
Clinical perspectives
What should the clinician do in considering use of vasopressin in patients who have septic shock? My current recommendations are to consider use of vasopressin in patients who have less severe septic shock because of the encouraging effects of vasopressin in these patients in VASST [52].
Should vasopressin levels be measured in patients who have septic shock? One obstacle is that vasopressin can only be measured by radioimmunoassay, which is relatively cumbersome and has too great a turnaround time for patients who have septic shock. I would therefore not recommend measurement of vasopressin levels (or copeptin levels, as already discussed on page 6 above) at this time for clinical purposes in patients who have septic shock. I would recommend that vasopressin be given as a continuous infusion not exceeding 0.03 units/minute, because that was the dose used in VASST.
Should vasopressin be used as first-line therapy in hypotensive septic patients? I believe this is a useful question for clinical research but not yet for clinical practice.
Should vasopressin and corticosteroids be given together? The potentially beneficial interaction of vasopressin infusion and corticosteroid treatment on mortality was discovered in a post hoc substudy of VASST and could be a false positive, and so is hypothesis-generating at this time. The Surviving Sepsis Guidelines [85] state that norepinephrine and vasopressin are similar and that vasopressin should be considered in patients who have septic shock.
Future research
Some themes for future research could include assessment of vasopressin as the first-line agent in septic shock, evaluation of the renal effects of vasopressin, further assessment of adverse effects of vasopressin versus norepinephrine, and use of the more specific AVPR1a agonist. Preliminary reports suggest that an AVPR1a agonist causes less third-spacing of fluid (vascular leak syndrome), could reduce release of the procoagulant protein von Willebrand factor (released by stimulation of AVPR2), and decreases mortality compared with vasopressin in two different models of septic shock.
Conclusions
Vasopressin stimulates a family of receptors (AVPR1a, AVPR1b, and AVPR2). AVPR1a is responsible for the vasoconstriction associated with vasopressin. Sepsis may downregulate all three vasopressin receptors. Vasopressin levels increase early in septic shock because hypotension is the most potent stimulus for release of stored vasopressin. Vasopressin levels then decline rapidly - due to depletion of stored vasopressin.
Responses to vasopressin and norepinephrine are modified in sepsis and, furthermore, the effects of vasopressin differ in different vascular beds in sepsis. Vasopressin infusion increases blood pressure, decreases norepinephrine dose requirements, and improves renal function. Previous clinical studies of vasopressin in septic shock, however, were small or not controlled. Adverse effects of vasopressin include mesenteric ischemia, myocardial ischemia, digital ischemia, skin necrosis, and hyponatremia.
VASST, a randomized controlled trial of vasopressin versus norepinephrine in septic shock, was performed to address uncertainties regarding efficacy and safety of vasopressin in septic shock. There was no significant difference between vasopressin-treated and norepinephrine-treated groups in 28-day mortality (35.4% versus 39.3%, P = 0.26; ARR = -2.9 to 10.7%) or in organ dysfunction rates. Vasopressin may have decreased mortality in patients who had less severe septic shock in VASST. Serious adverse event rates were similar in the vasopressin and norepinephrine groups.
Vasopressin therefore appears equally as safe as norepinephrine in septic shock, and may improve mortality in less severe septic shock. There have been remarkably few large studies of interactions of the commonly used therapies in critical care and the effects on mortality. Low-dose vasopressin infusion plus corticosteroids significantly decreased 28-day mortality compared with corticosteroids plus norepinephrine (44.7 to 35.9%, P = 0.03; P = 0.008 interaction statistic). This interaction is potentially important and requires further study.
Summary
Low-dose vasopressin may be effective in patients already receiving norepinephrine who have less severe septic shock - such as patients with modest norepinephrine infusion (5 to 15 μg/minute) or low serum lactate levels. Additional studies such as large randomized controlled trials are necessary to address questions regarding higher doses of vasopressin, use of vasopressin as the first agent in septic shock, use of terlipressin and other more specific AVPR1a agonists, and the interaction of vasopressin and corticosteroids in septic shock.
Abbreviations
- ACTH:
-
adrenocorticotropic hormone
- ARR:
-
absolute risk reduction
- IFN:
-
interferon
- IL:
-
interleukin
- MAP:
-
mean arterial pressure
- TNF:
-
tumor necrosis factor
- VASST:
-
Vasopressin and Septic Shock Trial.
References
Thibonnier M, Auzan C, Madhun Z, Wilkins P, Berti-Mattera L, Clauser E: Molecular cloning, sequencing, and functional expression of a cDNA encoding the human V1a vasopressin receptor. J Biol Chem. 1994, 269: 3304-3310.
Thibonnier M, Graves MK, Wagner MS, Chatelain N, Soubrier F, Corvol P, Willard HF, Jeunemaitre X: Study of V(1)-vascular vasopressin receptor gene microsatellite polymorphisms in human essential hypertension. J Mol Cell Cardiol. 2000, 324: 557-564.
Berrada K, Plesnicher CL, Luo X, Thibonnier M: Dynamic interaction of human vasopressin/oxytocin receptor subtypes with G protein-coupled receptor kinases and protein kinase C after agonist stimulation. J Biol Chem. 2000, 275: 27229-27237.
Okamura T, Ayajiki K, Fujioka H, Toda N: Mechanisms underlying arginine vasopressin-induced relaxation in monkey isolated coronary arteries. J Hypertens. 1999, 17: 673-678. 10.1097/00004872-199917050-00011.
Evora PR, Pearson PJ, Schaff HV: Arginine vasopressin induces endotheliumdependent vasodilatation of the pulmonary artery. V1-receptor-mediated production of nitric oxide. Chest. 1993, 103: 1241-1245. 10.1378/chest.103.4.1241.
Russ RD, Walker BR: Role of nitric oxide in vasopressinergic pulmonary vasodilatation. Am J Physiol. 1992, 262 (3 Pt 2): H743-H747.
Sai Y, Okamura T, Amakata Y, Toda N: Comparison of responses of canine pulmonary artery and vein to angiotensin II, bradykinin and vasopressin. Eur J Pharmacol. 1995, 282: 235-241. 10.1016/0014-2999(95)00343-J.
