
Abstract
Sepsis still represents an important clinical and economic challenge for intensive care units. Severe complications like multi-organ failure with high mortality and the lack of specific diagnostic tools continue to hamper the development of improved therapies for sepsis. Fundamental questions regarding the cellular pathogenesis of experimental and clinical sepsis remain unresolved. According to experimental data, inhibiting macrophage migration inhibitory factor, high-mobility group box protein 1 (HMGB1), and complement factor C5a and inhibiting the TREM-1 (triggering receptor expressed on myeloid cells 1) signaling pathway and apoptosis represent promising new therapeutic options. In addition, we have demonstrated that blocking the signal transduction pathway of receptor of advanced glycation endproducts (RAGE), a new inflammation-perpetuating receptor and a member of the immunglobulin superfamily, increases survival in experimental sepsis. The activation of RAGE by advanced glycation end-products, S100, and HMGB1 initiates nuclear factor kappa B and mitogen-activated protein kinase pathways. Importantly, the survival rate of RAGE knockout mice was more than fourfold that of wild-type mice in a septic shock model of cecal ligation and puncture (CLP). Additionally, the application of soluble RAGE, an extracellular decoy for RAGE ligands, improves survival in mice after CLP, suggesting that RAGE is a central player in perpetuating the innate immune response. Understanding the basic signal transduction events triggered by this multi-ligand receptor may offer new diagnostic and therapeutic options in patients with sepsis.
Similar content being viewed by others

Introduction
In the United States, sepsis is the main cause of death in non-cardiac intensive care units and is linked with increasing costs for patient care. Sepsis represents a range of disorders involving bacterial, fungal, or viral infections that can be disseminated by the bloodstream [1]. Epidemiological data from North America show an incidence of 3.0 cases per 1,000 persons. The overall mortality is approximately 50% in patients with severe septic shock [2]. Even high-priority engagement in sepsis research has led to only slight improvements in existing treatment strategies for sepsis. Currently, the detailed mechanisms linking the foreign bacterial agent (for example, in the bloodstream or in the abdomen) with the sophisticated ongoing transcription work of the cell nucleus are not completely understood.
The combined use of the pre-existing innate and inducible adaptive immune systems ensures that the host will be able to mount an appropriate immune response against different types of pathogenic agents [1]. The first line of defense is the innate immune system, which is characterized by non-clonally distributed leukocytes that react rapidly to microbial products without antigenic specificity. Host innate responses to bacterial or fungal infections are primarily mediated by neutrophils and monocytes/macrophages. These cells express germline-encoded pattern-recognition receptors (PRRs), which recognize certain invariable pathogen-associated molecular patterns, or PAMPs, shared by groups of micro-organisms. PRRs trigger signaling pathways that initiate an inflammatory response to infection [3]. Activating isoforms are truncated in their cytoplasmic tails and deliver stimulatory signals by associating with transmembrane adapter proteins, such as CD3γ, the γ-chain of Fc receptors, and DAP12 (also known as KARAP) [4]. Innate immune cells, however, also receive continuous off signals via inhibitory receptors that recognize ubiquitously expressed endogenous molecules. These receptors transmit their inhibitory signals through a cytoplasmatic immunoreceptor tyrosine-based inhibitory motif, or ITIM [5].
The balance between activating and inhibitory signals generated by the engagement of these receptors ultimately controls neutrophil- and macrophage-mediated phagocytosis, respiratory burst, and the release of proinflammatory cytokines. Under certain circumstances, an excessive inflammatory response to infectious agents can lead to septic shock. The disastrous endpoint of an overstimulated immune system is multiple organ failure as a result of endorgan damage. This process is characterized by the massive release of proinflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL)-1, macrophage migration inhibitory factor, and high-mobility group box protein 1 (HMGB1) [6, 7].
The receptor of advanced glycation endproducts (RAGE), a member of the immunglobulin superfamily, is involved in the signal transduction from pathogen substrates to cell activation during the onset and perpetuation of inflammation. Recent data suggest (a) that RAGE perpetuates and amplifies inflammation and (b) that targeting this receptor might attenuate hyperinflammation. Therefore, this multi-ligand receptor should be viewed as a PRR. Gaining further knowledge about the ligands and basic mechanisms of this receptor may offer new diagnostic and therapeutic options in patients with sepsis.
Receptor of advanced glycation endproducts
RAGE was first identified in lung tissue [8–10]. It is located on the basolateral membranes of alveolar epithelial type I cells [11], but RAGE mRNA has also been found in alveolar epithelial type II cells [12]. Originally, RAGE was identified as a receptor for advanced glycation endproducts (AGEs), explaining the choice of this name. AGEs are products of non-enzymatic glycation and oxidation of proteins, lipids, and other macromolecules that appear, in particular, under conditions of increased availability of reducing sugars and/or enhanced oxidative stress, especially when molecules turn over slowly and aldose levels are elevated [13, 14].
