
Abstract
Carbon monoxide (CO) is generated during incomplete combustion of carbon-containing compounds and leads to acute and chronic toxicity in animals and humans depending on the concentration and exposure time. In addition to exogenous sources, CO is also produced endogenously by the activity of heme oxygenases (HOs) and the physiological significance of HO-derived CO has only recently emerged. CO exerts vasoactive, anti-proliferative, anti-oxidant, anti-inflammatory and anti-apoptotic effects and contributes substantially to the important role of the inducible isoform HO-1 as a mediator of tissue protection and host defense. Exogenous application of low doses of gaseous CO might provide a powerful tool to protect organs and tissues under various stress conditions. Experimental evidence strongly suggests a beneficial effect under pathophysiological conditions such as organ transplantation, ischemia/reperfusion, inflammation, sepsis, or shock states. The cellular and molecular mechanisms mediating CO effects are only partially characterized. So far, only a few studies in humans are available, which, however, do not support the promising results observed in experimental studies. The protective effects of exogenous CO may strongly depend on the pathological condition, the mode, time point and duration of application, the administered concentration, and on the target tissue and cell. Differences in bioavailability of endogenous CO production and exogenous CO supplementation might also provide an explanation for the lack of protective effects observed in some experimental and clinical studies. Further randomized, controlled clinical studies are needed to clarify whether exogenous application of CO may turn into a safe and effective preventive and therapeutic strategy to treat pathophysiological conditions associated with inflammatory or oxidative stress.
Similar content being viewed by others


Carbon monoxide: exogenous sources and toxic effects
High concentrations of carbon monoxide (CO) are generated during incomplete combustion of carbon-containing compounds such as wood, coal, gas, oil, or tobacco. CO is a colorless and odorless gas that causes acute and chronic toxicity in humans and animals. CO mediates its toxic effects primarily by strongly binding to hemoglobin and forming carboxyhemoglobin (COHb), thereby reducing the oxygen-carrying capacity of the blood. The affinity of hemoglobin for CO is approximately 210 to 250 times that for oxygen [1]. Both decreased arterial oxygen content (impaired O2 binding to hemoglobin) and decreased tissue oxygen pressure (PO2; increased affinity of COHb for O2) lead to tissue hypoxia [2, 3]. There is a linear correlation between the inspired level of CO and arterial COHb levels [4]. Although the percentage of COHb in blood represents the best predictive marker for extrapolating the total amount of CO, COHb levels do not always correlate with the degree of injury and outcome [5]. COHb levels between 15 and 20% seem to be well tolerated in humans and are considered the 'biological threshold' above which severe CO-mediated injury is likely to occur [6]. In addition to hemoglobin, CO binding to other heme-containing proteins, such as cytochrome c oxidase (thus interfering with cellular respiration), catalase, or myoglobin, may partly contribute to the toxic effects.
The most vulnerable organs to CO-induced hypoxia are the heart and the brain because of their high metabolic rate [7]. The mild symptoms of acute CO poisoning are often non-specific and include headache, nausea, vomiting, dizziness, and fatigue, which may progress to confusion, tachypnea, tachycardia, impaired vision and hearing, convulsions, loss of consciousness, finally leading to death when immediate and adequate treatment is not available. The amount of CO inhaled and/or the exposure time are the most critical factors that determine the severity of CO poisoning. In addition, children and older adults are more susceptible and may have more severe symptoms [8]. Predisposing conditions for CO toxicity have been described, such as cardiovascular disorders (for example, coronary heart disease), chronic obstructive pulmonary disease (COPD), or anemia [9]. Heavy smokers may have more severe symptoms since their COHb levels are already elevated.
Carbon monoxide appears to be the leading cause of injury and death due to poisoning worldwide [10]. Since tissue hypoxia is the underlying mechanism of CO-induced injury, increasing the inspired oxygen concentration represents the treatment for CO poisoning. In severe poisoning, hyperbaric oxygen therapy is regarded as the therapy of choice [11]. Both normobaric and hyperbaric oxygen improve oxygen delivery by increasing the amount of oxygen dissolved in plasma and by reducing the half-life of COHb. However, the results from existing randomized, controlled trials of hyperbaric versus normobaric oxygen in the treatment of acute CO poisoning provide conflicting results regarding the effectiveness of hyperbaric oxygen for the prevention of neurological symptoms [12]. An ongoing phase IV randomized clinical trial investigates important clinical outcomes (for example, 6-week cognitive sequelae) of patients with acute CO poisoning randomized to receive either one or three hyperbaric oxygen treatments [13]. The estimated study completion date is May 2009. If treatment of CO poisoning is timely, most patients are able to recover, but even with adequate treatment CO poisoning may result in permanent memory loss or brain damage. For the long-term sequelae of acute CO poisoning, only symptomatic therapy is available. Chronic exposure to CO may lead to myocardial hypertrophy [14].
Functions of endogenous carbon monoxide production
Coburn and colleagues [15] demonstrated that CO is endogenously produced in animals and humans. The vast majority of endogenous CO is derived from the oxidative breakdown of heme by microsomal heme oxygenases (HOs). HO catalyzes the first and rate-limiting step in heme degradation, yielding equimolar amounts of CO, iron, and biliverdin-IXα (Figure 1), which is further converted to bilirubin by biliverdin reductase [16]. Two isoforms of HO have been described, namely HO-1 [17, 18] and HO-2 [19, 20]. Furthermore, a third isoform has been found in rats [21], which represents a processed pseudogene derived from the gene for HO-2 [22]. HO-2 is constitutively expressed in many tissues, with high activity in testes, central nervous system, liver, kidney, and intestine. A basal expression of HO-1 is found in tissues that degrade senescent red blood cells, predominantly spleen, reticuloendothelial cells of the liver and bone marrow [23]. HO-1 is the inducible isoform, and induction of HO-1 gene expression occurs in response to a wide variety of endogenous and exogenous stimuli, such as chemical or physical stimuli, xenobiotics, hyperoxia, hypoxia, ischemia/reperfusion, inflammation, surgical procedures, or anesthetics [24–29].

Heme oxygenase pathway. Heme oxygenase catalyzes the rate-limiting step in the degradation of heme leading to the generation of equimolar amounts of free iron, biliverdin and carbon monoxide.
The critical role of HO-1 under physiological conditions was demonstrated in the first described case of human HO-1 deficiency. The boy in this case presented with severe growth retardation, persistent hemolytic anemia, and severe, persistent endothelial damage [30] and died at the age of 6 years [31]. Over the past decade the function of HO-1 has expanded from a heme-degrading enzyme to a key mediator of tissue protection and host defense, and its cytoprotective effects have been described in vivo and in vitro [24, 25, 28, 32–42].
The products of the HO pathway – CO, iron, and biliverdin/bilirubin – have long been regarded solely as waste products. Recently, the unique biological functions of the products and their contribution to the protective effects of the HO system have attracted great interest. Thus, the HO system has different functions: besides the breakdown of heme, a pro-oxidant [43], it produces cytoprotective substances, and the inducibility of HO-1 renders it a powerful endogenous cytoprotective system.