Kim SJ, Young LJ, Gonen D, Veenstra-VanderWeele J, Courchesne R, Courchesne E, Lord C, Leventhal BL, Cook EH, Insel TR: Transmission disequilibrium testing of arginine vasopressin receptor 1A (AVPR1A) polymorphisms in autism. Mol Psychiatry. 2002, 7: 503-507. 10.1038/sj.mp.4001125.
Knafo A, Israel S, Darvasi A, Bachner-Melman R, Uzefovsky F, Cohen L, Feldman E, Lerer E, Laiba E, Raz Y, Nemanov L, Gritsenko I, Dina C: Individual differences in allocation of funds in the dictator game associated with length of the arginine vasopressin 1a receptor RS3 promoter region and correlation between RS3 length and hippocampal mRNA. Genes Brain Behav. 2008, 7: 266-275. 10.1111/j.1601-183X.2007.00341.x.
Bucher M, Hobbhahn J, Taeger K, Kurtz A: Cytokine-mediated downregulation of vasopressin V(1A) receptors during acute endotoxemia in rats. Am J Physiol Regul Integr Comp Physiol. 2002, 282: R979-R984.
Roth BL, Spitzer JA: Altered hepatic vasopressin and alpha 1-adrenergic receptors after chronic endotoxin infusion. Am J Physiol. 1987, 252 (5 Pt 1): E699-E702.
Barrett LK, Orie NN, Taylor V, Stidwill RP, Clapp LH, Singer M: Differential effects of vasopressin and norepinephrine on vascular reactivity in a long-term rodent model of sepsis. Crit Care Med. 2007, 35: 2337-2343. 10.1097/01.CCM.0000281861.72907.17.
Antoni FA, Holmes MC, Makara GB, Karteszi M, Laszlo FA: Evidence that the effects of arginine-8-vasopressin (AVP) on pituitary corticotropin (ACTH) release are mediated by a novel type of receptor. Peptides. 1984, 5: 519-522. 10.1016/0196-9781(84)90080-9.
Hernando F, Schoots O, Lolait SJ, Burbach JP: Immunohistochemical localization of the vasopressin V1b receptor in the rat brain and pituitary gland: anatomical support for its involvement in the central effects of vasopressin. Endocrinology. 2001, 142: 1659-1668. 10.1210/en.142.4.1659.
Lolait SJ, Stewart LQ, Jessop DS, Young WS, O'Carroll AM: The hypothalamic-pituitary-adrenal axis response to stress in mice lacking functional vasopressin V1b receptors. Endocrinology. 2007, 148: 849-856.
de Keyzer Y, Lenne F, Auzan C, Jegou S, Rene P, Vaudry H, Kuhn JM, Luton JP, Clauser E, Bertagna X: The pituitary V3 vasopressin receptor and the corticotroph phenotype in ectopic ACTH syndrome. J Clin Invest. 1996, 97: 1311-1318. 10.1172/JCI118547.
O'Connor PM, Cowley AW: Vasopressin-induced nitric oxide production in rat inner medullary collecting duct is dependent on V2 receptor activation of the phosphoinositide pathway. Am J Physiol Renal Physiol. 2007, 293: F526-F532. 10.1152/ajprenal.00052.2007.
Park F, Zou AP, Cowley AW: Arginine vasopressin-mediated stimulation of nitric oxide within the rat renal medulla. Hypertension. 1998, 32: 896-901.
Martin K, Borgel D, Lerolle N, Feys HB, Trinquart L, Vanhoorelbeke K, Deckmyn H, Legendre P, Diehl JL, Baruch D: Decreased ADAMTS-13 (a disintegrin-like and metalloprotease with thrombospondin type 1 repeats) is associated with a poor prognosis in sepsis-induced organ failure. Crit Care Med. 2007, 35: 2375-2382. 10.1097/01.CCM.0000284508.05247.B3.
Ono T, Mimuro J, Madoiwa S, Soejima K, Kashiwakura Y, Ishiwata A, Takano K, Ohmori T, Sakata Y: Severe secondary deficiency of von Willebrand factorcleaving protease (ADAMTS13) in patients with sepsis-induced disseminated intravascular coagulation: its correlation with development of renal failure. Blood. 2006, 107: 528-534. 10.1182/blood-2005-03-1087.
Schmidt C, Hocherl K, Kurt B, Bucher M: Role of nuclear factor-κBdependent induction of cytokines in the regulation of vasopressin V1A-receptors during cecal ligation and puncture-induced circulatory failure. Crit Care Med. 2008, 36: 2363-2372. 10.1097/CCM.0b013e318180b51d.
Grinevich V, Knepper MA, Verbalis J, Reyes I, Aguilera G: Acute endotoxemia in rats induces down-regulation of V2 vasopressin receptors and aquaporin-2 content in the kidney medulla. Kidney Int. 2004, 65: 54-62. 10.1111/j.1523-1755.2004.00378.x.
Qahwash IM, Cassar CA, Radcliff RP, Smith GW: Bacterial lipopolysaccharideinduced coordinate downregulation of arginine vasopressin receptor V3 and corticotropin-releasing factor receptor 1 messenger ribonucleic acids in the anterior pituitary of endotoxemic steers. Endocrine. 2002, 18: 13-20. 10.1385/ENDO:18:1:13.
Thibonnier M, Conarty DM, Preston JA, Plesnicher CL, Dweik RA, Erzurum SC: Human vascular endothelial cells express oxytocin receptors. Endocrinology. 1999, 140: 1301-1309. 10.1210/en.140.3.1301.
Gutkowska J, Jankowski M, Mukaddam-Daher S, McCann SM: Oxytocin is a cardiovascular hormone. Braz J Med Biol Res. 2000, 33: 625-633.
Indrambarya T, Boyd JH, Wang Y, McConechy M, Walley KR: Low-dose vasopressin infusion results in increased mortality and cardiac dysfunction following ischemia-reperfusion injury in mice. Crit Care. 2009, 13: R98-10.1186/cc7930.
Mori M, Tsushima H, Matsuda T: Antidiuretic effects of ATP induced by microinjection into the hypothalamic supraoptic nucleus in water-loaded and ethanol-anesthetized rats. Jpn J Pharmacol. 1994, 66: 445-450. 10.1254/jjp.66.445.
Guo W, Sun J, Xu X, Bunstock G, He C, Xiang Z: P2X receptors are differentially expressed on vasopressin- and oxytocin-containing neurons in the supraoptic and paraventricular nuclei of rat hypothalamus. Histochem Cell Biol. 2009, 131: 29-41. 10.1007/s00418-008-0493-9.
