
Abstract
The pathogenesis of sepsis-induced multiple organ failure may crucially depend on the development of mitochondrial dysfunction and consequent cellular energetic failure. According to this hypothesis, interventions aimed at preventing or reversing mitochondrial damage may have major clinical relevance, although the timing of such interventions will be critical to both ensuring benefit and avoiding harm. Early correction of tissue hypoxia, strict control of glycaemia, and modulation of oxidative and nitrosative stress may afford protection during the initial, acute systemic inflammatory response. The regulated induction of a hypometabolic state resembling hibernation may protect the cells from dying once energy failure has developed, allowing the possibility of functional recovery. Repair of damaged organelles through stimulation of mitochondrial biogenesis and reactivation of cellular metabolism may accelerate resolution of the multiple organ failure syndrome.
Similar content being viewed by others
Introduction
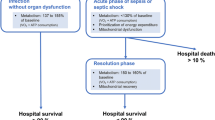
Sepsis is the systemic inflammatory response to infection and represents a major cause of morbidity and mortality in patients admitted to intensive care units (ICUs) [1]. However, despite decades of research, the pathophysiology of sepsis remains incompletely understood. A critical limitation of tissue oxygen delivery due to macrocirculatory or microcirculatory failure may play a role, especially in the early phase of the disease process before resuscitation has been initiated. Nonetheless, a growing body of evidence suggests that multiple organ failure (MOF) may develop during sepsis mainly as a consequence of impaired cellular oxygen utilization. Supportive data in patients include the following findings: total body oxygen consumption falls progressively with increasing severity of sepsis [2]; skeletal muscle tissue oxygen tension is abnormally high but normalizes during the recovery phase [3]; necrotic and apoptotic cell death is minimal, if it occurs at all, in most dysfunctioning organs [4]; and organs with limited regenerative capabilities, such as kidney, are usually able to recover to such an extent that long-term support is usually not needed [5]. Sepsis-induced MOF may thus be related to a potentially reversible impairment in cellular function rather than any permanent structural damage.
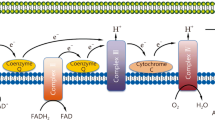
The mitochondrion is the powerhouse of the cell [6]. Cellular energy production depends on three interconnected pathways: glycolysis within the cytoplasm, the Krebs cycle and the electron transport chain within the mitochondria (Figure 1). Glycolysis is a sequence of reactions that degrade glucose to pyruvate. In the presence of oxygen, pyruvate and other fuel molecules such as fatty acids and amino acids enter the mitochondria, where they are completely oxidized within the Krebs cycle. The reduced nicotinamide (NADH) and flavin (FADH2) adenine dinucleotides transfer electrons to the respiratory enzyme complexes located in the inner mitochondrial membrane (electron transport chain) for the process of ATP generation by oxidative phosphorylation. NADH donates electrons specifically to complex I whereas FADH2 reduces complex II. The electrons then flow via coenzyme Q (ubiquinone) to complex III, and are then transported via cytochrome C to reach complex IV (cytochrome oxidase). At this final stage, oxygen is reduced to water. Electron transfer through complexes I, III and IV generates a proton gradient across the inner mitochondrial membrane that is used by ATP synthase (complex V) to generate energy by phosphorylating ADP. The complete oxidation of one molecule of glucose yields 30–36 molecules of ATP, two of which come from glycolysis and two from the Krebs cycle. Glycolysis can also occur in the absence of oxygen. However, when oxygen is lacking, pyruvate can no longer be further oxidized within the mitochondria and is thus metabolized to lactate within the cytoplasm. Glycolysis represents a much less efficient metabolic pathway compared with the Krebs cycle and oxidative phosphorylation, because there is net synthesis of only two molecules of ATP per molecule of glucose [7].

Schematic representation of oxidative phosphorylation within the mitochondria. Electrons donated from NADH and FADH2 pass down the electron transport chain with oxygen being the terminal acceptor at complex IV. This movement of electrons results in a shift of protons across the inner mitochondrial membrane, generating the energy necessary for ATP synthase to produce ATP from ADP. FADH2, flavin adenine dinucleotide, reduced; NADH, nicotinamide adenine dinucleotide, reduced.