Bilirubin has been described as a potent endogenous anti-oxidant [44] with potential clinical implications [45]. Free iron exhibits oxidizing capacities, although the iron released during heme degradation stimulates the synthesis of ferritin [46], which sequesters unbound iron, thereby serving as an additional anti-oxidant [47]. The observation that CO can weakly activate soluble guanylate cyclase (sGC), thereby stimulating the production of cGMP, suggested an important role of CO as an intracellular messenger molecule, thus acting in a similar way to nitric oxide [48, 49]. The functions of CO as a neural messenger have since been described [50]. Vasoactive effects of CO have been reported in the pulmonary vasculature [51] and in the liver [37, 52], where CO acts to maintain portal venous vascular tone in a relaxed state [37]. In addition to the biological functions of CO under physiological conditions, the substantial contribution of CO to the protective effects of induced HO activity has recently been recognized and includes vasoactive, anti-oxidative, anti-inflammatory, anti-apoptotic, and anti-proliferative properties. Thus, CO has advanced from a toxic waste product to a physiological regulator and the importance of endogenously derived CO to control homeostasis under both physiological and pathophysiological conditions is increasingly recognized in every organ system and cell type.
Although different mechanisms explaining the effects of CO have been described, the exact underlying signaling mechanisms and precise molecular targets of CO are only partially elucidated. Effects mediated by CO-induced activation of sGC/cGMP include inhibition of platelet activation and aggregation, smooth muscle relaxation, vasoactive effects, inhibition of cellular proliferation, and effects on neurotransmission [37, 49–56]. cGMP-independent mechanisms of vasoregulation have also been suggested. CO may directly activate calcium-dependent potassium channels, thus mediating dilation of blood vessels [57]. Recent evidence suggests an important role of CO as a signaling molecule in modulating mitogen-activated protein kinases (MAPKs), especially p38 MAPK in response to oxidative stress and inflammation (reviewed in [58, 59]). CO-mediated activation of p38 MAPK has been shown to exert anti-inflammatory [60], anti-apoptotic, and anti-proliferative effects [61, 62]. Downstream target molecules of CO-dependent p38 MAPK activation have been identified, namely heat shock protein 70 and caveolin-1 [61, 62]. Zhang and colleagues [63] demonstrated that the anti-apoptotic effects of CO involve both phosphatidylinositol 3-kinase/Akt and p38 MAPK signaling pathways in endothelial cells in a model of anoxia-reoxygenation injury. In hepatocytes, CO activated nuclear factor-κB (NF-κB) through a mechanism that involves reactive oxygen species-induced Akt phosphorylation and protected against cell death [64]. Figure 2 provides a simplified overview of the described CO-mediated signal transduction pathways.

Carbon monoxide signal transduction pathways. CO, carbon monoxide; HSF, heat shock factor; HSP, heat shock protein; MAPK, mitogen-activated protein kinase; NFκB, nuclear factor-κB; NO, nitric oxide; sGC, soluble guanylate cyclase.
Therapeutic applications of carbon monoxide
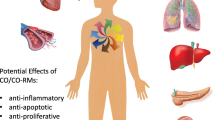
The observation that induction of HO-1 gene expression under pathological conditions plays an important role in organ preservation strongly suggests that CO might be substantially involved in mediating these effects. This is supported by the observation in models of HO-1 deficiency or after blockade of HO activity that the protective effects of induction of HO-1 are mimicked by low amounts of exogenous CO [54, 59, 65]. However, pre-induction of the HO-1 system by exogenous stimuli to induce local CO release or exogenous application of CO to potentiate the endogenous protective effects may be challenging. To increase the availability of CO, different approaches have been developed, including induction of HO-1 gene expression with pharmacological and genetic strategies, inhalation of low doses of CO, and application of CO-releasing molecules. Figure 3 briefly summarizes the protective effects and the potential therapeutic applications of CO in a variety of disorders and diseases of different organ systems.

Protective effects and potential therapeutic applications of carbon monoxide. ALI, acute lung injury; ARDS, acute respiratory distress syndrome; CO, carbon monoxide; I/R, ischemia/reperfusion.
Induction of HO-1 gene expression
Strategies to induce HO-1 as a protective mechanism against a subsequent stress event include pharmacological approaches such as volatile anesthetics [40] or heme derivatives [32, 33], and genetic approaches [39] as well as the use of other inducers as described above. Long-term overexpression of HO-1 by targeted gene transfer has become a powerful tool to investigate the specific role of the HO-1 enzyme [66]. The amount of CO released by the induced activity of HO-1 is unknown. In addition, induction of HO-1 increases the concentration of all products of the pathway, and the contribution of CO to the observed protective effects is difficult to evaluate.
Exogenous application of carbon monoxide
Inhalation of CO represents a novel therapeutic approach and exerts both local effects on the lungs and systemic effects. The challenge remains to reach safe and effective concentrations in target tissues without producing deleterious effects caused by CO-mediated tissue hypoxia. The tolerance to CO exposure has been investigated in rodents and conflicting results have been obtained: while continuous application of 500 ppm CO for 2 years had no deleterious effects [67], 200 ppm for 20 h per day over 14 days induced myocardial hypertrophy [14].
The CO-releasing properties of transition metal carbonyls were first described by Herrman [68]. Motterlini and his group have developed CO-releasing molecules (CO-RMs) as a new strategy to deliver defined amounts of CO for therapeutic applications [6, 69] without significantly affecting COHb levels [70]. In particular, the synthesis of a water-soluble compound might be promising. So far, only experimental data are available. The use of CO-RMs to characterize CO-mediated cytoprotection has been reviewed by Foresti and colleagues [6].
Preclinical experimental studies
In most experimental models, acute rather than chronic inhalation of CO is applied (10 to 1,000 ppm for 1 to 24 h). Depending on the concentration, different exposure times are required to reach COHb equilibrium [71]. CO inhalation has been shown to be protective in experimental inflammatory and non-inflammatory disease models (reviewed in [6, 25, 72–75]). The majority of studies investigating the effects of low amounts of inhaled CO concentrate on disease models in the lungs. In addition to local effects in the lungs, inhaled CO is also able to affect systemic organ dysfunction.
Lung
The protective effects of inhaled CO have been investigated in models of acute lung injury, acute respiratory distress syndrome (ARDS), ischemia/reperfusion, asthma, and remote lung injury. The first in vivo evidence to suggest a therapeutic potential of low dose gaseous CO was provided by Otterbein and colleagues [76]. Rats exposed to low concentrations of CO exhibited a significant attenuation of hyperoxia-induced lung injury and increased survival. CO exposure exerted anti-inflammatory and anti-apoptotic effects. The molecular mechanisms of the observed inhibition of pro-inflammatory cytokines involve the MKK3/p38 MAPK pathway [77]. In contrast, low levels of CO were not protective in a similar rat model of hyperoxic acute lung injury [4]. Inhalation of CO attenuated the development of hypoxia-induced pulmonary artery hypertension in rats, presumably through activation of Ca2+-activated K+ channels [78] and was also able to reverse established pulmonary hypertension [79]. Inhalation of CO for 6 h after intratracheal injection of acidic solution in mice reduced early neutrophil recruitment without affecting chemokine levels in bronchoalveolar fluid [80]. The pathomechanisms of allergen-induced asthma include inflammation and bronchoconstriction. In ovalbumin-induced asthma, CO treatment of mice for 2 h before aerosol challenge led to a specific reduction of the pro-inflammatory cytokine IL-5 while other pro-inflammatory or anti-inflammatory cytokines were unaffected [81]. In the same model of inflammation, Ameredes and colleagues [82] showed a CO-induced, cGMP-dependent reduction of airway hyper-responsiveness.
In experimental models of lung ischemia and reperfusion, including transplantation, inhaled CO has anti-inflammatory and anti-apoptotic effects [54, 63, 83–86]. The p38 MAPK pathway and downstream target genes, such as that for early growth response-1 (Egr-1), seem to play important roles in mediating the CO effects [84].