Knott TK, Velazquez-Marrero C, Lemos JR: ATP elicits inward currents in isolated vasopressinergic neurohypophysial terminals via P2X2 and P2X3 receptors. Pflugers Arch. 2005, 450: 381-389. 10.1007/s00424-005-1471-x.
Knott TK, Marrero HG, Custer EE, Lemos JR: Endogenous ATP potentiates only vasopressin secretion from neurohypophysial terminals. J Cell Physiol. 2008, 217: 155-161. 10.1002/jcp.21485.
Song Z, Vijayaraghavan S, Sladek CD: ATP increases intracellular calcium in supraoptic neurons by activation of both P2X and P2Y purinergic receptors. Am J Physiol Regul Integr Comp Physiol. 2007, 292: R423-R431.
Song Z, Gomes DA, Stevens W: Role of purinergic P2Y1 receptors in regulation of vasopressin and oxytocin secretion. Am J Physiol Regul Integr Comp Physiol. 2009, 297: R478-R484. 10.1152/ajpregu.00163.2009.
Troadec JD, Thirion S, Nicaise G, Lemos JR, Dayanithi G: ATP-evoked increases in [Ca2+]i and peptide release from rat isolated neurohypophysial terminals via a P2X2 purinoceptor. J Physiol. 1998, 511 (Pt 1): 89-103.
Buller KM, Khanna S, Sibbald JR, Day TA: Central noradrenergic neurons signal via ATP to elicit vasopressin responses to haemorrhage. Neuroscience. 1996, 73: 637-642. 10.1016/0306-4522(96)00156-X.
Sperlagh B, Mergl Z, Juranyi Z, Vizi ES, Makara GB: Local regulation of vasopressin and oxytocin secretion by extracellular ATP in the isolated posterior lobe of the rat hypophysis. J Endocrinol. 1999, 160: 343-350. 10.1677/joe.0.1600343.
Kapoor JR, Sladek CD: Purinergic and adrenergic agonists synergize in stimulating vasopressin and oxytocin release. J Neurosci. 2000, 20: 8868-8875.
Troadec JD, Thirion S: Multifaceted purinergic regulation of stimulussecretion coupling in the neurohypophysis. Neuro Endocrinol Lett. 2002, 23: 273-280.
Xiang Z, He C, Burnstock G: P2X5 receptors are expressed on neurons containing arginine vasopressin and nitric oxide synthase in the rat hypothalamus. Brain Res. 2006, 1099: 56-63. 10.1016/j.brainres.2006.04.126.
Sladek CD, Song Z: Regulation of vasopressin release by co-released neurotransmitters: mechanisms of purinergic and adrenergic synergism. Prog Brain Res. 2008, 170: 93-107.
Gomes DA, Song Z, Stevens W, Sladek CD: Sustained stimulation of vasopressin and oxytocin release by ATP and phenylephrine requires recruitment of desensitization-resistant P2X purinergic receptors. Am J Physiol Regul Integr Comp Physiol. 2009, 297: R940-R949. 10.1152/ajpregu.00358.2009.
Espallergues J, Solovieva O, Techer V, Bauer K, Alonso G, Vincent A, Hussy N: Synergistic activation of astrocytes by ATP and norepinephrine in the rat supraoptic nucleus. Neuroscience. 2007, 148: 712-723. 10.1016/j.neuroscience.2007.03.043.
Song Z, Sladek CD: Does conversion of ATP to adenosine terminate ATP-stimulated vasopressin release from hypothalamo-neurohypophyseal explants?. Brain Res. 2005, 1047: 105-111. 10.1016/j.brainres.2005.04.025.
Cha SH, Sekine T, Endou H: P2 purinoceptor localization along rat nephron and evidence suggesting existence of subtypes P2Y1 and P2Y2. Am J Physiol. 1998, 274 (6 Pt 2): F1006-F1014.
Wildman SS, Boone M, Peppiatt-Wildman CM, Contreras-Sanz A, King BF, Shirley DG, Deen PM, Unwin RJ: Nucleotides downregulate aquaporin 2 via activation of apical P2 receptors. J Am Soc Nephrol. 2009, 20: 1480-1490. 10.1681/ASN.2008070686.
Rieg T, Bundey RA, Chen Y, Deschenes G, Junger W, Insel PA, Vallon V: Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB J. 2007, 21: 3717-3726. 10.1096/fj.07-8807com.
Zenteno-Savin T, Sada-Ovalle I, Ceballos G, Rubio R: Effects of arginine vasopressin in the heart are mediated by specific intravascular endothelial receptors. Eur J Pharmacol. 2000, 410: 15-23. 10.1016/S0014-2999(00)00853-0.
Wilson MF, Brackett DJ, Tompkins P, Benjamin B, Archer LT, Hinshaw LB: Elevated plasma vasopressin concentrations during endotoxin and E. coli shock. Adv Shock Res. 1981, 6: 15-26.
Landry DW, Levin HR, Gallant EM, Ashton RC, Seo S, D'Alessandro D, Oz MC, Oliver JA: Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation. 1997, 95: 1122-1125.
Landry DW, Levin HR, Gallant EM, Seo S, D'Alessandro D, Oz MC, Oliver JA: Vasopressin pressor hypersensitivity in vasodilatory septic shock. Crit Care Med. 1997, 25: 1279-1282. 10.1097/00003246-199708000-00012.
Sharshar T, Blanchard A, Paillard M, Raphael JC, Gajdos P, Annane D: Circulating vasopressin levels in septic shock. Crit Care Med. 2003, 31: 1752-1758. 10.1097/01.CCM.0000063046.82359.4A.
Sharshar T, Carlier R, Blanchard A, Feydy A, Gray F, Paillard M, Raphael JC, Gajdos P, Annane D: Depletion of neurohypophyseal content of vasopressin in septic shock. Crit Care Med. 2002, 30: 497-500. 10.1097/00003246-200203000-00001.
Russell JA, Walley KR, Singer J, Gordon AC, Hebert PC, Cooper DJ, Holmes CL, Mehta S, Granton JT, Storms MM, Cook DJ, Presneill JJ, Ayers D: Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008, 358: 877-887. 10.1056/NEJMoa067373.