Because mitochondria utilize more than 90% of total body oxygen consumption to produce ATP, the abnormalities in oxygen consumption described during sepsis are likely to be associated with evidence of mitochondrial dysfunction. Studies conducted during the early phase of sepsis (within the first few hours) have produced conflicting results. Nonetheless, mitochondrial structure and function were consistently shown to be impaired in a severity-dependent manner in animal models lasting at least 12–16 hours [8]. Of note, ATP levels were variably affected, depending on the balance between energy production and consumption, the model and possibly the tissue under investigation. In septic shock patients studied within 24 hours of ICU admission, the degree of skeletal muscle mitochondrial dysfunction was associated with the severity of the disease [9]. In this work, tissue ATP levels were significantly lower in nonsurvivors than in an orthopaedic surgical control population, but they were maintained in those who survived sepsis.
A reduction in energy consumption implies a reduction in cellular metabolism, which manifests clinically as organ dysfunction. Rather than being viewed negatively as 'failure', an alternative paradigm may be advanced whereby this metabolic shutdown represents an adaptive cellular strategy [10]. In the face of persisting mitochondrial dysfunction and reduced ATP production, the cell may shift its focus to survival rather than aiming to continue normal functioning.
The pathogenesis of mitochondrial dysfunction during sepsis is likely to be highly complex. Nitric oxide (NO), reactive oxygen species and other inflammatory mediators are produced in excess and can directly inhibit mitochondrial respiration. NO competes with oxygen in binding to cytochrome oxidase (complex IV), thereby decreasing the activity of the enzyme. This will block the electron transport chain and lead to overproduction of superoxide. Superoxide will react with NO to generate peroxynitrite and other nitrogen species that are able to alter the structure and function of several other mitochondrial proteins, notably complex I [11]. Early cellular hypoxia may favour the competitive NO-mediated inhibition of cytochrome oxidase, contributing to the earlier, if not greater, development of mitochondrial dysfunction [12].
Endocrine changes that occur during sepsis are also likely to play a role. Among others, thyroid and sex hormones, insulin, glucocorticoids and leptin positively modulate mitochondrial energy production, protein synthesis and biogenesis [13–17]. Increased incidences of the low tri-iodothyronine (T3) syndrome, hypogonadism, insulin resistance, adrenal insufficiency and decreased circulating leptin levels in nonsurvivors compared with survivors have been reported during prolonged sepsis and critical illness [18, 19]. Accordingly, depletion of respiratory complex proteins has been described in the diaphragm in a rat model of sepsis [20].
A further mechanism could be represented by the down-regulated synthesis of new mitochondrial protein. In human volunteers, administration of bacterial endotoxin decreased blood leucocyte expression of mitochondrial respiratory chain complexes and ATP synthase genes [21].
Assuming that the pathogenesis of MOF during sepsis is contingent on development of mitochondrial dysfunction and cellular energetic failure, recovery is likely to occur when damaged organelles are repaired or replaced. Preliminary results have shown an association between progressive improvement in mitochondrial respiration and organ function in patients who survive their episode of septic shock [22].
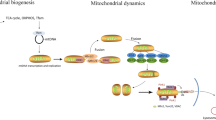
Strategies aimed at preventing or reversing mitochondrial dysfunction and cellular energetic failure may thus represent a new therapeutic option in the treatment of sepsis (Figure 2).

Hypothesized role of mitochondria in the development of MOF and subsequent recovery. Potential therapeutic interventions are illustrated at the appropriate steps. MOF, multiple organ failure.
Prevention and early reversal of mitochondrial dysfunction
Mitochondrial dysfunction in sepsis can occur even with aggressive fluid resuscitation [23] and adequate tissue oxygenation [24, 25]. Derangement in liver metabolism possibly due to mitochondrial damage was recently reported in a hyperdynamic, normotensive, mechanically ventilated, antibiotic-treated septic animal model, despite preserved microvascular perfusion [26]. Nonetheless, early cellular hypoxia can further limit aerobic production of ATP and contribute to the development of energy failure.