RAGE expression occurs both constitutively and inducibly, depending upon cell type and developmental stage. Whereas RAGE is constitutively expressed during embryonal development, RAGE expression is downregulated in adult life. Known exceptions are skin and lung, which constitutively express RAGE throughout life [15]. However, downregulated cells can be induced to express RAGE in situations in which inflammatory mediators and ligands accumulate [16, 17]. The activation of RAGE initiates nuclear factor kappa B (NF-κB) [18, 19] and mitogen-activated protein kinase (MAPK) pathways [20]. Additionally, RAGE-mediated cellular stimulation promotes increased expression of the receptor itself. This positive feedback loop, characterized by ligand-receptor interaction followed by increased expression of the receptor, suggests that RAGE functions as a propagation and perpetuation factor: the two-hit model of RAGE engagement is based on this finding [21]. The transcription factors regulating RAGE in this setting include specificity protein-1, activator protein-2, NF-κB, and NF-IL-6 [22]. Takada and colleagues [23] reported that matrix metalloproteinase-9 (gelatinase B) plays a critical role in concordant expression, at least in human pancreatic cancer cells.
Localization and structure of RAGE
The gene for RAGE is located on chromosome 6 near the major histocompatibility complex III in humans and mice, in the proximity of genes encoding TNF, lymphotoxin, and the homebox gene HOX12 [24, 25]. The extracellular domain of RAGE consists of one V-type immunglobulin domain followed by two C-type immunglobulin domains. The V-type domain, in particular, interacts with the potential extracellular ligands [9, 10, 19, 20]. The rest of the molecule is a single transmembrane-spanning domain completed by a 43-amino acid, highly charged cytosolic tail. This cytosolic tail lacks known signaling motifs such as phosphorylation sites or kinase domains. Hofmann and colleagues [26] showed that the cytosolic tail is essential for signal transduction of RAGE because a truncated form of RAGE, in which the cytosolic tail is deleted, can bind both ligands and the wild-type receptor but does not mediate any cellular activation. In the rat lung, extracellular signal-regulated kinase 1/2 (ERK-1/2) was shown to bind intracellularly to the cytoplasmic tail of RAGE, suggesting that ERK may play a role in RAGE signaling through interaction with RAGE [27]. The existence of truncated and partly secreted RAGE isoforms from the same gene implies that the pre-mRNA of RAGE in humans can be subjected to alternative splicing [13]. In contrast, the truncated isoforms in mice seem to be produced by carboxyl-terminal truncation [28].
Although only little is known about the physiologic role of RAGE, it may fit with the concept of pleiotropic antagonism [29]. This concept of an evolutionary basis for the development of age-related diseases postulates that genes that are beneficial during the reproductive phase of life may become deleterious to development later on. Formerly, this interest was mainly focused on the role of RAGE in chronic diseases. Particularly under pathologic conditions, RAGE is up-regulated in blood vessels, neurons, and transformed epithelia and is involved in several chronic diseases, such as rheumatoid arthritis, diabetes, inflammatory kidney disease, arteriosclerosis, inflammatory bowel disease, neurodegenerative disorders (especially Alzheimer disease), and wound-healing disorders [14].
RAGE interactions with its ligands in acute inflammation and sepsis
RAGE is a multi-ligand receptor and interacts with different structures to transmit a signal into the cell and recognizes three-dimensional structures rather than specific amino acid sequences. Therefore, RAGE seems to fulfill the requirements of a PRR. As a member of the immunoglobulin superfamily, it interacts with a diverse class of ligands, including AGEs [9, 10], S100/calgranulins [26], HMGB1 [30], amyloid β-peptide [31], amyloid A [32], leukocyte adhesion receptors [33], prions [34], Escherichia coli curli operons [35], and β-sheet fibrils [14].
The AGE-RAGE interaction
AGEs are a heterogeneous group of compounds produced by non-enzymatic glycation and oxidation of proteins and lipids that exhibit characteristic absorbance and fluorescence properties, Nε-(carboxymethyl)lysine being a highly reactive AGE [36, 37]. They are protease-resistant and can cause irreversible tissue damage. AGEs can bind to various cellular surface receptors and thereby induce post-receptor signaling, activation of transcription factors, and gene expression in vitro and in vivo. Several receptors that bind AGEs, including AGE-R1, AGE-R2, AGE-R3, the scavenger receptor II, and RAGE, have been identified [38]. Binding of AGEs (and other ligands) to RAGE generates intracellular reactive oxygen species and depletes antioxidant defense mechanisms at the same time [39, 40]. As a result, AGEs binding to RAGE, reduced glutathione, and ascorbic acid are diminished.
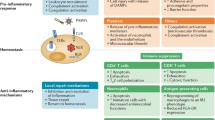
Depletion of glutathione leads to reduced glyoxalase-1 recycling and decreased in situ activity. Glyoxalase-1, however, has an important role in reducing the cellular AGE load [38, 41]. Furthermore, the myeloperoxidase in human phagocytes generates Nε-(carboxymethyl)lysine at sites of inflammation and thus sustains cellular activation via RAGE [36]. AGE-RAGE interaction activates intracellular signal transduction pathways, such as the ERK-1/2 kinases [27], the p38 MAPK, the stress-activated protein kinase/c-jun N-terminal kinase (SAPK/JNK) kinases [30, 42], rho-GTPases, phosphoinositide 3-kinases, JAK/STAT (Janus kinase/signal transducer and activator of transcription) pathway [17, 43, 44], and the NF-κB pathway [14] (Figure 1). In addition to activating the NF-κB pathway, triggering RAGE induces de novo p65 mRNA synthesis; this results in a growing pool of transcriptionally active NF-κBp65, which appears to overwhelm endogenous autoregulatory feedback inhibitory loops [18]. However, no proximal signaling events directly downstream of the receptor have been discovered yet. Only direct binding of ERK to the intracellular domain of RAGE has been demonstrated thus far [27].