Jochberger S, Dorler J, Luckner G, Mayr VD, Wenzel V, Ulmer H, Morgenthaler NG, Hasibeder WR, Dunser MW: The vasopressin and copeptin response to infection, severe sepsis, and septic shock. Crit Care Med. 2009, 37: 476-482. 10.1097/CCM.0b013e3181957532.
Carnio EC, Moreto V, Giusti-Paiva A, Antunes-Rodrigues J: Neuro-immuneendocrine mechanisms during septic shock: role for nitric oxide in vasopressin and oxytocin release. Endocr Metab Immune Disord Drug Targets. 2006, 6: 137-142.
Correa PB, Pancoto JA, de Oliveira-Pelegrin GR, Carnio EC, Rocha MJ: Participation of iNOS-derived NO in hypothalamic activation and vasopressin release during polymicrobial sepsis. J Neuroimmunol. 2007, 183: 17-25. 10.1016/j.jneuroim.2006.10.021.
Moreto V, Stabile AM, Antunes-Rodrigues J, Carnio EC: Role of hemeoxygenase pathway on vasopressin deficiency during endotoxemic shock-like conditions. Shock. 2006, 26: 472-476. 10.1097/01.shk.0000230301.86139.6a.
Morgenthaler NG, Muller B, Struck J, Bergmann A, Redl H, Christ-Crain M: Copeptin, a stable peptide of the arginine vasopressin precursor, is elevated in hemorrhagic and septic shock. Shock. 2007, 28: 219-226. 10.1097/SHK.0b013e318033e5da.
Muller B, Morgenthaler N, Stolz D, Schuetz P, Muller C, Bingisser R, Bergmann A, Tamm M, Christ-Crain M: Circulating levels of copeptin, a novel biomarker, in lower respiratory tract infections. Eur J Clin Invest. 2007, 37: 145-152. 10.1111/j.1365-2362.2007.01762.x.
Jochberger S, Morgenthaler NG, Mayr VD, Luckner G, Wenzel V, Ulmer H, Schwarz S, Hasibeder WR, Friesenecker BE, Dunser MW: Copeptin and arginine vasopressin concentrations in critically ill patients. J Clin Endocrinol Metab. 2006, 91: 4381-4386. 10.1210/jc.2005-2830.
Jochberger S, Mayr VD, Luckner G, Wenzel V, Ulmer H, Schmid S, Knotzer H, Pajk W, Hasibeder W, Friesenecker B, Mayr AG, Dunser MW: Serum vasopressin concentrations in critically ill patients. Crit Care Med. 2006, 34: 293-299. 10.1097/01.CCM.0000198528.56397.4F.
Lin IY, Ma HP, Lin AC, Chong CF, Lin CM, Wang TL: Low plasma vasopressin/norepinephrine ratio predicts septic shock. Am J Emerg Med. 2005, 23: 718-724. 10.1016/j.ajem.2005.02.055.
Luckner G, Dunser MW, Jochberger S, Mayr VD, Wenzel V, Ulmer H, Schmid S, Knotzer H, Pajk W, Hasibeder W, Mayr AG, Friesenecker B: Arginine vasopressin in 316 patients with advanced vasodilatory shock. Crit Care Med. 2005, 33: 2659-2666. 10.1097/01.CCM.0000186749.34028.40.
Luckner G, Mayr VD, Jochberger S, Wenzel V, Ulmer H, Hasibeder WR, Dunser MW: Comparison of two dose regimens of arginine vasopressin in advanced vasodilatory shock. Crit Care Med. 2007, 35: 2280-2285. 10.1097/01.CCM.0000281853.50661.23.
Lodha R, Vivekanandhan S, Sarthi M, Kabra SK: Serial circulating vasopressin levels in children with septic shock. Pediatr Crit Care Med. 2006, 7: 220-224. 10.1097/01.PCC.0000216414.00362.81.
Scharte M, Meyer J, Van Aken H, Bone HG: Hemodynamic effects of terlipressin (a synthetic analog of vasopressin) in healthy and endotoxemic sheep. Crit Care Med. 2001, 29: 1756-1760. 10.1097/00003246-200109000-00017.
Nakajima Y, Baudry N, Duranteau J, Vicaut E: Effects of vasopressin, norepinephrine, and L-arginine on intestinal microcirculation in endotoxemia. Crit Care Med. 2006, 34: 1752-1757. 10.1097/01.CCM.0000218812.73741.6C.
Knotzer H, Maier S, Dunser MW, Hasibeder WR, Hausdorfer H, Brandner J, Torgersen C, Ulmer H, Friesenecker B, Iannetti C, Pajik W: Arginine vasopressin does not alter mucosal tissue oxygen tension and oxygen supply in an acute endotoxemic pig model. Intensive Care Med. 2006, 32: 170-174. 10.1007/s00134-005-2858-z.
Leone M, Boyle WA: Decreased vasopressin responsiveness in vasodilatory septic shock-like conditions. Crit Care Med. 2006, 34: 1126-1130. 10.1097/01.CCM.0000206466.56669.BE.
Kang CH, Kim WG: The effect of vasopressin on organ blood flow in an endotoxin-induced rabbit shock model. J Invest Surg. 2006, 19: 361-369. 10.1080/08941930600985702.
Dubois MJ, De Backer D, Creteur J, Anane S, Vincent JL: Effect of vasopressin on sublingual microcirculation in a patient with distributive shock. Intensive Care Med. 2003, 29: 1020-1023.
Kienbaum P, Prante C, Lehmann N, Sander A, Jalowy A, Peters J: Alterations in forearm vascular reactivity in patients with septic shock. Anaesthesia. 2008, 63: 121-128. 10.1111/j.1365-2044.2007.05286.x.
Russell JA: Vasopressin in vasodilatory and septic shock. Curr Opin Crit Care. 2007, 13: 383-391. 10.1097/MCC.0b013e328263885e.
Dunser MW, Mayr AJ, Ulmer H, Ritsch N, Knotzer H, Pajk W, Luckner G, Mutz NJ, Hasibeder WR: The effects of vasopressin on systemic hemodynamics in catecholamine-resistant septic and postcardiotomy shock: a retrospective analysis. Anesth Analg. 2001, 93: 7-13.