Optimization of oxygen delivery can ameliorate cellular energetic failure provided that mitochondria retain their ability to produce energy. Patients with severe sepsis or septic shock whose global oxygen delivery was optimized early after admission to an emergency room experienced better outcomes than did conventionally managed patients [27]. Conversely, no benefit [28] or even harm [29] was reported when a similar approach was adopted after admission to ICU, when organ failure had already become established. The same intervention, performed at different time points, had very different clinical impacts. In the early phase, when the cellular energetic machinery is still likely to be functional and oxygen supply may represent a limiting factor, reversal of tissue hypoxia may ameliorate the impending cellular energetic failure and reduce the incidence/severity of organ dysfunction. In a later phase, when mitochondrial damage has occurred and the cell has become intrinsically unable to utilize oxygen to produce ATP, a similar strategy may not provide any benefit. Lack of improvement in oxygen consumption despite a re-established oxygen supply has been associated with unfavourable outcomes in patients with sepsis syndrome or septic shock [30].
Hyperglycaemia and insulin resistance are common among critically ill patients and represent an additional potential threat to mitochondrial integrity. Acute hyperglycaemia can dramatically increase the production of reactive oxygen species in normal bovine aortic endothelial cells [31]. Moreover, insulin stimulates mitochondrial protein synthesis and oxidative phosphorylation [15]. Maintenance of normo-glycaemia with intensive insulin therapy during critical illness has been demonstrated to preserve hepatocyte mitochondrial ultrastructure and function [32] and improve outcome in both medical and surgical intensive care patients [33, 34].
Reactive oxygen and nitrogen species are over-produced during sepsis, whereas mitochondrial antioxidants (reduced glutathione and manganese superoxide) are depleted. The membrane permeable glutathione ethyl ester can protect complex I from oxidative and nitrosative damage in an early phase [35]. Manganese-based superoxide dismutase mimetics may exert a similar protective effect, scavenging superoxide anions and preventing them from further reacting with NO to generate peroxynitrite within the mitochondria [36].
Prevention of cellular energetic failure in the presence of mitochondrial dysfunction
Once permanent mitochondrial dysfunction has developed, cellular optimization of any residual ability to produce energy and/or reduce metabolic requirements may prevent the ATP level from dropping below the threshold that stimulates initiation of cell death pathways.
Electron donors that are able to 'bypass' defective components of the respiratory chain may help in attaining the former objective. Within the inner mitochondrial membrane, complex II works in parallel with complex I, albeit to a lesser extent, transferring electrons from FADH2 produced during the oxidation of succinate to coenzyme Q. Unlike complex I, the activity of complex II is relatively preserved during sepsis [9, 23, 37]. When complex I is inhibited, the administration of succinate may increase electron flow through the respiratory chain and thus increase generation of ATP, provided that any inhibition of the electron transport chain distal to complex II has not become rate-limiting. Preliminary data from our laboratory confirm this action. In two different animal models of sepsis, the infusion of succinate dimethyl ester prevented the fall in liver ATP content [38] and prolonged survival time [39].
Another possible strategy that could be pursued in the face of a severe and extended impairment in mitochondrial energy production is to reduce cellular energetic expenditure. Hibernating and aestivating animals reduce their metabolic rate in the face of climate change or drought. Similarly, oxygen-conforming organisms such as turtles and frogs can tolerate prolonged periods of hypoxia by suppressing ATP turnover [40]. Humans do not hibernate or aestivate and have only a limited tolerance to inadequate oxygenation. Nonetheless, patients with chronic coronary artery disease frequently develop a myocardial contractile dysfunction – termed myocardial hibernation – that may represent an adaptive response to ischaemia, rather than depend on an ongoing energetic deficit, which will recover on restoration of adequate perfusion [41].
Mechanism(s) governing hibernation remain to be clarified. Carbon monoxide and NO may mediate the active decrease in energy demand that occurs in cells that lack oxygen [42, 43]. The natural peptide 'hibernation induction trigger', its synthetic analogue [D-Ala2, D-Leu5] enkephalin (DADLE) and other δ-opioids can also reduce cellular metabolism and protect organs against ischaemia [44]. Rapid induction of profound cerebral hypothermia in animals that do not normally hibernate may guarantee protection during prolonged cardio-circulatory arrest [45]. Mice exposed to hydrogen sulphide experience a dramatic decrease in their metabolic rate: within 6 hours, oxygen consumption and carbon dioxide production drop by around 90%, and body core temperature approaches that of the environment [46]. Such a suspended animation-like state fully reverses when the hydrogen sulphide is discontinued, without any permanent behavioural or functional damage. It is conceivable that, even during sepsis, induced hibernation may protect the organism from prolonged energetic failure and enable faster recovery on resolution of the inflammatory insult. Some caveats do need to be applied. For example, the hyperthermic response to infection activates the expression of cytoprotective heat shock proteins and may therefore be considered beneficial [47]. Therapeutic induction of hibernation may remove this intrinsic protective mechanism with potentially deleterious results.