Receptor of advanced glycation endproducts (RAGE)-mediated signal transduction. AGE, advanced glycation endproduct; C, C-type immunglobulin domain; ERK-1/2, extracellular signal-regulated kinase 1/2; HMGB1, high-mobility group box protein 1; ICAM-1, intercellular adhesion molecule 1; IκB, inhibitor of kappa B; IKK, inhibitor of kappa B kinase; JAK, Janus kinase; JNK, c-jun N-terminal kinase; MAC-1, macrophage-1 antigen; NF-κB, nuclear factor kappa B; P13K, phosphoinositide 3-kinase; ROS, reactive oxygen species; SAPK, stress-activated protein kinase; STAT, signal transducer and activator of transcription; V, V-type immunglobulin domain; VCAM-1, vascular cell adhesion molecule 1; VLA-4, very late antigen 4.
NF-κB is frequently present in sepsis, hyperglycemia, and oxidative stress. As already described, these conditions favor formation of 'advanced glycation endproducts', which can trigger RAGE and subsequently lead to sustained inflammation. Intensive insulin therapy interferes with this pathway and therefore may explain, in part, why this treatment modality is effective.
Relevance of RAGE-S100/calgranulin interaction
In addition to binding AGEs, RAGE binds proteins of the S100/calgranulin family, including S100-A12, also known as extracellular newly identified RAGE-binding protein (ENRAGE), and S100B [18, 26, 45]. Most of the S100/calgranulins are encoded on human chromosome 1q21 and represent a family of multiple members that have important intracellular properties that are linked to homeostatic properties, such as calcium binding [46, 47].
S100/calgranulin members such as S100A12 and S100B activate endothelial cells, macrophages, smooth muscle cells, and peripheral blood mononuclear cells (including T cells) via RAGE, thus triggering activation of signaling cascades and generation of cytokines and proinflammatory adhesion molecules [26, 48]. In addition, S100P stimulates cell proliferation and survival via RAGE [49]. However, whereas nano-molar concentrations of S100B induce trophic effects in RAGE-expressing cells, micromolar concentrations promote apoptosis, likely through oxidant stress [50]. RAGE-S100 interactions have been implicated in inflammation, too, since binding of S100A12 from the S100/calgranulin family to RAGE in murine macrophages resulted in the elaboration of IL-1β, TNF-α, and IL-2 [26]. Furthermore, EN-RAGE induced intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) expression on endothelial cells. EN-RAGE also decreased NF-κB activation and pro-inflammatory cytokine expression by blocking RAGE engagement. Intravenous infusion of EN-RAGE into mice enhanced VCAM-1 expression in the lungs, which was abrogated by soluble RAGE (sRAGE), neutralizing anti-EN-RAGE or anti-RAGE monoclonal antibody and lending support to the in vitro findings. In addition, treatment with sRAGE in murine models in vivo strongly diminished delayed-type hypersensitivity (DTH) and inflammatory colitis [26].
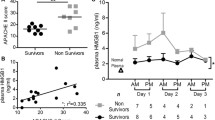
The precise mechanism by which transcription and translation of S100/calgranulins are regulated is still poorly understood. However, there is evidence that these molecules are released by activated monocytes, promoting the presence of S100/calgranulin at sites of inflammation [26, 47]. Interaction of these polypeptides and RAGE, therefore, might represent a proximal step in the cascade of events perpetuating inflammation. We [51] demonstrated that S100 species are increased in septic patients. However, we did not observe any significant difference between survivors and non-survivors.
Amphoterin/HMGB1 as RAGE ligand
Amphoterin, one of the HMGB DNA-binding proteins (amphoterin is another name for HMGB1), also acts as a signal-transducing ligand of RAGE. HMGB1, encoded on human chromosome 13q12-13, is a nuclear protein present in almost all eukaryotic cells. It stabilizes nucleosome function and acts as a transcription factor-like protein that regulates the expression of several genes [52]. The non-histone chromosomal protein HMGB1 not only has intracellular functions, but also may exist extracellularly and on the surface of cells, especially on migrating cells in neuronal development and tumors [43, 53, 54]. It is secreted as a cytokine by activated macrophages, mature dendritic cells, and natural killer cells in response to cell stimulation [55]. Active release is observed after acetylation in the nucleus, blocking re-entry into the nucleus by interacting with the nuclear-importer protein complex [56]. Thereafter, cytosolic HMGB1 migrates to cytoplasmic secretory vesicles, where it is released into the immunological synapse or into the extracellular space. Together with S100 [26], heat shock proteins [57], ATP [58], and uric acid [59], HMGB1 [60] is one of the main prototypes of the group of so-called damage-associated molecular pattern molecules: all of these molecules are released in response to infection or other inflammatory stimuli, especially during tissue damage (for example, by necrotic cells). Whereas HMGB1 is released from the nucleus, the other molecules are localized in the cytosol.