Dunser MW, Mayr AJ, Stallinger A, Ulmer H, Ritsch N, Knotzer H, Pajk W, Mutz NJ, Hasibeder WR: Cardiac performance during vasopressin infusion in postcardiotomy shock. Intensive Care Med. 2002, 28: 746-751. 10.1007/s00134-002-1265-y.
Dunser MW, Mayr AJ, Ulmer H, Knotzer H, Sumann G, Pajk W, Friesenecker B, Hasibeder WR: Arginine vasopressin in advanced vasodilatory shock: a prospective, randomized, controlled study. Circulation. 2003, 107: 2313-2319. 10.1161/01.CIR.0000066692.71008.BB.
Holmes CL, Walley KR, Chittock DR, Lehman T, Russell JA: The effects of vasopressin on hemodynamics and renal function in severe septic shock: a case series. Intensive Care Med. 2001, 27: 1416-1421. 10.1007/s001340101014.
Malay MB, Ashton RC, Landry DW, Townsend RN: Low-dose vasopressin in the treatment of vasodilatory septic shock. J Trauma. 1999, 47: 699-703. 10.1097/00005373-199910000-00014.
Patel BM, Chittock DR, Russell JA, Walley KR: Beneficial effects of short-term vasopressin infusion during severe septic shock. Anesthesiology. 2002, 96: 576-582. 10.1097/00000542-200203000-00011.
Hiltebrand LB, Krejci V, Jakob SM, Takala J, Sigurdsson GH: Effects of vasopressin on microcirculatory blood flow in the gastrointestinal tract in anesthetized pigs in septic shock. Anesthesiology. 2007, 106: 1156-1167. 10.1097/01.anes.0000267599.02140.86.
Dunser MW, Mayr AJ, Tur A, Pajk W, Barbara F, Knotzer H, Ulmer H, Hasibeder WR: Ischemic skin lesions as a complication of continuous vasopressin infusion in catecholamine-resistant vasodilatory shock: incidence and risk factors. Crit Care Med. 2003, 31: 1394-1398. 10.1097/01.CCM.0000059722.94182.79.
Aninat C, Seguin P, Descheemaeker PN, Morel F, Malledant Y, Guillouzo A: Catecholamines induce an inflammatory response in human hepatocytes. Crit Care Med. 2008, 36: 848-854. 10.1097/CCM.0B013E31816532BE.
American College of Chest Physicians/Society of Critical Care Medicine: Consensus conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992, 20: 864-874. 10.1097/00003246-199206000-00025.
Bone RC, Sibbald WJ, Sprung CL: The ACCP-SCCM consensus conference on sepsis and organ failure. Chest. 1992, 101: 1481-1483. 10.1378/chest.101.6.1481.
Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ: Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992, 101: 1644-1655. 10.1378/chest.101.6.1644.
Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, Reinhart K, Angus DC, Brun-Buisson C, Beale R, Calandra T, Dhainaut JF, Gerlach H: Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008, 36: 296-327. 10.1097/01.CCM.0000298158.12101.41.
Sprung CL, Annane D, Keh D, Moreno R, Singer M, Freivogel K, Weiss YG, Benbenishty J, Kalenka A, Forst H, Laterre PF, Reinhart K, Cuthbertson BH: Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008, 358: 111-124. 10.1056/NEJMoa071366.
Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, Peterson E, Tomlanovich M: Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001, 345: 1368-1377. 10.1056/NEJMoa010307.
Hall LG, Oyen LJ, Taner CB, Cullinane DC, Baird TK, Cha SS, Sawyer MD: Fixeddose vasopressin compared with titrated dopamine and norepinephrine as initial vasopressor therapy for septic shock. Pharmacotherapy. 2004, 24: 1002-1012. 10.1592/phco.24.11.1002.36139.
Russell JA, Walley KR, Gordon AC, Cooper DJ, Hebert PC, Singer J, Holmes CL, Mehta S, Granton JT, Storms MM, Cook DJ, Presneill JJ: Interaction of vasopressin infusion, corticosteroid treatment, and mortality of septic shock. Crit Care Med. 2009, 37: 811-818. 10.1097/CCM.0b013e3181961ace.
Rabadan-Diehl C, Aguilera G: Glucocorticoids increase vasopressin V1b receptor coupling to phospholipase C. Endocrinology. 1998, 139: 3220-3226. 10.1210/en.139.7.3220.
Ertmer C, Bone HG, Morelli A, Van Aken H, Erren M, Lange M, Traber DL, Westphal M: Methylprednisolone reverses vasopressin hyporesponsiveness in ovine endotoxemia. Shock. 2007, 27: 281-288. 10.1097/01.shk.0000235140.97903.90.
Lauand F, Ruginsk SG, Rodrigues HL, Reis WL, de Castro M, Elias LL, Antunes-Rodrigues J: Glucocorticoid modulation of atrial natriuretic peptide, oxytocin, vasopressin and Fos expression in response to osmotic, angiotensinergic and cholinergic stimulation. Neuroscience. 2007, 147: 247-257. 10.1016/j.neuroscience.2007.04.021.
Pietranera L, Saravia F, Roig P, Lima A, De Nicola AF: Mineralocorticoid treatment upregulates the hypothalamic vasopressinergic system of spontaneously hypertensive rats. Neuroendocrinology. 2004, 80: 100-110. 10.1159/000081314.
Aguilera G, Rabadan-Diehl C: Vasopressinergic regulation of the hypothalamic-pituitary-adrenal axis: implications for stress adaptation. Regul Pept. 2000, 96: 23-29. 10.1016/S0167-0115(00)00196-8.
Tanoue A, Ito S, Honda K, Oshikawa S, Kitagawa Y, Koshimizu TA, Mori T, Tsujimoto G: The vasopressin V1b receptor critically regulates hypothalamic-pituitary-adrenal axis activity under both stress and resting conditions. J Clin Invest. 2004, 113: 302-309.
Annane D: Vasopressin plus corticosteroids: the shock duo!. Crit Care Med. 2009, 37: 1126-1127. 10.1097/CCM.0b013e31819879c1.
Dunser MW, Hasibeder WR, Wenzel V: Vasopressin in septic shock. N Engl J Med. 2008, 358: 2736-author reply 7-8
Westphal M, Freise H, Kehrel BE, Bone HG, Van Aken H, Sielenkamper AW: Arginine vasopressin compromises gut mucosal microcirculation in septic rats. Crit Care Med. 2004, 32: 194-200. 10.1097/01.CCM.0000104201.62736.12.