Mechanical ventilation may cause profound lung injury and inflammatory responses. Dolinay and colleagues [87] described a CO-mediated suppression of tumor necrosis factor (TNF)-alpha release and neutrophil recruitment and postulated an involvement of the p38 MAPK pathway. A study in knock-out mice suggests a key role of Egr-1 as a pro-inflammatory regulator in ventilator-induced lung injury. Moreover, peroxysome proliferator-activated receptor-gamma, an anti-inflammatory nuclear regulator, seems to be involved in the protective effects of CO [88].
In addition to attenuating local lung injury, CO also protects against remote lung injury. After ischemia and reperfusion of the lower extremities, CO significantly reduced ischemia/reperfusion-induced acute lung injury [89]. Pretreatment with inhaled CO reduced pulmonary inflammatory response and provided anti-apoptotic effects in a model of cardiopulmonary bypass in pigs [90].
Liver
Effects of CO on the liver have been investigated in models of inflammation- and ischemia/reperfusion-induced hepatocellular injury as well as in burn injury. TNF-alpha-induced hepatocyte cell death in mice was prevented by CO inhalation. CO-induced activation of NF-κB and inducible nitric oxide synthase and nitric oxide-induced HO-1 expression were required for the protective effects [91]. In addition, CO-stimulated liver ATP generation through the activation of sGC was a prerequisite for CO to protect against TNF-alpha-induced apoptosis [92]. In models of liver ischemia and reperfusion, HO-1 induction plays an important role in maintaining hepatocellular integrity [38] and induction of HO-1 before (low flow) ischemia can attenuate the subsequent hepatic injury [32, 40]. A role for CO in preventing hypoxia-induced decreases in hepatocyte ATP levels was postulated in a mouse model of hemorrhagic shock and resuscitation [93]. In cold ischemia reperfusion associated with liver transplantation, CO inhalation suppressed the inflammatory response. Downregulation of MEK/ERK1/2 seems to play a role in mediating the protective effects while the NF-κB signaling pathway does not seem to be affected [94]. CO-RM-liberated CO attenuates liver injury in burn mice by mechanisms involving downregulation of pro-inflammatory mediators and suppression of the pro-adhesive phenotype of endothelial cells [95, 96].
Intestine
The protective effects of CO in the intestine have been investigated in a variety of animal models of postoperative ileus and cold ischemia/reperfusion injury associated with transplantation. The development of postoperative ileus may occur after mild manipulation of the small bowel during surgery, which initiates an inflammatory response within the intestinal muscularis [97] that is characterized by the release of pro-inflammatory mediators, increased expression of adhesion molecules on the vascular endothelium, and recruitment of leukocytes from the systemic circulation [98, 99]. Inhalation of CO significantly attenuated the surgically induced molecular inflammatory response and the associated decline in gastrointestinal contractility that is characteristic of postoperative ileus [100, 101]. Similar effects could be observed after intraperitoneal injection of CO-saturated Ringer's lactate solution, possibly in a sGC-dependent manner [102].
Nakao and colleagues [103] provide a large body of evidence that inhaled CO is also protective by improving post-transplant motility and attenuating the inflammatory cytokine response in the syngeneic rat transplant model. In addition, CO is anti-apoptotic and significantly improves animal survival [104]. Similar protective results can be achieved after storage of grafts in University of Wisconsin solution saturated with CO [105].
Vascular diseases
Short-term administration of CO has been shown to be protective against vascular injury. CO rescued the pro-thrombotic phenotype of Hmox1 deficiency during oxidative stress [106]. Intravenous injection of CO-saturated saline produced vasodilatation and improved microvascular hemodynamics in a hamster skinfold window chamber preparation, possibly via increased cardiac output and local cGMP content [107]. Otterbein and colleagues [55] described a beneficial effect of inhaled CO in preventing arteriosclerotic lesions that occur following aorta transplantation.
Heart
Experimental models of heart transplantation or cardiopulmonary bypass have been used to investigate CO effects on accompanying organ injury. CO reduced ischemia/reperfusion injury and cardiac rejection of mouse to rat cardiac transplants via anti-apoptotic, anti-inflammatory and vasodilatory mechanisms, and suppression of platelet aggregation and fibrinolysis [65]. Treatment of the donor (CO inhalation) and graft (CO-saturated storage solution) but not the recipient protected against ischemia/reperfusion injury via anti-apoptotic mechanisms [108]. In contrast, low-dose CO inhalation of the recipient after transplantation effectively ameliorated heart allograft rejection via downregulation of pro-inflammatory mediators [109].
In a clinically relevant model of cardiopulmonary bypass surgery in pigs, treatment with CO improved cardiac energetics, prevented edema formation and apoptosis, and facilitated recovery [110]. In a rat model of ischemia/reperfusion injury induced by occlusion of the left anterior descending coronary artery, pre-exposure to CO significantly reduced infarct size and migration of macrophages into infarct areas. In addition, TNF-alpha expression was reduced. The protective effects were mediated by CO-induced activation of p38 MAPK, protein kinase B (Akt), endothelial nitric oxide synthase, and cGMP in the myocardium [111].
Kidney
Most of the studies of CO effects in kidneys concentrate on models of cold ischemia/reperfusion injury in transplantation. Ischemia/reperfusion injury of kidney grafts is one of the major deleterious factors affecting successful renal transplantation. Renal ischemia/reperfusion injury causes delayed graft function and plays a significant role in the development of chronic allograft nephropathy [112, 113]. Exposure to low concentrations of CO prevented fibroinflammatory changes associated with chronic allograft nephropathy and preserved long-term renal allograft function [114]. Storage of kidneys with cold preservation solutions containing CO-RMs also improved their function upon reperfusion [115]. Hypoxia-inducible factor-1-mediated upregulation of vascular endothelial growth factor seems to contribute to the protective mechanisms [116]. Nakao and colleagues [117] provide evidence that prevention of cytochrome P450 degradation, maintenance of normal intracellular heme levels and a reduction of lipid peroxidation participate in the protective effects of CO-RMs during storage of kidney grafts.
Systemic inflammation
As a model of systemic inflammation, lipopolysaccharide (LPS)-induced inflammatory response and organ injury has widely been used to study protective COmediated effects. In rodents and pigs injected with LPS, inhalation of CO leading to 14.08 ± 1.34% COHb significantly reduced LPS-induced cytokine response [118, 119] and improved long-term survival [120]. Further mechanisms of CO-mediated protection against LPS-induced multiple injury in rats have been described and include anti-oxidative, anti-inflammatory and anti-apoptotic effects, and up-regulation of HO-1 expression [121]. In contrast, in a randomized, controlled study in pigs, CO exposure did not alter LPS-induced levels of pro- and anti-inflammatory cytokines [122]. The lack of protective effects observed in this study might possibly be explained by the low level of COHb measured (5% compared to 14%) [118].
Clinical studies
While a large body of experimental evidence suggests the potential of low amounts of inhaled CO to protect the lungs and systemic organs and tissues against oxidative and inflammatory insults, only a few studies on therapeutic applications of CO inhalation in humans have been published.
In a randomized, double-blinded, placebo-controlled, two-way cross-over trial experimental endotoxemia was induced in healthy volunteers by injection of 2 ng/kg LPS. The potential anti-inflammatory effects of CO inhalation were investigated by inhalation of 500 ppm CO (leading to an increase in COHb from 1.2% to 7%) versus synthetic air as a placebo for 1 h. CO inhalation had no effect on the inflammatory response as measured by systemic cytokine production (TNF-alpha, IL-6, IL-8, IL-1α and IL-1β) [123]. In this study, no adverse side effects of CO inhalation were observed.