Cellular migration, invasion, and proliferation are enhanced when RAGE is engaged in tumor cells via HMGB1 [43]. A COOH-terminal motif in HMGB1 (amino acids 150 to 183) seems to be responsible for RAGE binding [61]. HMGB1 has a propagating role in inflammatory responses [6, 62] and seems to be an important RAGE ligand in sepsis and acute inflammation [52, 63, 64]. Recent studies have shown that the monocyte-derived HMGB1 is a late-acting cytokine mediator of endotoxin lethality. In animal experiments, the time-dependent induction of HMGB1 release by macrophage cultures could be detected 8 hours after lipopolysaccharide (LPS) stimulation. Furthermore, endotoxemia leads to a systemic increase in HMGB1 levels in mice [6, 64]. Systemic HMGB1 levels were also measured during endotoxemia in the serum of mice after injection of LPS. HMGB1 was first detected in serum after 8 hours and increased to a plateau from 16 to 32 hours after LPS stimulation [6]. Interestingly, this delay is one of the typical observations in patients with sepsis, when clinical signs appear several hours after the first infection-associated cytokines are detected in the bloodstream, and opens a therapeutic window. Examinations in healthy volunteers and septic patients showed (a) no HMGB1 in the serum of healthy humans, (b) dramatically increased HMGB1 levels in septic patients, and (c) markedly higher HMGB1 levels in non-survivors of septic shock than in patients who survived [6].
HMGB1 amplifies the cytokine cascade during systemic inflammation [62, 64]. In addition, HMGB1 seems to be an autocrine/paracrine regulator of monocyte invasion, involving RAGE through the endothelium [65]. The proinflammatory activity of HMGB1 is exerted by the B-box of the protein. When this HMGB1 B-box was added to enterocytic monolayers, intenstinal permeability increased [66]. These effects were strongly diminished in the presence of an anti-RAGE antibody, suggesting a significant role of RAGE in HMGB1-initiated pathogenic events. Using bone marrow macrophages from RAGE knockout (KO) mice, Kokkola and colleagues [67] recently provided formal proof that a major component of HMGB1 action on cells is mediated via RAGE. As a response to HMGB1 stimulation, macrophages from RAGE KO mice produced significantly lower amounts of TNF, IL-1β, and IL-6. However, cytokine production was not totally abrogated in RAGE-/- macrophages, although there was a significant difference from that of wild-type macrophages. In addition, phosphorylation of p38, p44/42, or SAPK/JNK kinases was similar to that of wild-type macrophages and macrophages from IL-1 RI KO mice. These data clearly indicate that RAGE is a major receptor for HMGB1 but that HMGB1 also exerts important effects via different receptors such as Toll-like receptor (TLR)-2 and TLR-4 [68].
Park and colleagues [68] showed that interactions of HMGB1 with TLR-2 and TLR-4 represent early events after macrophage exposure to HMGB1. In contrast, they found only scant evidence of binding between HMGB1 and RAGE in their experiments. Recently, Yu and colleagues [69] demonstrated that neutralizing antibodies against TLR-4, but not TLR-2 or RAGE, dose-dependently attenuated HMGB1-induced IL-8 release in human whole blood. The interaction of HMGB1 and TLR-4 seems to be important in liver ischemia/reperfusion also [70]. Interestingly, the N-terminal domain of thrombomodulin sequesters HMGB1, preventing HMGB1 from binding to RAGE [63]. Furthermore, a soluble form of this N-terminal domain of thrombomodulin protected mice from LPS-induced lethal shock. This survival benefit was observed in both wild-type and RAGE KO mice. Importantly, lethality in RAGE KO mice was only 50%, as compared with 100% in wild-type mice, after administration of a high dose of LPS. Neutralizing HMGB1 reduced the lethality in RAGE KO mice to zero, further confirming that HMGB1 exerts its deleterious effects not only via RAGE.
In vivo, administration of blocking antibodies to HMGB1 resulted in an improved survival in rodents subjected to high-dose LPS [6]. In an animal model of LPS-induced acute lung injury (ALI), the administration of anti-HMGB1, notably both before and after endotoxin administration, reduced the typical signs of lung damage in acute inflammation, such as neutrophil accumulation and lung edema [71]. These results were supported by Ueno and colleagues [72], who found that concentrations of HMGB1 were increased in plasma and lung epithelial lining fluid of patients with ALI. Extracellular HMGB1 may play a key role in the pathogenesis of clinical and experimental ALI. However, it is also expressed in healthy airways, which suggests that it plays a physiologic role in the lung as well [72]. Data have shown that anti-HMGB1 antibodies protect against sepsis in an animal model of cecal ligation and puncture (CLP), even when antibody administration is delayed by 24 hours [73]. These studies indicate that anti-HMGB1 antiserum may be a new, potential therapeutic target, as survival improved greatly in LPS- and CLP-treated mice [6, 73]. The observation that administration of blocking antibodies to HMGB1 protected mice from lethal septicemia strongly suggests that the engagement of cell surface receptors such as RAGE by HMGB1 might play an important role in mediating the pathogenic effects of HMGB1 [6].
Wang and colleagues [74] demonstrated that nicotinic stimulation prevents activation of the NF-κB pathway and inhibits HMGB1 secretion through a specific nicotinic anti-inflammatory pathway. In conclusion, acetylcholine seems to be the first known inhibitor of HMGB1 released from human macrophages. Nevertheless, it has not yet been formally proven that direct interaction of HMGB1-RAGE contributes to sepsis lethality, and other interactions such as HMGB1-TLR-2 and -TLR-4 are also important.
Potential clinical perspectives
Engagement of RAGE by its ligands results in sustained NF-κB activation [14] in all cell types studied so far, particularly mononuclear phagocytes and vascular endothelium [75]. Sustained cellular activation leads to cellular dysfunction and tissue destruction. When sRAGE used as a decoy, RAGE-neutralizing antibodies, and a dominant-negative receptor have been used, RAGE has been shown to be involved in different chronic disease models.