Martikainen TJ, Tenhunen JJ, Uusaro A, Ruokonen E: The effects of vasopressin on systemic and splanchnic hemodynamics and metabolism in endotoxin shock. Anesth Analg. 2003, 97: 1756-1763. 10.1213/01.ANE.0000087039.60041.2E.
Klinzing S, Simon M, Reinhart K, Bredle DL, Meier-Hellmann A: High-dose vasopressin is not superior to norepinephrine in septic shock. Crit Care Med. 2003, 31: 2646-2650. 10.1097/01.CCM.0000094260.05266.F4.
Leone M, Albanese J, Delmas A, Chaabane W, Garnier F, Martin C: Terlipressin in catecholamine-resistant septic shock patients. Shock. 2004, 22: 314-319. 10.1097/01.shk.0000136097.42048.bd.
Leone M, Boutiere-Albanese B, Valette S, Camoin-Jau L, Barrau K, Albanese J, Martin C, Dignat-George F: Cell adhesion molecules as a marker reflecting the reduction of endothelial activation induced by glucocorticoids. Shock. 2004, 21: 311-314. 10.1097/00024382-200404000-00004.
O'Brien A, Clapp L, Singer M: Terlipressin for norepinephrine-resistant septic shock. Lancet. 2002, 359: 1209-1210. 10.1016/S0140-6736(02)08225-9.
Albanese J, Leone M, Delmas A, Martin C: Terlipressin or norepinephrine in hyperdynamic septic shock: a prospective, randomized study. Crit Care Med. 2005, 33: 1897-1902. 10.1097/01.CCM.0000178182.37639.D6.
Asfar P, Pierrot M, Veal N, Moal F, Oberti F, Croquet V, Douay O, Gallois Y, Saumet JL, Alquier P, Cales P: Low-dose terlipressin improves systemic and splanchnic hemodynamics in fluid-challenged endotoxic rats. Crit Care Med. 2003, 31: 215-220. 10.1097/00003246-200301000-00033.
Asfar P, Hauser B, Ivanyi Z, Ehrmann U, Kick J, Albicini M, Vogt J, Wachter U, Bruckner UB, Radermacher P, Bracht H: Low-dose terlipressin during longterm hyperdynamic porcine endotoxemia: effects on hepatosplanchnic perfusion, oxygen exchange, and metabolism. Crit Care Med. 2005, 33: 373-380. 10.1097/01.CCM.0000152253.45901.FB.
Morelli A, Rocco M, Conti G, Orecchioni A, De Gaetano A, Cortese G, Coluzzi F, Vernaglione E, Pelaia P, Pietropaoli P: Effects of terlipressin on systemic and regional haemodynamics in catecholamine-treated hyperkinetic septic shock. Intensive Care Med. 2004, 30: 597-604. 10.1007/s00134-003-2094-3.
Westphal M, Stubbe H, Sielenkamper AW, Ball C, Van Aken H, Borgulya R, Bone HG: Effects of titrated arginine vasopressin on hemodynamic variables and oxygen transport in healthy and endotoxemic sheep. Crit Care Med. 2003, 31: 1502-1508. 10.1097/01.CCM.0000063042.15272.84.
Westphal M, Stubbe H, Sielenkamper AW, Borgulya R, Van Aken H, Ball C, Bone HG: Terlipressin dose response in healthy and endotoxemic sheep: impact on cardiopulmonary performance and global oxygen transport. Intensive Care Med. 2003, 29: 301-308.
Pesaturo AB, Jennings HR, Voils SA: Terlipressin: vasopressin analog and novel drug for septic shock. Ann Pharmacother. 2006, 40: 2170-2177. 10.1345/aph.1H373.
Morelli A, Ertmer C, Lange M, Dunser M, Rehberg S, Van Aken H, Pietropaoli P, Westphal M: Effects of short-term simultaneous infusion of dobutamine and terlipressin in patients with septic shock: the DOBUPRESS study. Br J Anaesth. 2008, 100: 494-503. 10.1093/bja/aen017.
Lange M, Ertmer C, Westphal M: Vasopressin vs. terlipressin in the treatment of cardiovascular failure in sepsis. Intensive Care Med. 2008, 34: 821-832. 10.1007/s00134-007-0946-y.
Bernadich C, Bandi JC, Melin P, Bosch J: Effects of F-180, a new selective vasoconstrictor peptide, compared with terlipressin and vasopressin on systemic and splanchnic hemodynamics in a rat model of portal hypertension. Hepatology. 1998, 27: 351-356. 10.1002/hep.510270206.
Forsling ML, Aziz LA, Miller M, Davies R, Donovan B: Conversion of triglycylvasopressin to lysine-vasopressin in man. J Endocrinol. 1980, 85: 237-244. 10.1677/joe.0.0850237.
Fabian M, Forsling ML, Jones JJ, Pryor JS: The clearance and antidiuretic potency of neurohypophysial hormones in man, and their plasma binding and stability. J Physiol. 1969, 204: 653-668.
Nilsson G, Lindblom P, Ohlin M, Berling R, Vernersson E: Pharmacokinetics of terlipressin after single i.v. doses to healthy volunteers. Drugs Exp Clin Res. 1990, 16: 307-314.
Klein M, Weksler N, Borer A, Koyfman L, Kesslin J, Gurman GM: Terlipressin facilitates transport of septic patients treated with norepinephrine. Isr Med Assoc J. 2006, 8: 691-693.
Solanki P, Chawla A, Garg R, Gupta R, Jain M, Sarin SK: Beneficial effects of terlipressin in hepatorenal syndrome: a prospective, randomized placebocontrolled clinical trial. J Gastroenterol Hepatol. 2003, 18: 152-156. 10.1046/j.1440-1746.2003.02934.x.
Sanyal AJ, Boyer T, Garcia-Tsao G, Regenstein F, Rossaro L, Appenrodt B, Blei A, Gulberg V, Sigal S, Teuber P: A randomized, prospective, double-blind, placebo-controlled trial of terlipressin for type 1 hepatorenal syndrome. Gastroenterology. 2008, 134: 1360-1368. 10.1053/j.gastro.2008.02.014.