This study is in contrast to the above described results obtained in most experimental models of endotoxemia. Possible explanations for this discrepancy could be that blood from different species has different affinities for CO, different COHb half-lives, different hemoglobin CO saturation points (different COHb levels at the same CO concentration), or different basic physiologies, such as heart rate.
COPD is characterized by an inflammatory and oxidative stress response. Furthermore, COPD is accompanied by increased COHb levels that correlate with exhaled CO [124]. However, the endogenous CO release might not be sufficient to protect against the development and progression of COPD. In a randomized, placebo-controlled, cross-over study 20 ex-smoking patients with stable COPD were examined to assess safety, feasibility, and potential anti-inflammatory effects of CO inhalation. Inhalation of 100 to 125 ppm CO for 2 h per day on 4 consecutive days led to a maximal individual COHb level of 4.5%. In two patients, exacerbations of COPD occurred during or after the CO inhalation period; otherwise the treatment was well tolerated. The primary study endpoint was sputum neutrophil counts. Although there was a trend towards reduction in sputum eosinophils and improvement of bronchial responsiveness, no significant therapeutic effects were observed [125]. The results of this pilot study are interesting, since they provide some evidence for a potential therapeutic use of inhaled CO. However, whether CO inhalation increases the risk of COPD exacerbations needs to be determined.
One clinical study investigating the effects of low amounts of inhaled CO is currently in progress [126]. A single blinded, randomized, placebo controlled phase I study in healthy subjects investigates the potential of inhaled carbon monoxide in preventing lung inflammatory responses following local endotoxin instillation. The study is ongoing, but currently not recruiting participants.
Conclusion
CO has long been regarded solely as a toxic environmental or endogenous waste product. In addition to cytoprotective properties of endogenous CO, recent evidence strongly suggests protective effects of low concentrations of exogenous CO under pathophysiological conditions such as organ transplantation, ischemia/reperfusion, inflammation, sepsis, or shock states. Studies in humans are scarce and so far do not support the promising results observed in pre-clinical experimental studies. A potential beneficial effect of exogenous CO may highly depend on the pathological condition, the mode, time point and duration of application, the administered concentration, and on the target tissue. Further randomized, controlled clinical trials are needed to clarify whether exogenous application of CO, either by inhalation or intravenous application of CO-RMs, may become a safe and effective preventive and therapeutic tool to treat pathophysiological conditions associated with inflammatory or oxidative stress.
Note
This article is part of a review series on Gaseous mediators, edited by Peter Radermacher.
Other articles in the series can be found online at http://ccforum.com/series/gaseous_mediators
Abbreviations
- CO:
-
carbon monoxide
- COHb:
-
carboxyhemoglobin
- COPD:
-
chronic obstructive pulmonary disease
- CO-RM:
-
carbon monoxide-releasing molecule
- HO:
-
heme oxygenase
- IL:
-
interleukin
- LPS:
-
lipopolysaccharide
- MAPK:
-
mitogen-activated protein kinase
- NF-κB:
-
nuclear factor-κB
- sGC:
-
soluble guanylate cyclase
- TNF:
-
tumor necrosis factor.
References
Rodkey FL, O'Neal JD, Collison HA, Uddin DE: Relative affinity of hemoglobin S and hemoglobin A for carbon monoxide and oxygen. Clin Chem 1974, 20: 83-84.
Stewart RD: The effect of carbon monoxide on humans. Annu Rev Pharmacol 1975, 15: 409-423. 10.1146/annurev.pa.15.040175.002205
Piantadosi CA: Biological chemistry of carbon monoxide. Antioxid Redox Signal 2002, 4: 259-270. 10.1089/152308602753666316
Clayton CE, Carraway MS, Suliman HB, Thalmann ED, Thalmann KN, Schmechel DE, Piantadosi CA: Inhaled carbon monoxide and hyperoxic lung injury in rats. Am J Physiol Lung Cell Mol Physiol 2001, 281: L949-L957.
Hampson NB, Hauff NM: Carboxyhemoglobin levels in carbon monoxide poisoning: do they correlate with the clinical picture? Am J Emerg Med 2008, 26: 665-669. 10.1016/j.ajem.2007.10.005
Foresti R, Bani-Hani MG, Motterlini R: Use of carbon monoxide as a therapeutic agent: promises and challenges. Intensive Care Med 2008, 34: 649-658. 10.1007/s00134-008-1011-1
Turino GM: Effect of carbon monoxide on the cardiorespiratory system. Carbon monoxide toxicity: physiology and biochemistry. Circulation 1981, 63: 253A-259A.
Van Meter KW: Carbon monoxide poisoning. In Emergency Medicine. Edited by: Tintinalli JE, Kelen GD, Stapczynski JS. New York: McGraw Hill: New York; 2003:1238-1242.
Allred EN, Bleecker ER, Chaitman BR, Dahms TE, Gottlieb SO, Hackney JD, Pagano M, Selvester RH, Walden SM, Warren J: Short-term effects of carbon monoxide exposure on the exercise performance of subjects with coronary artery disease. N Engl J Med 1989, 321: 1426-1432.
Carbon monoxide-related deaths – United States, 1999–2004 MMWR Morb Mortal Wkly Rep 2007, 56: 1309-1312.
Thom SR: Hyperbaric-oxygen therapy for acute carbon monoxide poisoning. N Engl J Med 2002, 347: 1105-1106. 10.1056/NEJMe020103
Juurlink DN, Buckley NA, Stanbrook MB, Isbister GK, Bennett M, McGuigan MA: Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev 2005, CD002041.
One vs. Three Hyperbaric Oxygen Treatments for Acute Carbon Monoxide Poisoning (1V3CORCT)[http://www.clinicaltrials.gov/ct2/show/NCT00465855?term=co+poisoning&rank=1]
Loennechen JP, Nilsen OG, Arbo I, Aadahl P, Nilsen T, Waldum HL, Sandvik AK, Ellingsen O: Chronic exposure to carbon monoxide and nicotine: endothelin ET(A) receptor antagonism attenuates carbon monoxide-induced myocardial hypertrophy in rat. Toxicol Appl Pharmacol 2002, 178: 8-14. 10.1006/taap.2001.9300
Coburn RF, Blakemore WS, Forster RE: Endogenous carbon monoxide production in man. J Clin Invest 1963, 42: 1172-1178. 10.1172/JCI104802
Tenhunen R, Marver HS, Schmid R: The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA 1968, 61: 748-755. 10.1073/pnas.61.2.748
Maines MD, Kappas A: Cobalt induction of hepatic heme oxygenase; with evidence that cytochrome P-450 is not essential for this enzyme activity. Proc Natl Acad Sci USA 1974, 71: 4293-4297. 10.1073/pnas.71.11.4293
Yoshida T, Takahashi S, Kikuchi G: Partial purification and reconstitution of the heme oxygenase system from pig spleen microsomes. J Biochem 1974, 75: 1187-1191.
Maines MD, Trakshel GM, Kutty RK: Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem 1986, 261: 411-419.
Trakshel GM, Kutty RK, Maines MD: Purification and characterization of the major constitutive form of testicular heme oxygenase. The noninducible isoform. J Biol Chem 1986, 261: 11131-11137.