RAGE also has a critical role in acute inflammation. A resulting deleterious inflammatory response after ischemia/reperfusion of the liver has been associated with RAGE engagement in mice. The problem of ischemia/reperfusion is clinically relevant for liver transplantion or resection. In an animal model of total hepatic ischemia, blocking RAGE by administering sRAGE increased survival and caused fewer histological alterations in treated animals, which is in line with a decrease in RAGE-induced signaling and activation of transcription factors [76]. Furthermore, blocking RAGE significantly increased survival after massive liver resection [77].
We have clarified the role of RAGE in sepsis, DTH, and autoimmune encephalomyelitis (EAE) [75]. Several studies investigating the role of RAGE in inflammatory diseases used sRAGE to bind extracellulary potential RAGE ligands [26, 43, 78]. However, not only does sRAGE scavenge the ligands and prevent them from interacting with RAGE, but these ligands may be able to engage further receptor types and transduce completely different signaling pathways. To overcome this problem, RAGE KO mice were studied. In the setting of EAE, which served as a model to test the role of RAGE in the adaptive immune response, no differences between wild-type and RAGE KO mice could be detected [75]. Interestingly, RAGE transgenic mice showed significantly enhanced clinical EAE scores. In addition, RAGE KO mice and wild-type species developed the same inflammatory response in a second model (DTH). At first glance, these results seemed to be in contrast to previous data from DTH experiments with sRAGE, in which the inflammatory response in mice pretreated with sRAGE was lower [26]. The application of sRAGE in wild-type and RAGE KO mice, however, reduced the inflammatory response in mice in the DTH model [75]. These findings support the concept that the effects of sRAGE in DTH were not caused mainly by preventing ligand engagement of the cell-bound RAGE [75]. Thus, RAGE ligands exert their effects in these diseases through different receptors. Likely candidates to bind one or more of these ligands are known AGE receptors AGE-R1, AGE-R2, AGE-R3, and CD36 as well as newly identified AGE-binding proteins such as N-glycans, ezrin, and megalin [14].
In contrast to the minor role of RAGE in adaptive immunity, the most interesting finding was that RAGE KO mice were protected from lethal septic shock as compared with wild-type controls. In a CLP model, largely dependent on the innate immune response, RAGE KO mice were also better protected from lethal septic shock than were wild-type controls (80% versus 20%). To confirm the critical role of RAGE in sepsis and to exclude artifacts by gene deletion, RAGE KO mice were crossed into tie2-RAGE mice overexpressing RAGE in the vasculature. Mortality in these mice was similar to that in wild-type mice.
To test whether blocking of RAGE signaling pathways by sRAGE might be a therapeutic option, we injected sRAGE into wild-type mice, which resulted in an improved survival (40% versus 17%) compared with untreated control animals. NF-κB activation was more strongly induced in lungs of wild-type mice than in RAGE KO mice, suggesting a main contributing role in reducing mortality in RAGE KO mice and one that fits the high RAGE expression in the lung. In conclusion, these findings show that RAGE KO mice have, at least in part, a normal adaptive immune system. In contrast, the PRR RAGE displays a central role in the innate immune system, with an impact on perpetuation of the immune response [75].
In addition, we found that RAGE serves as a novel counter-receptor for the leukocyte β2 integrin Mac-1 (CD11b/CD18) and (to a lesser extent) p150,95 (CD11c/CD18), being directly involved in leukocyte recruitment in vitro and in vivo [33]. This leukocyte recruitment via RAGE is enhanced in the presence of S100 proteins. Thus, RAGE acts as an endothelial adhesion receptor, promoting leukocyte recruitment and subsequent inflammation by direct binding of leukocytes, and enhances expression of VCAM-1 and ICAM-1 after engagement of RAGE [14, 26]. These findings are consistent with the fact that histological examination showed fewer inflammatory cells adherent to the peritoneum of RAGE KO mice after CLP as compared with that of wild-type mice [75]. Remarkably, RAGE-/-seems to have a moderate proinflammatory phenotype because C-reactive protein, basal NF-κB activation, and cytokine levels were slightly increased. In conclusion, the crucial role of RAGE in experimental sepsis not only provides strong evidence for its perpetuating role in the innate immune response, but may open further opportunities to develop novel approaches for treating septicemia.
A number of studies, including clinical investigations, have shown that genetic variants of RAGE might be of further interest [79, 80]. These variants are found on coding/translational sequences as well as in the transcriptional regulatory elements. Hofmann and colleagues [81] found that the variant form of RAGE enhances binding and cytokine production compared with wild-type animals. What kind of cellular consequences these genetic polymorphisms have and what clinical relevance these polymorphisms will have remain to be determined in further studies.
Conclusion
This review summarizes the current knowledge on RAGE, a new inflammation-perpetuating receptor, which plays a pivotal role in sepsis. It is involved in signal transduction from pathogen substrates to cell activation during the onset of inflammation and perpetuates the immune response. Targeting this receptor might attenuate hyperinflammation. Essentially, understanding the basic signal transduction of RAGE may offer new diagnostic and therapeutic options in patients with sepsis.