Martin-Llahi M, Pepin MN, Guevara M, Diaz F, Torre A, Monescillo A, Soriano G, Terra C, Fabrega E, Arroyo V, Rhodes J, Gines P: Terlipressin and albumin vs albumin in patients with cirrhosis and hepatorenal syndrome: a randomized study. Gastroenterology. 2008, 134: 1352-1359. 10.1053/j.gastro.2008.02.024.
Gordon AC, Russell JA, Walley KR, Singer J, Ayers D, Storms MM, Holmes CL, Hebert PC, Cooper DJ, Mehta S, Granton JT, Cook DJ, Presneill JJ: The effects of vasopressin on acute kidney injury in septic shock. Intensive Care Medicine. 2009, 36: 83-91.
Morelli A, Ertmer C, Rehberg S, Lange M, Orecchioni A, Cecchini V, Bachetoni A, D'Alessandro M, Van Aken H, Pietropaoli P, Westphal M: Continuous terlipressin versus vasopressin infusion in septic shock (TERLIVAP): a randomized, controlled pilot study. Crit Care. 2009, 13: R130-10.1186/cc7990.
Rehberg S, Ertmer C, Kohler G, Spiegel HU, Morelli A, Lange M, Moll K, Schlack K, Van Aken H, Su F, Vincent JL, Westphal M: Role of arginine vasopressin and terlipressin as first-line vasopressor agents in fulminant ovine septic shock. Intensive Care Med. 2009, 35: 1286-1296. 10.1007/s00134-009-1470-z.
Michel F, Thomachot L, David M, Nicaise C, Vialet R, Di Marco JN, Lagier P, Martin C: Continuous low-dose infusion of terlipressin as a rescue therapy in meningococcal septic shock. Am J Emerg Med. 2007, 25: 863.e1-2. 10.1016/j.ajem.2007.02.019.
Lange M, Morelli A, Ertmer C, Koehler G, Broking K, Hucklenbruch C, Bone HG, Van Aken H, Traber DL, Westphal M: Continuous versus bolus infusion of terlipressin in ovine endotoxemia. Shock. 2007, 28: 623-629.
Broking K, Lange M, Morelli A, Ertmer C, Aken HV, Luecke M, Rehberg S, Bowering N, Bone HG, Traber DL, Westphal M: Employing dobutamine as a useful agent to reverse the terlipressin-linked impairments in cardiopulmonary hemodynamics and global oxygen transport in healthy and endotoxemic sheep. Shock. 2008, 29: 71-77.
Ryckwaert F, Virsolvy A, Fort A, Murat B, Richard S, Guillon G, Colson PH: Terlipressin, a provasopressin drug exhibits direct vasoconstrictor properties: consequences on heart perfusion and performance. Crit Care Med. 2009, 37: 876-881. 10.1097/CCM.0b013e31819b8199.
Ouattara A, Landi M, Le Manach Y, Lecomte P, Leguen M, Boccara G, Coriat P, Riou B: Comparative cardiac effects of terlipressin, vasopressin, and norepinephrine on an isolated perfused rabbit heart. Anesthesiology. 2005, 102: 85-92. 10.1097/00000542-200501000-00016.
Noguera I, Medina P, Segarra G, Martinez MC, Aldasoro M, Vila JM, Lluch S: Potentiation by vasopressin of adrenergic vasoconstriction in the rat isolated mesenteric artery. Br J Pharmacol. 1997, 122: 431-438. 10.1038/sj.bjp.0701397.
Urge J, Sincl F, Prochazka V, Urbanek K: Terlipressin-induced ventricular arrhythmia. Scand J Gastroenterol. 2008, 43: 1145-1148. 10.1080/00365520801905304.
Siami S, Bailly-Salin J, Polito A, Porcher R, Blanchard A, Haymann JP, Laborde K, Maxime V, Boucly C, Carlier R, Annane D, Sharshar T: Osmoregulation of vasopressin secretion is altered in the postacute phase of septic shock. Crit Care Med. 2010, 38: 1962-1969.
Bauer SR, Aloi JJ, Ahrens CL, Yeh JY, Culver DA, Reddy AJ: Discontinuation of vasopressin before norepinephrine increases the incidence of hypotension in patients recovering from septic shock: a retrospective cohort study. J Crit Care. 2010, 25: 362.e7-362.e11. 10.1016/j.jcrc.2009.10.005.
Holt DB, Delaney RR, Uyehara CF: Effects of combination dobutamine and vasopressin therapy on microcirculatory blood flow in a porcine model of severe endotoxic shock. J Surg Res. 2009, [Epub ahead of print]
Rehberg S, Ertmer C, Vincent JL, Spiegel HU, Köhler G, Erren M, Lange M, Morelli A, Seisel J, Su F, Van Aken H, Traber DL, Westphal M: Effects of combined arginine vasopressin and levosimendan on organ function in ovine septic shock. Crit Care Med. 2010, 38: 2016-2023.
Nakada T, Russell JA, Wellman H, Boyd JH, Nakada E, Thain KR, Thair SA, Hirasawa H, Oda S, Walley KR: Leucyl/cystinyl aminopeptidase (LNPEP) gene variants in septic shock. Chest. 2011, 139: 1042-1049. 10.1378/chest.10-2517.
Wagener G, Kovalevskaya G, Minhaz M, Mattis F, Emond JC, Landry DW: Vasopressin deficiency and vasodilatory state in end-stage liver disease. J Cardiothorac Vasc Anesth. 2010.
Rehberg S, Ertmer C, Lange M, Morelli A, Whorton E, Dünser M, Strohhäcker AK, Lipke E, Kampmeier TG, Van Aken H, Traber DL, Westphal M: Role of selective V2-receptor-antagonism in septic shock: a randomized, controlled, experimental study. Crit Care. 2010, 14: R200-10.1186/cc9320.
Sharshar T, Carlier R, Blanchard A, Feyde A, Gray F, Paillard M, Raphael JC, Gajdos P, Annane D: Depletion of neurohypophyseal content of vasopressin in septic shock. Crit Care Med. 2002, 30: 497-500. 10.1097/00003246-200203000-00001.
Sharshar T, Blanchard A, Paillard M, Raphael JC, Gajdos P, Annane D: Circulating vasopressin levels in septic shock. Crit Care Med. 2003, 31: 1752-1758. 10.1097/01.CCM.0000063046.82359.4A.