The converse may also hold true. Premature stimulation of cellular metabolism before mitochondria have regained their ability to respond adequately in terms of energy production may lead to cellular compromise. Examples of harmful therapeutic approaches that may be invoked are the use of high-dose dobutamine [29], thyroxine [48] and growth hormone [49].
Resolution of mitochondrial dysfunction: arousal from 'hibernation'
Repair and replacement of damaged mitochondria are probably controlled at a transcriptional level, but proximal steps in the signalling pathway still need to be elucidated. NO was recently suggested to play a major role. Long-term exposure to a low concentration of the gas triggered expression of transcriptional factors that regulate mito-chondrial proliferation and significantly increased mito-chondrial mass in different cells in culture [50]. NO exerts different actions depending on the rate, amount and site of production. The large quantity synthesized by the inducible isoform of nitric oxide synthase (NOS) during the acute inflammatory response to sepsis blocks mitochondrial respiration and can be cytotoxic. On the other hand, the smaller amounts of NO produced by the specific constitutive endothelial NOS may trigger mitochondrial biogenesis in a later phase. Nitration also dramatically accelerates mito-chondrial protein turnover, from days to hours [51]. Taken together, these results suggest that recovery from mito-chondrial dysfunction may depend on a NO-dependent signalling pathway. Specific inhibition of inducible NOS during sepsis may represent a potential therapeutic strategy [52–55], although dose selection will be critical. This is pertinent to the dose-related increase in mortality reported in a phase III trial of a nonspecific NOS inhibitor in septic shock patients [56]. Indeed, the overall negative outcome of this study camouflages the survival benefit seen with low doses.
Hormones may play an equally important role. Thyroid hormones stimulate mitochondrial activity. Injection of T3 in hypothyroid rats upregulated mitochondrial biogenesis-related transcription factors [57]. In contrast to the acute response, persistently low circulating levels of T3 during the prolonged phase of critical illness may be due to neuroendocrine dysfunction [18]. Replacement hormonal therapy given at the right time, when cells have regained the ability to both restore mitochondrial activity and increase metabolic rate, may beneficially arouse the cell and promote earlier organ recovery. However, as described above, thyroxine supplementation may prove dangerous [48], and so the right conditions must be in place.
Other hormones that could be considered in the treatment of sepsis are leptin and oestrogen. Leptin is a hormone secreted by adipose tissue. It regulates food intake and energy balance to maintain constancy of total body fat mass. In diabetic fatty rats, ectopic hyperleptinaemia triggered mitochondrial proliferation, transforming white adipocytes into mitochondria-rich, fat-oxidizing cells [17]. Administration of oestrogen or antiandrogen agents after trauma/haemorrhage also increased mitochondrial enzyme activities, protein synthesis and ATP levels relative to those in sham-operated controls [58].
A further biological equivalent to sepsis-induced hibernation is bacterial dormancy. This is a reversible, low-growth state well recognized in mycobacteria such as Mycobacterium tuberculosis. Micrococcus luteus can be aroused from its quiescent phase by an endogenous protein named 'resuscitation promoting factor' [59]. As mitochondria descend from a bacterial endosymbiont, the identification and application of a similar protein that can specifically stimulate mitochondrial activity may well yield beneficial results.
Conclusion
Mitochondrial dysfunction occurs during sepsis and may play a major role in the development of MOF.
Prevention and correction of mitochondrial dysfunction and cellular energetic failure represent novel strategies that may improve clinical outcomes of septic patients. Timing of any intervention appears to be critical and the possibly adaptive role of some changes currently viewed as pathological must be considered. The regulated induction of a hypometabolic state resembling hibernation may help the cell in facing a reduced capacity to generate energy. The stimulation of mitochondrial activity and biogenesis during the late phase of sepsis may accelerate the recovery process. This increasing insight into underlying mechanisms promises to be an exciting era of novel therapeutic developments.