McCoubrey WK Jr, Huang TJ, Maines MD: Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur J Biochem 1997, 247: 725-732. 10.1111/j.1432-1033.1997.00725.x
Hayashi S, Omata Y, Sakamoto H, Higashimoto Y, Hara T, Sagara Y, Noguchi M: Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336: 241-250. 10.1016/j.gene.2004.04.002
Maines MD: The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 1997, 37: 517-554. 10.1146/annurev.pharmtox.37.1.517
Bauer M, Bauer I: Heme oxygenase-1: redox regulation and role in the hepatic response to oxidative stress. Antioxid Redox Signal 2002, 4: 749-758. 10.1089/152308602760598891
Ryter SW, Alam J, Choi AM: Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 2006, 86: 583-650. 10.1152/physrev.00011.2005
Loboda A, Jazwa A, Grochot-Przeczek A, Rutkowski AJ, Cisowski J, Agarwal A, Jozkowicz A, Dulak J: Heme oxygenase-1 and the vascular bed: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 2008, 10: 1767-1812. 10.1089/ars.2008.2043
Ferrandiz ML, Devesa I: Inducers of heme oxygenase-1. Curr Pharm Des 2008, 14: 473-486. 10.2174/138161208783597399
Bauer M, Huse K, Settmacher U, Claus RA: The heme oxygenasecarbon monoxide system: regulation and role in stress response and organ failure. Intensive Care Med 2008, 34: 640-648. 10.1007/s00134-008-1010-2
Bauer I, Wanner GA, Rensing H, Alte C, Miescher EA, Wolf B, Pannen BH, Clemens MG, Bauer M: Expression pattern of heme oxygenase isoenzymes 1 and 2 in normal and stress-exposed rat liver. Hepatology 1998, 27: 829-838. 10.1002/hep.510270327
Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, Ohta K, Kasahara Y, Koizumi S: Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest 1999, 103: 129-135. 10.1172/JCI4165
Kawashima A, Oda Y, Yachie A, Koizumi S, Nakanishi I: Heme oxygenase-1 deficiency: the first autopsy case. Hum Pathol 2002, 33: 125-130. 10.1053/hupa.2002.30217
Kubulus D, Rensing H, Paxian M, Thierbach JT, Meisel T, Redl H, Bauer M, Bauer I: Influence of heme-based solutions on stress protein expression and organ failure after hemorrhagic shock. Crit Care Med 2005, 33: 629-637. 10.1097/01.CCM.0000156295.48075.49
Kubulus D, Mathes A, Pradarutti S, Raddatz A, Heiser J, Pavlidis D, Wolf B, Bauer I, Rensing H: Hemin arginate-induced heme oxygenase 1 expression improves liver microcirculation and mediates an anti-inflammatory cytokine response after hemorrhagic shock. Shock 2008, 29: 583-590. 10.1097/SHK.0b013e318157e526
Mersmann J, Tran N, Zacharowski PA, Grotemeyer D, Zacharowski K: Rosiglitazone is cardioprotective in a murine model of myocardial I/R. Shock 2008, 30: 64-68. 10.1097/SHK.0b013e31815dbdc3
Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, Rosenberg ME: Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest 1992, 90: 267-270. 10.1172/JCI115847
Otterbein L, Sylvester SL, Choi AM: Hemoglobin provides protection against lethal endotoxemia in rats: the role of heme oxygenase-1. Am J Respir Cell Mol Biol 1995, 13: 595-601.
Pannen BH, Kohler N, Hole B, Bauer M, Clemens MG, Geiger KK: Protective role of endogenous carbon monoxide in hepatic microcirculatory dysfunction after hemorrhagic shock in rats. J Clin Invest 1998, 102: 1220-1228. 10.1172/JCI3428
Rensing H, Bauer I, Datene V, Patau C, Pannen BH, Bauer M: Differential expression pattern of heme oxygenase-1/heat shock protein 32 and nitric oxide synthase-II and their impact on liver injury in a rat model of hemorrhage and resuscitation. Crit Care Med 1999, 27: 2766-2775. 10.1097/00003246-199912000-00027
Rossler OG, Bauer I, Chung HY, Thiel G: Glutamate-induced cell death of immortalized murine hippocampal neurons: neuroprotective activity of heme oxygenase-1, heat shock protein 70, and sodium selenite. Neurosci Lett 2004, 362: 253-257. 10.1016/j.neulet.2004.03.033
Schmidt R, Tritschler E, Hoetzel A, Loop T, Humar M, Halverscheid L, Geiger KK, Pannen BH: Heme oxygenase-1 induction by the clinically used anesthetic isoflurane protects rat livers from ischemia/reperfusion injury. Ann Surg 2007, 245: 931-942. 10.1097/01.sla.0000256891.45790.4d
Tullius SG, Nieminen-Kelhä M, Buelow R, Reutzel-Selke A, Martins PN, Pratschke J, Bachmann U, Lehmann M, Southard D, Iyer S, Schmidbauer G, Sawitzki B, Reinke P, Neuhaus P, Volk HD: Inhibition of ischemia/reperfusion injury and chronic graft deterioration by a single-donor treatment with cobalt-protoporphyrin for the induction of heme oxygenase-1. Transplantation 2002, 74: 591-598. 10.1097/00007890-200209150-00001
Applegate LA, Luscher P, Tyrrell RM: Induction of heme oxygenase: a general response to oxidant stress in cultured mammalian cells. Cancer Res 1991, 51: 974-978.
Balla G, Vercellotti GM, Muller-Eberhard U, Eaton J, Jacob HS: Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab Invest 1991, 64: 648-655.
Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN: Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235: 1043-1046. 10.1126/science.3029864
Djousse L, Rothman KJ, Cupples LA, Levy D, Ellison RC: Effect of serum albumin and bilirubin on the risk of myocardial infarction (the Framingham Offspring Study). Am J Cardiol 2003, 91: 485-488. 10.1016/S0002-9149(02)03256-3
Eisenstein RS, Garcia-Mayol D, Pettingell W, Munro HN: Regulation of ferritin and heme oxygenase synthesis in rat fibroblasts by different forms of iron. Proc Natl Acad Sci USA 1991, 88: 688-692. 10.1073/pnas.88.3.688
Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, Eaton JW, Vercellotti GM: Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem 1992, 267: 18148-18153.
Furchgott RF, Jothianandan D: Endothelium-dependent and -independent vasodilation involving cyclic GMP: relaxation induced by nitric oxide, carbon monoxide and light. Blood Vessels 1991, 28: 52-61.
Morita T, Mitsialis SA, Koike H, Liu Y, Kourembanas S: Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J Biol Chem 1997, 272: 32804-32809. 10.1074/jbc.272.52.32804
Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH: Carbon monoxide: a putative neural messenger. Science 1993, 259: 381-384. 10.1126/science.7678352
Cardell LO, Ueki IF, Stjärne P, Agusti C, Takeyama K, Lindén A, Nadel JA: Bronchodilatation in vivo by carbon monoxide, a cyclic GMP related messenger. Br J Pharmacol 1998, 124: 1065-1068. 10.1038/sj.bjp.0701878
Suematsu M, Kashiwagi S, Sano T, Goda N, Shinoda Y, Ishimura Y: Carbon monoxide as an endogenous modulator of hepatic vascular perfusion. Biochem Biophys Res Commun 1994, 205: 1333-1337. 10.1006/bbrc.1994.2811
Brune B, Ullrich V: Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol Pharmacol 1987, 32: 497-504.