Abbreviations
- AGE:
-
= advanced glycation endproduct
- ALI:
-
= acute lung injury
- CLP:
-
= cecal ligation and puncture
- DTH:
-
= delayed-type hypersensitivity
- EAE:
-
= experimental allergic (or autoimmune) encephalomyelitis
- EN-RAGE:
-
= extracellular newly identified receptor of advanced glycation endproducts-binding protein
- ERK-1/2:
-
= extracellular signal-regulated kinase 1/2
- HMGB1:
-
= high-mobility group box protein 1
- ICAM-1:
-
= intercellular adhesion molecule 1
- IL:
-
= interleukin
- JNK:
-
= c-jun N-terminal kinase
- KO:
-
= knockout
- LPS:
-
= lipopolysaccharide
- MAPK:
-
= mitogen-activated protein kinase
- NF-κB:
-
= nuclear factor kappa B
- PRR:
-
= pattern-recognition receptor
- RAGE:
-
= receptor of advanced glycation endproducts
- SAPK:
-
= stress-activated protein kinase
- sRAGE:
-
= soluble receptor of advanced glycation endproducts
- TLR:
-
= Toll-like receptor
- TNF:
-
= tumor necrosis factor
- VCAM-1:
-
= vascular cell adhesion molecule 1.
References
Weigand MA, Horner C, Bardenheuer HJ, Bouchon A: The systemic inflammatory response syndrome. Best Pract Res Clin Anaesthesiol 2004, 18: 455-475. 10.1016/j.bpa.2003.12.005
Martin GS, Mannino DM, Eaton S, Moss M: The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 2003, 348: 1546-1554. 10.1056/NEJMoa022139
Aderem A, Ulevitch RJ: Toll-like receptors in the induction of the innate immune response. Nature 2000, 406: 782-787. 10.1038/35021228
Vely F, Vivier E: Conservation of structural features reveals the existence of a large family of inhibitory cell surface receptors and noninhibitory/activatory counterparts. J Immunol 1997, 159: 2075-2077.
Colonna M: TREMs in the immune system and beyond. Nat Rev Immunol 2003, 3: 445-453. 10.1038/nri1106
Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et al.: HMGB-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285: 248-251. 10.1126/science.285.5425.248
Riedemann NC, Guo RF, Ward PA: Novel strategies for the treatment of sepsis. Nat Med 2003, 9: 517-524. 10.1038/nm0503-517
Lisanti MP, Scherer PE, Vidugiriene J, Tang Z, Hermanowski-Vosatka A, Tu YH, Cook RF, Sargiacomo M: Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J Cell Biol 1994, 126: 111-126. 10.1083/jcb.126.1.111
Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A: Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 1992, 267: 14998-15004.
Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, Esposito C, Hegarty H, Hurley W, Clauss M, et al.: Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem 1992, 267: 14987-14997.
Shirasawa M, Fujiwara N, Hirabayashi S, Ohno H, Iida J, Makita K, Hata Y: Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells 2004, 9: 165-174. 10.1111/j.1356-9597.2004.00712.x
Katsuoka F, Kawakami Y, Arai T, Imuta H, Fujiwara M, Kanma H, Yamashita K: Type II alveolar epithelial cells in lung express receptor for advanced glycation end products (RAGE) gene. Biochem Biophys Res Commun 1997, 238: 512-516. 10.1006/bbrc.1997.7263
Schlueter C, Hauke S, Flohr AM, Rogalla P, Bullerdiek J: Tissue-specific expression patterns of the RAGE receptor and its soluble forms–a result of regulated alternative splicing? Biochim Biophys Acta 2003, 1630: 1-6.
Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP: Understanding RAGE, the receptor for advanced glycation end products. J Mol Med 2005, 83: 876-886. 10.1007/s00109-005-0688-7
Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A, et al.: Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol 1993, 143: 1699-1712.
Huttunen HJ, Fages C, Rauvala H: Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem 1999, 274: 19919-19924. 10.1074/jbc.274.28.19919
Thornalley PJ: Glutathione-dependent detoxification of alpha-oxoaldehydes by the glyoxalase system: involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chem Biol Interact 1998, 111: 137-151. 10.1016/S0009-2797(97)00157-9
Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Kloting I, et al.: Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001, 50: 2792-2808. 10.2337/diabetes.50.12.2792
Sousa MM, Yan SD, Stern D, Saraiva MJ: Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Lab Invest 2000, 80: 1101-1110.
Degryse B, Bonaldi T, Scaffidi P, Muller S, Resnati M, Sanvito F, Arrigoni G, Bianchi ME: The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol 2001, 152: 1197-1206. 10.1083/jcb.152.6.1197
Chavakis T, Bierhaus A, Nawroth PP: RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes Infect 2004, 6: 1219-1225. 10.1016/j.micinf.2004.08.004
Li J, Schmidt AM: Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem 1997, 272: 16498-16506. 10.1074/jbc.272.26.16498
Takada M, Hirata K, Ajiki T, Suzuki Y, Kuroda Y: Expression of receptor for advanced glycation end products (RAGE) and MMP-9 in human pancreatic cancer cells. Hepatogastroenterology 2004, 51: 928-930.