Abbreviations
- FADH2 = flavin adenine dinucleotide:
-
reduced
- ICU = :
-
intensive care unit
- MOF = :
-
multiple organ failure
- NADH=:
-
nicotinamide adenine dinucleotide reduced
- NO = :
-
nitric oxide
- NOS = :
-
nitric oxide synthase
- T3= :
-
tri-iodothyronine.
References
Padkin A, Goldfrad C, Brady AR, Young D, Black N, Rowan K: Epidemiology of severe sepsis occurring in the first 24 hrs in intensive care units in England, Wales, and Northern Ireland. Crit Care Med 2003, 31: 2332-2338. 10.1097/01.CCM.0000085141.75513.2B
Kreymann G, Grosser S, Buggisch P, Gottschall C, Matthaei S, Greten H: Oxygen consumption and resting metabolic rate in sepsis, sepsis syndrome, and septic shock. Crit Care Med 1993, 21: 1012-1019.
Boekstegers P, Weidenhofer S, Kapsner T, Werdan K: Skeletal muscle partial pressure of oxygen in patients with sepsis. Crit Care Med 1994, 22: 640-650.
Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE: Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med 1999, 27: 1230-1251. 10.1097/00003246-199907000-00002
Noble JS, MacKirdy FN, Donaldson SI, Howie JC: Renal and respiratory failure in Scottish ICUs. Anaesthesia 2001, 56: 124-129. 10.1046/j.1365-2044.2001.01841.x
Rich P: Chemiosmotic coupling: the cost of living. Nature 2003, 421: 583. 10.1038/421583a
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P: Molecular Biology of the Cell. New York: Garland Publishing; 2002.
Singer M, Brealey D: Mitochondrial dysfunction in sepsis. Biochem Soc Symp 1999, 66: 149-166.
Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M: Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360: 219-223. 10.1016/S0140-6736(02)09459-X
Singer M, De Santis V, Vitale D, Jeffcoate W: Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet 2004, 364: 545-548. 10.1016/S0140-6736(04)16815-3
Liaudet L, Soriano FG, Szabo C: Biology of nitric oxide signaling. Crit Care Med 2000, 28: N37-N52. 10.1097/00003246-200004001-00005
Frost MT, Wang Q, Moncada S, Singer M: Hypoxia accelerates nitric oxide-dependent inhibition of mitochondrial complex I in activated macrophages. Am J Physiol Regul Integr Comp Physiol 2005, 288: R394-R400.
Weitzel JM, Iwen KA, Seitz HJ: Regulation of mitochondrial biogenesis by thyroid hormone. Exp Physiol 2003, 88: 121-128. 10.1113/eph8802506
Stirone C, Duckles SP, Krause DN, Procaccio V: Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol 2005, 68: 959-965. 10.1124/mol.105.014662
Stump CS, Short KR, Bigelow ML, Schimke JM, Nair KS: Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc Natl Acad Sci USA 2003, 100: 7996-8001. 10.1073/pnas.1332551100
Scheller K, Sekeris CE: The effects of steroid hormones on the transcription of genes encoding enzymes of oxidative phosphorylation. Exp Physiol 2003, 88: 129-140. 10.1113/eph8802507
Orci L, Cook WS, Ravazzola M, Wang MY, Park BH, Montesano R, Unger RH: Rapid transformation of white adipocytes into fat-oxidizing machines. Proc Natl Acad Sci USA 2004, 101: 2058-2063. 10.1073/pnas.0308258100
Van den Berghe G, de Zegher F, Bouillon R: Clinical review 95: acute and prolonged critical illness as different neuroendocrine paradigms. J Clin Endocrinol Metab 1998, 83: 1827-1834. 10.1210/jc.83.6.1827
Bornstein SR, Licinio J, Tauchnitz R, Engelmann L, Negrao AB, Gold P, Chrousos GP: Plasma leptin levels are increased in survivors of acute sepsis: associated loss of diurnal rhythm, in cortisol and leptin secretion. J Clin Endocrinol Metab 1998, 83: 280-283. 10.1210/jc.83.1.280
Callahan LA, Supinski GS: Sepsis induces diaphragm electron transport chain dysfunction and protein depletion. Am J Respir Crit Care Med 2005, 172: 861-868. 10.1164/rccm.200410-1344OC
Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, et al.: A network-based analysis of systemic inflammation in humans. Nature 2005, 437: 1032-1037. 10.1038/nature03985
Brealey DA, Hargreaves I, Heales S, Land J, Smolenski R, Singer M: Recovery from organ failure is associated with improved mitochondrial function in septic patients [abstract]. Intensive Care Med 2003, 29: S134.