Le Clerc F, Walter-Nicolet E, Leteurtre S, Noizet O, Sadik A, Cremer R, Fourier C: Admission plasma vasopressin levels in children with meningococcal septic shock. Intensive Care Med. 2003, 29: 1339-1344. 10.1007/s00134-003-1868-y.
Kruger S, Ewig S, Kunde J: C-terminal provasopressin (copeptin) in patients with community-acquired pneumonia - influence of antibiotic pretreatment. Results from the German competence network (CAPNETZ). J Antimicrob Chemother. 2009, 64: 159-162. 10.1093/jac/dkp148.
de Kruif MD, Lemaire LC, Giebelen IA, Struck J, Morgenthaler NG, Papassotiriou J, Elliott PJ, van der Poll T: The influence of corticosteroids on the release of novel biomarkers in human endotoxemia. Intensive Care Med. 2008, 34: 518-522. 10.1007/s00134-007-0955-x.
Acknowledgements
Support for VASST from the Canadian Institutes of Health Research (grant number MCT 44152). Registration: ISRCTN94845869.
In addition to the author, the following persons and institutions participated in the VASST trial: Writing Committee - J.A. Russell (chair), K.R. Walley,
A.C. Gordon, C. Holmes Boulton, J.T. Granton, P.C. Hebert, D.J. Cooper, S. Mehta, J. Singer, D.J. Cook, J.J. Presneill, M.M. Storms; Executive Committee - J.A. Russell (chair), K.R. Walley, C.L. Holmes Boulton, J.T. Granton, P.C. Hebert, D.J. Cooper, S. Mehta, J. Singer, A.C. Gordon, M.M. Storms (project coordinator), S. Jones (administrative assistant); Management Committee - J.A. Russell (chair), M.M. Storms (project coordinator), K.R. Walley, C.L. Holmes Boulton, J. Singer, A.C. Gordon, S. Jones (administrative assistant); DSMB - G.R. Bernard (chair), A.S. Slutsky, G.A Wells; CIHR - A. Gasparini; Data Management - Singer, B. Savage, D. Ayers, R. Woods, K. Wu, M. Maralit; Monitoring - L. Smith, K. Foley, A. Suri, M. Steinberg, B. Howe, P. Galt, A. Higgins, M.M. Storms; Laboratory - M.E. LeBlanc, A.M. Sutherland, A. Sham, A. McLeod.
Clinical centers and VASST investigators
Canada
British Columbia: St Paul's Hospital - J.A. Russell, K.R. Walley, D.R. Dorscheid, M. Hameed, L. Lazosky, S. Helderweirt, K. Foley, C. Honeyman, T. Terins; Vancouver General Hospital - D. Chittock, J. Ronco, L. Smith, S. Logie; Royal Jubilee Hospital - G. Wood, F. Auld; Kelowna General Hospital - C. Holmes Boulton, V. Stedham, M. Mantle, L. Fox, G.D. McCauley, J.D. Rolf, C. Erbacher; Richmond General Hospital - G. Martinka, S. Goulding, S. Silverwood, L. Leung; Royal Columbian Hospital - S. Keenan, J. Murray, M. Van Osch.
Manitoba: St Boniface Hospital - B. Light, M. Dominique; Winnipeg Health Science Centre - P. Gray, R. Stimpson, S. Rosser, D. Bell, W. Janz.
Ontario: Ottawa Hospital, General Campus - P.C. Hebert, T. McArdle, I. Watpool; UHN - Toronto General & Toronto Western Hospitals - J.T. Granton, M. Steinberg, A. Matte-Martyn; St Joseph's Hospital - D.J Cook, E. McDonald, F. Clarke, A. Tkaczyk, N. Zytaruk; Mount Sinai Hospital - S. Mehta, T. Stewart, A. Suri, C. Martinez-Motta, R. MacDonald, V. Sivanantham; Ottawa Hospital, Civic Campus - R. Hodder, J. Foxall, M. Lewis; St Michael's Hospital - M. Ward, C. Dos Santos, J. Friedrich, D. Scales, O. Smith, I. DeCampos, A. Richards, H. Michalopoulos, U. Bakshi; Sunnybrook and Women's College Health Science Centre - W. Sibbald, T. Smith, K. Code, B. Bojilov, C. Dale, M. Keogh; Hamilton Health Sciences Centre - M. Meade, L. Hand; London Health Sciences Centre - C. Martin, J. Kehoe, V. Binns; Sudbury Regional Hospital - M. Mehta, M. McGuire; Charles LeMoyne Hospital - G. Poirier, L. Provost; Windsor - Hotel Dieu Hospital - J. Muscedere, C. Diemer.
Australia
Victoria: Alfred Hospital - D.J. Cooper, V. Pellegrino, A. Hilton, S. Morrison, C. Weatherburn, K. Moulden, C. Harry; Royal Melbourne - J.J. Presneill, M. S Robertson, J.F. Cade, B.D. Howe, D.K. Barge, C.A. Boyce, F. Healy; Monash Medical Center - C. Wright, D. Weyandt, J. Barrett, C. Walker, P. Galt, S. Burton. Western Australia: Royal Perth Hospital - G. Dobb, S. Perryman, J. Chamberlain, L. Thomas.
South Australia: Flinders Medical Centre - A. Bersten, L. Daly, T. Hunt, D. Wood.
USA
Mayo Hospital - B. Patel, J. Larson, M. Rady, G. LeBrun, E. Boyd, R. Rush.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
JAR reports holding stock in Sirius Genomics Incorporated, which has submitted patents owned by the University of British Columbia and licensed to Sirius Genomics, that are related to the genetics of vasopressin, NIK and protein C. The University of British Columbia has also submitted a patent related to the use of vasopressin in septic shock. JAR is an inventor on these patents. JAR reports receiving consulting fees from Ferring, which manufactures vasopressin, from Astra Zeneca which manufactures anti-TNF, from Sirius Genomics Inc., and from Biocritica which sells activated protein C in the USA. JAR reports receiving grant support from Sirius Genomics, Novartis, Ferring, and Eli Lilly. JAR has received speaking honoraria from Pfizer and Eli Lilly.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Russell, J.A. Bench-to-bedside review: Vasopressin in the management of septic shock. Crit Care 15, 226 (2011). https://doi.org/10.1186/cc8224
Published:
DOI: https://doi.org/10.1186/cc8224