Fujita T, Toda K, Karimova A, Yan SF, Naka Y, Yet SF, Pinsky DJ: Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med 2001, 7: 598-604. 10.1038/87929
Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, Usheva A, Stachulak C, Bodyak N, Smith RN, Csizmadia E, Tyagi S, Akamatsu Y, Flavell RJ, Billiar TR, Tzeng E, Bach FH, Choi AM, Soares MP: Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med 2003, 9: 183-190. 10.1038/nm817
Utz J, Ullrich V: Carbon monoxide relaxes ileal smooth muscle through activation of guanylate cyclase. Biochem Pharmacol 1991, 41: 1195-1201. 10.1016/0006-2952(91)90658-R
Wang R, Wu L, Wang Z: The direct effect of carbon monoxide on KCa channels in vascular smooth muscle cells. Pflugers Arch 1997, 434: 285-291. 10.1007/s004240050398
Ryter SW, Otterbein LE, Morse D, Choi AM: Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance. Mol Cell Biochem 2002, 234–235: 249-263. 10.1023/A:1015957026924
Kim HP, Ryter SW, Choi AM: CO as a cellular signaling molecule. Annu Rev Pharmacol Toxicol 2006, 46: 411-449. 10.1146/annurev.pharmtox.46.120604.141053
Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM: Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 2000, 6: 422-428. 10.1038/74680
Kim HP, Wang X, Zhang J, Suh GY, Benjamin IJ, Ryter SW, Choi AM: Heat shock protein-70 mediates the cytoprotective effect of carbon monoxide: involvement of p38 beta MAPK and heat shock factor-1. J Immunol 2005, 175: 2622-2629.
Kim HP, Wang X, Nakao A, Kim SI, Murase N, Choi ME, Ryter SW, Choi AM: Caveolin-1 expression by means of p38beta mitogen-activated protein kinase mediates the antiproliferative effect of carbon monoxide. Proc Natl Acad Sci USA 2005, 102: 11319-11324. 10.1073/pnas.0501345102
Zhang X, Shan P, Alam J, Fu XY, Lee PJ: Carbon monoxide differentially modulates STAT1 and STAT3 and inhibits apoptosis via a phosphatidylinositol 3-kinase/Akt and p38 kinase-dependent STAT3 pathway during anoxia-reoxygenation injury. J Biol Chem 2005, 280: 8714-8721. 10.1074/jbc.M408092200
Kim HS, Loughran PA, Rao J, Billiar TR, Zuckerbraun BS: Carbon monoxide activates NF-kappaB via ROS generation and Akt pathways to protect against cell death of hepatocytes. Am J Physiol Gastrointest Liver Physiol 2008, 295: G146-G152. 10.1152/ajpgi.00105.2007
Sato K, Balla J, Otterbein L, Smith RN, Brouard S, Lin Y, Csizmadia E, Sevigny J, Robson SC, Vercellotti G, Choi AM, Bach FH, Soares MP: Carbon monoxide generated by heme oxygenase-1 suppresses the rejection of mouse-to-rat cardiac transplants. J Immunol 2001, 166: 4185-4194.
Abraham NG, Asija A, Drummond G, Peterson S: Heme oxygenase-1 gene therapy: recent advances and therapeutic applications. Curr Gene Ther 2007, 7: 89-108. 10.2174/156652307780363134
Stupfel M, Bouley G: Physiological and biochemical effects on rats and mice exposed to small concentrations of carbon monoxide for long periods. Ann N Y Acad Sci 1970, 174: 342-368. 10.1111/j.1749-6632.1970.tb49799.x
Herrmann WA: 100 years of metal carbonyls: a serendipitous chemical discovery of major scientific and industrial impact. J Organomet Chem 1990, 383: 21-44. 10.1016/0022-328X(90)85120-N
Motterlini R: Carbon monoxide-releasing molecules (CO-RMs): vasodilatory, anti-ischaemic and anti-inflammatory activities. Biochem Soc Trans 2007, 35: 1142-1146. 10.1042/BST0351142
Motterlini R, Mann BE, Foresti R: Therapeutic applications of carbon monoxide-releasing molecules. Expert Opin Investig Drugs 2005, 14: 1305-1318. 10.1517/13543784.14.11.1305
Stewart RD, Peterson JE, Baretta ED, Bachand RT, Hosko MJ, Herrmann AA: Experimental human exposure to carbon monoxide. Arch Environ Health 1970, 21: 154-164.
Wu L, Wang R: Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev 2005, 57: 585-630. 10.1124/pr.57.4.3
Schober P, Koch A, Zacharowski K, Loer SA: [Carbon monoxide: toxic molecule with antiinflammatory and cytoprotective properties]. Anasthesiol Intensivmed Notfallmed Schmerzther 2006, 41: 140-149. 10.1055/s-2006-925109
Hoetzel A, Schmidt R: [Carbon monoxide – poison or potential therapeutic?]. Anaesthesist 2006, 55: 1068-1079. 10.1007/s00101-006-1056-x
Hoetzel A, Dolinay T, Schmidt R, Choi AM, Ryter SW: Carbon monoxide in sepsis. Antioxid Redox Signal 2007, 9: 2013-2026. 10.1089/ars.2007.1762
Otterbein LE, Mantell LL, Choi AM: Carbon monoxide provides protection against hyperoxic lung injury. Am J Physiol 1999, 276: L688-L694.
Otterbein LE, Otterbein SL, Ifedigbo E, Liu F, Morse DE, Fearns C, Ulevitch RJ, Knickelbein R, Flavell RA, Choi AM: MKK3 mitogen-activated protein kinase pathway mediates carbon monoxide-induced protection against oxidant-induced lung injury. Am J Pathol 2003, 163: 2555-2563.
Dubuis E, Potier M, Wang R, Vandier C: Continuous inhalation of carbon monoxide attenuates hypoxic pulmonary hypertension development presumably through activation of BKCa channels. Cardiovasc Res 2005, 65: 751-761. 10.1016/j.cardiores.2004.11.007
Zuckerbraun BS, Chin BY, Wegiel B, Billiar TR, Czsimadia E, Rao J, Shimoda L, Ifedigbo E, Kanno S, Otterbein LE: Carbon monoxide reverses established pulmonary hypertension. J Exp Med 2006, 203: 2109-2119. 10.1084/jem.20052267
Nemzek JA, Fry C, Abatan O: Low-dose carbon monoxide treatment attenuates early pulmonary neutrophil recruitment after acid aspiration. Am J Physiol Lung Cell Mol Physiol 2008, 294: L644-L653. 10.1152/ajplung.00324.2007
Chapman JT, Otterbein LE, Elias JA, Choi AM: Carbon monoxide attenuates aeroallergen-induced inflammation in mice. Am J Physiol Lung Cell Mol Physiol 2001, 281: L209-L216.
Ameredes BT, Otterbein LE, Kohut LK, Gligonic AL, Calhoun WJ, Choi AM: Low-dose carbon monoxide reduces airway hyper-responsiveness in mice. Am J Physiol Lung Cell Mol Physiol 2003, 285: L1270-L1276.