Brealey D, Karyampudi S, Jacques TS, Novelli M, Stidwill R, Taylor V, Smolenski RT, Singer M: Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol 2004, 286: R491-R497.
VanderMeer TJ, Wang H, Fink MP: Endotoxemia causes ileal mucosal acidosis in the absence of mucosal hypoxia in a normodynamic porcine model of septic shock. Crit Care Med 1995, 23: 1217-1226. 10.1097/00003246-199507000-00011
Rosser DM, Stidwill RP, Jacobson D, Singer M: Oxygen tension in the bladder epithelium rises in both high and low cardiac output endotoxemic sepsis. J Appl Physiol 1995, 79: 1878-1882.
Albuszies G, Radermacher P, Vogt J, Wachter U, Weber S, Schoaff M, Georgieff M, Barth E: Effect of increased cardiac output on hepatic and intestinal microcirculatory blood flow, oxygenation, and metabolism in hyperdynamic murine septic shock. Crit Care Med 2005, 33: 2332-2338. 10.1097/01.CCM.0000182817.20977.E9
Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, Peterson E, Tomlanovich M: Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001, 345: 1368-1377. 10.1056/NEJMoa010307
Gattinoni L, Brazzi L, Pelosi P, Latini R, Tognoni G, Pesenti A, Fumagalli R: A trial of goal-oriented hemodynamic therapy in critically ill patients. SvO 2 Collaborative Group. N Engl J Med 1995, 333: 1025-1032. 10.1056/NEJM199510193331601
Hayes MA, Timmins AC, Yau EH, Palazzo M, Hinds CJ, Watson D: Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med 1994, 330: 1717-1722. 10.1056/NEJM199406163302404
Hayes MA, Timmins AC, Yau EH, Palazzo M, Watson D, Hinds CJ: Oxygen transport patterns in patients with sepsis syndrome or septic shock: influence of treatment and relationship to outcome. Crit Care Med 1997, 25: 926-936. 10.1097/00003246-199706000-00007
Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, et al.: Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404: 787-790. 10.1038/35008121
Vanhorebeek I, De Vos R, Mesotten D, Wouters PJ, De Wolf-Peeters C, Van den Berghe G: Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet 2005, 365: 53-59. 10.1016/S0140-6736(04)17665-4
Van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R: Intensive insulin therapy in the critically ill patients. N Engl J Med 2001, 345: 1359-1367. 10.1056/NEJMoa011300
Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters PJ, Milants I, Van WE, Bobbaers H, Bouillon R: Intensive insulin therapy in the medical ICU. N Engl J Med 2006, 354: 449-461. 10.1056/NEJMoa052521
Clementi E, Brown GC, Feelisch M, Moncada S: Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitro-sylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA 1998, 95: 7631-7636. 10.1073/pnas.95.13.7631
Salvemini D, Riley DP, Cuzzocrea S: SOD mimetics are coming of age. Nat Rev Drug Discov 2002, 1: 367-374. 10.1038/nrd796
Svistunenko DA, Davies N, Brealey D, Singer M, Cooper CE: Mitochondrial dysfunction in patients with severe sepsis: an EPR interrogation of individual respiratory chain components. Biochim Biophys Acta 2006, 1757: 262-272. 10.1016/j.bbabio.2006.03.007
Malaisse WJ, Nadi AB, Ladriere L, Zhang TM: Protective effects of succinic acid dimethyl ester infusion in experimental endotoxemia. Nutrition 1997, 13: 330-341.
Ferreira FL, Ladriere L, Vincent JL, Malaisse WJ: Prolongation of survival time by infusion of succinic acid dimethyl ester in a caecal ligation and perforation model of sepsis. Horm Metab Res 2000, 32: 335-336.
Hochachka PW, Buck LT, Doll CJ, Land SC: Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci USA 1996, 93: 9493-9498. 10.1073/pnas.93.18.9493
Heusch G, Schulz R: The biology of myocardial hibernation. Trends Cardiovasc Med 2000, 10: 108-114. 10.1016/S1050-1738(00)00058-X
Zuckerbraun BS, McCloskey CA, Gallo D, Liu F, Ifedigbo E, Otterbein LE, Billiar TR: Carbon monoxide prevents multiple organ injury in a model of hemorrhagic shock and resuscitation. Shock 2005, 23: 527-532.