Malherbe P, Richards JG, Gaillard H, Thompson A, Diener C, Schuler A, Huber G: cDNA cloning of a novel secreted isoform of the human receptor for advanced glycation end products and characterization of cells co-expressing cell-surface scavenger receptors and Swedish mutant amyloid precursor protein. Brain Res Mol Brain Res 1999, 71: 159-170. 10.1016/S0169-328X(99)00174-6
Sugaya K, Fukagawa T, Matsumoto K, Mita K, Takahashi E, Ando A, Inoko H, Ikemura T: Three genes in the human MHC class III region near the junction with the class II: gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int-3. Genomics 1994, 23: 408-419. 10.1006/geno.1994.1517
Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, et al.: RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97: 889-901. 10.1016/S0092-8674(00)80801-6
Ishihara K, Tsutsumi K, Kawane S, Nakajima M, Kasaoka T: The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett 2003, 550: 107-113. 10.1016/S0014-5793(03)00846-9
Hanford LE, Enghild JJ, Valnickova Z, Petersen SV, Schaefer LM, Schaefer TM, Reinhart TA, Oury TD: Purification and characterization of mouse soluble receptor for advanced glycation end products (sRAGE). J Biol Chem 2004, 279: 50019-50024. 10.1074/jbc.M409782200
Simm A, Bartling B, Silber RE: RAGE: a new pleiotropic antagonistic gene? Ann N Y Acad Sci 2004, 1019: 228-231. 10.1196/annals.1297.038
Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al.: The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem 1995, 270: 25752-25761. 10.1074/jbc.270.43.25752
Du Yan S, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW, Rajavashisth T, Chen X, Godman GC, Stern D, et al.: Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci U S A 1997, 94: 5296-5301. 10.1073/pnas.94.10.5296
Yan SD, Zhu H, Zhu A, Golabek A, Du H, Roher A, Yu J, Soto C, Schmidt AM, Stern D, et al.: Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat Med 2000, 6: 643-651. 10.1038/76216
Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, Nagashima M, Morser J, Arnold B, Preissner KT, et al.: The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med 2003, 198: 1507-1515. 10.1084/jem.20030800
Sasaki N, Takeuchi M, Chowei H, Kikuchi S, Hayashi Y, Nakano N, Ikeda H, Yamagishi S, Kitamoto T, Saito T, et al.: Advanced glycation end products (AGE) and their receptor (RAGE) in the brain of patients with Creutzfeldt-Jakob disease with prion plaques. Neurosci Lett 2002, 326: 117-120. 10.1016/S0304-3940(02)00310-5
Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ: Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 2002, 295: 851-855. 10.1126/science.1067484
Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, Hofmann M, Yan SF, Pischetsrieder M, Stern D, et al.: N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem 1999, 274: 31740-31749. 10.1074/jbc.274.44.31740
Anderson MM, Requena JR, Crowley JR, Thorpe SR, Heinecke JW: The myeloperoxidase system of human phagocytes generates Nepsilon-(carboxymethyl)lysine on proteins: a mechanism for producing advanced glycation end products at sites of inflammation. J Clin Invest 1999, 104: 103-113. 10.1172/JCI3042
Thornalley PJ: Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs. Cell Mol Biol (Noisy-le-grand) 1998, 44: 1013-1023.
Lander HM, Tauras JM, Ogiste JS, Hori O, Moss RA, Schmidt AM: Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem 1997, 272: 17810-17814. 10.1074/jbc.272.28.17810
Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL: Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab 2001, 280: E685-694.
Bierhaus A, Chevion S, Chevion M, Hofmann M, Quehenberger P, Illmer T, Luther T, Berentshtein E, Tritschler H, Muller M, et al.: Advanced glycation end product-induced activation of NF-kappaB is suppressed by alpha-lipoic acid in cultured endothelial cells. Diabetes 1997, 46: 1481-1490. 10.2337/diabetes.46.9.1481
Sorci G, Riuzzi F, Arcuri C, Giambanco I, Donato R: Amphoterin stimulates myogenesis and counteracts the antimyogenic factors basic fibroblast growth factor and S100B via RAGE binding. Mol Cell Biol 2004, 24: 4880-4894. 10.1128/MCB.24.11.4880-4894.2004
Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, et al.: Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000, 405: 354-360. 10.1038/35012626
Huang JS, Guh JY, Chen HC, Hung WC, Lai YH, Chuang LY: Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J Cell Biochem 2001, 81: 102-113. 10.1002/1097-4644(20010401)81:1<102::AID-JCB1027>3.0.CO;2-Y
Marenholz I, Heizmann CW, Fritz G: S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem Biophys Res Commun 2004, 322: 1111-1122. 10.1016/j.bbrc.2004.07.096
Donato R: S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol 2001, 33: 637-668. 10.1016/S1357-2725(01)00046-2
Schafer BW, Heinzmann CW: The S100 family of EF-hand calcium-binding proteins: functions and pathology. Trends Biochem Sci 1996, 21: 134-140.
Yan SSD, Wu ZY, Zhang HP, Furtado G, Chen X, Yan SF, Schmidt AM, Brown C, Stern A, LaFaille J, et al.: Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat Med 2003, 9: 287-293. 10.1038/nm831
Arumugam T, Simeone DM, Schmidt AM, Logsdon CD: S100P stimulates cell proliferation and survival via receptor for activated glycation end products (RAGE). J Biol Chem 2004, 279: 5059-5065. 10.1074/jbc.M310124200
Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H: Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem 2000, 275: 40096-40105. 10.1074/jbc.M006993200
Weigand MA, Volkmann M, Schmidt H, Martin E, Bohrer H, Bardenheuer HJ: Neuron-specific enolase as a marker of fatal outcome in patients with severe sepsis or septic shock. Anesthesiology 2000, 92: 905-907. 10.1097/00000542-200003000-00057
Lotze MT, Tracey KJ: High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 2005, 5: 331-342. 10.1038/nri1594
Rauvala H, Pihlaskari R: Isolation and some characteristics of an adhesive factor of brain that enhances neurite outgrowth in central neurons. J Biol Chem 1987, 262: 16625-16635.