Kohmoto J, Nakao A, Stolz DB, Kaizu T, Tsung A, Ikeda A, Shimizu H, Takahashi T, Tomiyama K, Sugimoto R, Choi AM, Billiar TR, Murase N, McCurry KR: Carbon monoxide protects rat lung transplants from ischemia-reperfusion injury via a mechanism involving p38 MAPK pathway. Am J Transplant 2007, 7: 2279-2290. 10.1111/j.1600-6143.2007.01940.x
Mishra S, Fujita T, Lama VN, Nam D, Liao H, Okada M, Minamoto K, Yoshikawa Y, Harada H, Pinsky DJ: Carbon monoxide rescues ischemic lungs by interrupting MAPK-driven expression of early growth response 1 gene and its downstream target genes. Proc Natl Acad Sci USA 2006, 103: 5191-5196. 10.1073/pnas.0600241103
Song R, Kubo M, Morse D, Zhou Z, Zhang X, Dauber JH, Fabisiak J, Alber SM, Watkins SC, Zuckerbraun BS, Otterbein LE, Ning W, Oury TD, Lee PJ, McCurry KR, Choi AM: Carbon monoxide induces cytoprotection in rat orthotopic lung transplantation via anti-inflammatory and anti-apoptotic effects. Am J Pathol 2003, 163: 231-242.
Zhang X, Shan P, Alam J, Davis RJ, Flavell RA, Lee PJ: Carbon monoxide modulates Fas/Fas ligand, caspases, and Bcl-2 family proteins via the p38alpha mitogen-activated protein kinase pathway during ischemia-reperfusion lung injury. J Biol Chem 2003, 278: 22061-22070. 10.1074/jbc.M301858200
Dolinay T, Szilasi M, Liu M, Choi AM: Inhaled carbon monoxide confers antiinflammatory effects against ventilator-induced lung injury. Am J Respir Crit Care Med 2004, 170: 613-620. 10.1164/rccm.200401-023OC
Hoetzel A, Dolinay T, Vallbracht S, Zhang Y, Kim HP, Ifedigbo E, Alber S, Kaynar AM, Schmidt R, Ryter SW, Choi AM: Carbon monoxide protects against ventilator-induced lung injury via PPAR-gamma and inhibition of Egr-1. Am J Respir Crit Care Med 2008, 177: 1223-1232. 10.1164/rccm.200708-1265OC
Boutros C, Zegdi R, Lila N, Cambillau M, Fornes P, Carpentier A, Fabini JN: Carbon monoxide can prevent acute lung injury observed after ischemia reperfusion of the lower extremities. J Surg Res 2007, 143: 437-442. 10.1016/j.jss.2007.02.013
Goebel U, Siepe M, Mecklenburg A, Stein P, Roesslein M, Schwer CI, Schmidt R, Doenst T, Geiger KK, Pahl HL, Schlensak C, Loop T: Carbon monoxide inhalation reduces pulmonary inflammatory response during cardiopulmonary bypass in pigs. Anesthesiology 2008, 108: 1025-1036. 10.1097/ALN.0b013e3181733115
Zuckerbraun BS, Billiar TR, Otterbein SL, Kim PK, Liu F, Choi AM, Bach FH, Otterbein LE: Carbon monoxide protects against liver failure through nitric oxide-induced heme oxygenase 1. J Exp Med 2003, 198: 1707-1716. 10.1084/jem.20031003
Tsui TY, Obed A, Siu YT, Yet SF, Prantl L, Schlitt HJ, Fan ST: Carbon monoxide inhalation rescues mice from fulminant hepatitis through improving hepatic energy metabolism. Shock 2007, 27: 165-171. 10.1097/01.shk.0000239781.71516.61
Zuckerbraun BS, McCloskey CA, Gallo D, Liu F, Ifedigbo E, Otterbein LE, Billiar TR: Carbon monoxide prevents multiple organ injury in a model of hemorrhagic shock and resuscitation. Shock 2005, 23: 527-532.
Kaizu T, Ikeda A, Nakao A, Tsung A, Toyokawa H, Ueki S, Geller DA, Murase N: Protection of transplant-induced hepatic ischemia/reperfusion injury with carbon monoxide via MEK/ERK1/2 pathway downregulation. Am J Physiol Gastrointest Liver Physiol 2008, 294: G236-G244. 10.1152/ajpgi.00144.2007
Sun BW, Chen ZY, Chen X, Liu C: Attenuation of leukocytes sequestration by carbon monoxide-releasing molecules: liberated carbon monoxide in the liver of thermally injured mice. J Burn Care Res 2007, 28: 173-181.
Sun BW, Sun Y, Sun ZW, Chen X: CO liberated from CORM-2 modulates the inflammatory response in the liver of thermally injured mice. World J Gastroenterol 2008, 14: 547-553. 10.3748/wjg.13.6127
Kalff JC, Carlos TM, Schraut WH, Billiar TR, Simmons RL, Bauer AJ: Surgically induced leukocytic infiltrates within the rat intestinal muscularis mediate postoperative ileus. Gastroenterology 1999, 117: 378-387. 10.1053/gast.1999.0029900378
Kalff JC, Hierholzer C, Tsukada K, Billiar TR, Bauer AJ: Hemorrhagic shock results in intestinal muscularis intercellular adhesion molecule (ICAM-1) expression, neutrophil infiltration, and smooth muscle dysfunction. Arch Orthop Trauma Surg 1999, 119: 89-93. 10.1007/s004020050363
Turler A, Schwarz NT, Turler E, Kalff JC, Bauer AJ: MCP-1 causes leukocyte recruitment and subsequently endotoxemic ileus in rat. Am J Physiol Gastrointest Liver Physiol 2002, 282: G145-G155.
Moore BA, Otterbein LE, Turler A, Choi AM, Bauer AJ: Inhaled carbon monoxide suppresses the development of postoperative ileus in the murine small intestine. Gastroenterology 2003, 124: 377-391. 10.1053/gast.2003.50060
Moore BA, Overhaus M, Whitcomb J, Ifedigbo E, Choi AM, Otterbein LE, Bauer AJ: Brief inhalation of low-dose carbon monoxide protects rodents and swine from postoperative ileus. Crit Care Med 2005, 33: 1317-1326. 10.1097/01.CCM.0000166349.76514.40
Nakao A, Schmidt J, Harada T, Tsung A, Stoffels B, Cruz RJ Jr, Kohmoto J, Peng X, Tomiyama K, Murase N, Bauer AJ, Fink MP: A single intraperitoneal dose of carbon monoxide-saturated ringer's lactate solution ameliorates postoperative ileus in mice. J Pharmacol Exp Ther 2006, 319: 1265-1275. 10.1124/jpet.106.108654
Nakao A, Moore BA, Murase N, Liu F, Zuckerbraun BS, Bach FH, Choi AM, Nalesnik MA, Otterbein LE, Bauer AJ: Immunomodulatory effects of inhaled carbon monoxide on rat syngeneic small bowel graft motility. Gut 2003, 52: 1278-1285. 10.1136/gut.52.9.1278
Nakao A, Kimizuka K, Stolz DB, Neto JS, Kaizu T, Choi AM, Uchiyama T, Zuckerbraun BS, Nalesnik MA, Otterbein LE, Murase N: Carbon monoxide inhalation protects rat intestinal grafts from ischemia/reperfusion injury. Am J Pathol 2003, 163: 1587-1598.