Rauvala H, Merenmies J, Pihlaskari R, Korkolainen M, Huhtala ML, Panula P: The adhesive and neurite-promoting molecule p30: analysis of the amino terminal sequence and production of antipeptide antibodies that detect p30 at the surface of neuroblastoma cells and of brain neurons. J Cell Biol 1988, 107: 2293-2305. 10.1083/jcb.107.6.2293
Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, Arnold B, Bianchi ME, Manfredi AA, Rovere-Querini P: Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol 2005, 174: 7506-7515.
Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME: Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 2003, 22: 5551-5560. 10.1093/emboj/cdg516
Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK: Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem 2002, 277: 15028-15034. 10.1074/jbc.M200497200
Schnurr M, Toy T, Stoitzner P, Cameron P, Shin A, Beecroft T, Davis ID, Cebon J, Maraskovsky E: ATP gradients inhibit the migratory capacity of specific human dendritic cell types: implications for P2Y11 receptor signaling. Blood 2003, 102: 613-620. 10.1182/blood-2002-12-3745
Shi Y, Evans JE, Rock KL: Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 2003, 425: 516-521. 10.1038/nature01991
Scaffidi P, Misteli T, Bianchi ME: Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418: 191-195. 10.1038/nature00858
Huttunen HJ, Fages C, Kuja-Panula J, Ridley AJ, Rauvala H: Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res 2002, 62: 4805-4811.
Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, et al.: High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 2000, 192: 565-570. 10.1084/jem.192.4.565
Abeyama K, Stern DM, Ito Y, Kawahara K, Yoshimoto Y, Tanaka M, Uchimura T, Ida N, Yamazaki Y, Yamada S, et al.: The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest 2005, 115: 1267-1274. 10.1172/JCI200522782
Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ: HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med 2001, 164: 1768-1773.
Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, Lepantalo M, Carpen O, Parkkinen J, Rauvala H: Regulation of monocyte migration by amphoterin (HMGB1). Blood 2004, 104: 1174-1182. 10.1182/blood-2003-10-3536
Sappington PL, Yang R, Yang H, Tracey KJ, Delude RL, Fink MP: HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology 2002, 123: 790-802. 10.1053/gast.2002.35391
Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, Nawroth P, Andersson U, Harris RA, Harris HE: RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol 2005, 61: 1-9. 10.1111/j.0300-9475.2005.01534.x
Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E: Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 2004, 279: 7370-7377. 10.1074/jbc.M306793200
Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H: HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006, 26: 174-179. 10.1097/01.shk.0000225404.51320.82
Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, et al.: The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med 2005, 201: 1135-1143. 10.1084/jem.20042614
Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ: HMG-1 as a mediator of acute lung inflammation. J Immunol 2000, 165: 2950-2954.
Ueno H, Matsuda T, Hashimoto S, Amaya F, Kitamura Y, Tanaka M, Kobayashi A, Maruyama I, Yamada S, Hasegawa N, et al.: Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med 2004, 170: 1310-1316. 10.1164/rccm.200402-188OC
Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, et al.: Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A 2004, 101: 296-301. 10.1073/pnas.2434651100
Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al-Abed Y, Wang H, Metz C, Miller EJ, et al.: Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 2004, 10: 1216-1221. 10.1038/nm1124
Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Grone HJ, Kurschus FC, Schmidt AM, et al.: Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest 2004, 113: 1641-1650. 10.1172/JCI200418704
Zeng S, Feirt N, Goldstein M, Guarrera J, Ippagunta N, Ekong U, Dun H, Lu Y, Qu W, Schmidt AM, et al.: Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology 2004, 39: 422-432. 10.1002/hep.20045
Cataldegirmen G, Zeng S, Feirt N, Ippagunta N, Dun H, Qu W, Lu Y, Rong LL, Hofmann MA, Kislinger T, et al.: RAGE limits regeneration after massive liver injury by coordinated suppression of TNF-alpha and NF-kappaB. J Exp Med 2005, 201: 473-484. 10.1084/jem.20040934
Hou FF, Ren H, Owen WF Jr, Guo ZJ, Chen PY, Schmidt AM, Miyata T, Zhang X: Enhanced expression of receptor for advanced glycation end products in chronic kidney disease. J Am Soc Nephrol 2004, 15: 1889-1896. 10.1097/01.ASN.0000131526.99506.F7
Hudson BI, Stickland MH, Grant PJ, Futers TS: Characterization of allelic and nucleotide variation between the RAGE gene on chromosome 6 and a homologous pseudogene sequence to its 5' regulatory region on chromosome 3: implications for polymorphic studies in diabetes. Diabetes 2001, 50: 2646-2651. 10.2337/diabetes.50.12.2646
Kankova K, Marova I, Zahejsky J, Muzik J, Stejskalova A, Znojil V, Vacha J: Polymorphisms 1704G/T and 2184A/G in the RAGE gene are associated with antioxidant status. Metabolism 2001, 50: 1152-1160. 10.1053/meta.2001.26757
Hofmann MA, Drury S, Hudson BI, Gleason MR, Qu W, Lu Y, Lalla E, Chitnis S, Monteiro J, Stickland MH, et al.: RAGE and arthritis: the G82S polymorphism amplifies the inflammatory response. Genes Immun 2002, 3: 123-135. 10.1038/sj.gene.6363861
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Bopp, C., Bierhaus, A., Hofer, S. et al. Bench-to-bedside review: The inflammation-perpetuating pattern-recognition receptor RAGE as a therapeutic target in sepsis. Crit Care 12, 201 (2008). https://doi.org/10.1186/cc6164
Published:
DOI: https://doi.org/10.1186/cc6164