Nakao A, Toyokawa H, Tsung A, Nalesnik MA, Stolz DB, Kohmoto J, Ikeda A, Tomiyama K, Harada T, Takahashi T, Yang R, Fink MP, Morita K, Choi AM, Murase N: Ex vivo application of carbon monoxide in University of Wisconsin solution to prevent intestinal cold ischemia/reperfusion injury. Am J Transplant 2006, 6: 2243-2255. 10.1111/j.1600-6143.2006.01465.x
True AL, Olive M, Boehm M, San H, Westrick RJ, Raghavachari N, Xu X, Lynn EG, Sack MN, Munson PJ, Gladwin MT, Nabel EG: Heme oxygenase-1 deficiency accelerates formation of arterial thrombosis through oxidative damage to the endothelium, which is rescued by inhaled carbon monoxide. Circ Res 2007, 101: 893-901. 10.1161/CIRCRESAHA.107.158998
Hangai-Hoger N, Tsai AG, Cabrales P, Suematsu M, Intaglietta M: Microvascular and systemic effects following top load administration of saturated carbon monoxide-saline solution. Crit Care Med 2007, 35: 1123-1132. 10.1097/01.CCM.0000259533.84180.C7
Akamatsu Y, Haga M, Tyagi S, Yamashita K, Graça-Souza AV, Ollinger R, Czismadia E, May GA, Ifedigbo E, Otterbein LE, Bach FH, Soares MP: Heme oxygenase-1-derived carbon monoxide protects hearts from transplant associated ischemia reperfusion injury. FASEB J 2004, 18: 771-772.
Nakao A, Toyokawa H, Abe M, Kiyomoto T, Nakahira K, Choi AM, Nalesnik MA, Thomson AW, Murase N: Heart allograft protection with low-dose carbon monoxide inhalation: effects on inflammatory mediators and alloreactive T-cell responses. Transplantation 2006, 81: 220-230. 10.1097/01.tp.0000188637.80695.7f
Lavitrano M, Smolenski RT, Musumeci A, Maccherini M, Slominska E, Di Florio E, Bracco A, Mancini A, Stassi G, Patti M, Giovannoni R, Froio A, Simeone F, Forni M, Bacci ML, D'Alise G, Cozzi E, Otterbein LE, Yacoub MH, Bach FH, Calise F: Carbon monoxide improves cardiac energetics and safeguards the heart during reperfusion after cardiopulmonary bypass in pigs. FASEB J 2004, 18: 1093-1095.
Fujimoto H, Ohno M, Ayabe S, Kobayashi H, Ishizaka N, Kimura H, Yoshida K, Nagai R: Carbon monoxide protects against cardiac ischemia-reperfusion injury in vivo via MAPK and Akt-eNOS pathways. Arterioscler Thromb Vasc Biol 2004, 24: 1848-1853. 10.1161/01.ATV.0000142364.85911.0e
Tullius SG, Heemann U, Hancock WW, Azuma H, Tilney NL: Long-term kidney isografts develop functional and morphologic changes that mimic those of chronic allograft rejection. Ann Surg 1994, 220: 425-432. 10.1097/00000658-199410000-00002
Gueler F, Gwinner W, Schwarz A, Haller H: Long-term effects of acute ischemia and reperfusion injury. Kidney Int 2004, 66: 523-527. 10.1111/j.1523-1755.2004.761_11.x
Neto JS, Nakao A, Toyokawa H, Nalesnik MA, Romanosky AJ, Kimizuka K, Kaizu T, Hashimoto N, Azhipa O, Stolz DB, Choi AM, Murase N: Low-dose carbon monoxide inhalation prevents development of chronic allograft nephropathy. Am J Physiol Renal Physiol 2006, 290: F324-F334. 10.1152/ajprenal.00026.2005
Sandouka A, Fuller BJ, Mann BE, Green CJ, Foresti R, Motterlini R: Treatment with CO-RMs during cold storage improves renal function at reperfusion. Kidney Int 2006, 69: 239-247. 10.1038/sj.ki.5000016
Faleo G, Neto JS, Kohmoto J, Tomiyama K, Shimizu H, Takahashi T, Wang Y, Sugimoto R, Choi AM, Stolz DB, Carrieri G, McCurry KR, Murase N, Nakao A: Carbon monoxide ameliorates renal cold ischemia-reperfusion injury with an upregulation of vascular endothelial growth factor by activation of hypoxia-inducible factor. Transplantation 2008, 85: 1833-1840. 10.1097/TP.0b013e31817c6f63
Nakao A, Faleo G, Shimizu H, Nakahira K, Kohmoto J, Sugimoto R, Choi AM, McCurry KR, Takahashi T, Murase N: Ex vivo carbon monoxide prevents cytochrome P450 degradation and ischemia/reperfusion injury of kidney grafts. Kidney Int 2008, 74: 1009-1016. 10.1038/ki.2008.342
Mazzola S, Forni M, Albertini M, Bacci ML, Zannoni A, Gentilini F, Lavitrano M, Bach FH, Otterbein LE, Clement MG: Carbon monoxide pretreatment prevents respiratory derangement and ameliorates hyperacute endotoxic shock in pigs. FASEB J 2005, 19: 2045-2047.
Bilban M, Bach FH, Otterbein SL, Ifedigbo E, d'Avila JC, Esterbauer H, Chin BY, Usheva A, Robson SC, Wagner O, Otterbein LE: Carbon monoxide orchestrates a protective response through PPARgamma. Immunity 2006, 24: 601-610. 10.1016/j.immuni.2006.03.012
Sarady JK, Zuckerbraun BS, Bilban M, Wagner O, Usheva A, Liu F, Ifedigbo E, Zamora R, Choi AM, Otterbein LE: Carbon monoxide protection against endotoxic shock involves reciprocal effects on iNOS in the lung and liver. FASEB J 2004, 18: 854-856.
Liu SH, Xu XR, Ma K, Xu B: Protection of carbon monoxide inhalation on lipopolysaccharide-induced multiple organ injury in rats. Chin Med Sci J 2007, 22: 169-176.
Aberg AM, Abrahamsson P, Johansson G, Haney M, Winso O, Larsson JE: Does carbon monoxide treatment alter cytokine levels after endotoxin infusion in pigs? A randomized controlled study. J Inflamm (Lond) 2008, 5: 13. 10.1186/1476-9255-5-13
Mayr FB, Spiel A, Leitner J, Marsik C, Germann P, Ullrich R, Wagner O, Jilma B: Effects of carbon monoxide inhalation during experimental endotoxemia in humans. Am J Respir Crit Care Med 2005, 171: 354-360. 10.1164/rccm.200404-446OC
Yasuda H, Yamaya M, Nakayama K, Ebihara S, Sasaki T, Okinaga S, Inoue D, Asada M, Nemoto M, Sasaki H: Increased arterial carboxyhemoglobin concentrations in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005, 171: 1246-1251. 10.1164/rccm.200407-914OC
Bathoorn E, Slebos DJ, Postma DS, Koeter GH, van Oosterhout AJ, Toorn M, Boezen HM, Kerstjens HA: Anti-inflammatory effects of inhaled carbon monoxide in patients with COPD: a pilot study. Eur Respir J 2007, 30: 1131-1137. 10.1183/09031936.00163206
Carbon Monoxide to Prevent Lung Inflammation[http://www.clinicaltrials.gov/ct2/show/NCT00094406?term=inhaled+carbon+monoxide+lung+inflammatory+responses+endotoxin&rank=1]
Acknowledgements
Supported by grants from the Deutsche Forschungsgemeinschaft (DFG PA 533/3-2 and PA 533/4-1), from the Else-Kroener-Fresenius Stiftung (A68/06), and from the University of Saarland (HOMFOR A/2003/42).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Bauer, I., Pannen, B.H. Bench-to-bedside review: Carbon monoxide – from mitochondrial poisoning to therapeutic use. Crit Care 13, 220 (2009). https://doi.org/10.1186/cc7887
Published:
DOI: https://doi.org/10.1186/cc7887