Teodoro RO, O'Farrell PH: Nitric oxide-induced suspended animation promotes survival during hypoxia. EMBO J 2003, 22: 580-587. 10.1093/emboj/cdg070
Su TP: Delta opioid peptide[D-Ala(2), D-Leu(5)]enkephalin promotes cell survival. J Biomed Sci 2000, 7: 195-199.
Behringer W, Safar P, Wu X, Kentner R, Radovsky A, Kochanek PM, Dixon CE, Tisherman SA: Survival without brain damage after clinical death of 60–120 mins in dogs using suspended animation by profound hypothermia. Crit Care Med 2003, 31: 1523-1531. 10.1097/01.CCM.0000063450.73967.40
Blackstone E, Morrison M, Roth MB: H 2 S induces a suspended animation-like state in mice. Science 2005, 308: 518. 10.1126/science.1108581
Bruemmer-Smith S, Stuber F, Schroeder S: Protective functions of intracellular heat-shock protein (HSP) 70-expression in patients with severe sepsis. Intensive Care Med 2001, 27: 1835-1841. 10.1007/s00134-001-1131-3
Acker CG, Singh AR, Flick RP, Bernardini J, Greenberg A, Johnson JP: A trial of thyroxine in acute renal failure. Kidney Int 2000, 57: 293-298. 10.1046/j.1523-1755.2000.00827.x
Takala J, Ruokonen E, Webster NR, Nielsen MS, Zandstra DF, Vundelinckx G, Hinds CJ: Increased mortality associated with growth hormone treatment in critically ill adults. N Engl J Med 1999, 341: 785-792. 10.1056/NEJM199909093411102
Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, et al.: Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 2003, 299: 896-899. 10.1126/science.1079368
Elfering SL, Haynes VL, Traaseth NJ, Ettl A, Giulivi C: Aspects, mechanism, and biological relevance of mitochondrial protein nitration sustained by mitochondrial nitric oxide synthase. Am J Physiol Heart Circ Physiol 2004, 286: H22-H29. 10.1152/ajpheart.00766.2003
Liaudet L, Rosselet A, Schaller MD, Markert M, Perret C, Feihl F: Nonselective versus selective inhibition of inducible nitric oxide synthase in experimental endotoxic shock. J Infect Dis 1998, 177: 127-132.
Unno N, Wang H, Menconi MJ, Tytgat SH, Larkin V, Smith M, Morin MJ, Chavez A, Hodin RA, Fink MP: Inhibition of inducible nitric oxide synthase ameliorates endotoxin-induced gut mucosal barrier dysfunction in rats. Gastroenterology 1997, 113: 1246-1257. 10.1053/gast.1997.v113.pm9322519
King CJ, Tytgat S, Delude RL, Fink MP: Ileal mucosal oxygen consumption is decreased in endotoxemic rats but is restored toward normal by treatment with aminoguanidine. Crit Care Med 1999, 27: 2518-2524. 10.1097/00003246-199911000-00032
Matejovic M, Krouzecky A, Martinkova V, Rokyta R Jr, Kralova H, Treska V, Radermacher P, Novak I: Selective inducible nitric oxide synthase inhibition during long-term hyperdynamic porcine bacteremia. Shock 2004, 21: 458-465. 10.1097/00024382-200405000-00010
Lopez A, Lorente JA, Steingrub J, Bakker J, McLuckie A, Willatts S, Brockway M, Anzueto A, Holzapfel L, Breen D, et al.: Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Crit Care Med 2004, 32: 21-30. 10.1097/01.CCM.0000105581.01815.C6
Weitzel JM, Radtke C, Seitz HJ: Two thyroid hormone-mediated gene expression patterns in vivo identified by cDNA expression arrays in rat. Nucleic Acids Res 2001, 29: 5148-5155. 10.1093/nar/29.24.5148
Hsieh YC, Yang S, Choudhry MA, Yu HP, Bland KI, Schwacha MG, Chaudry IH: Flutamide restores cardiac function after trauma-hemorrhage via an estrogen-dependent pathway through upregulation of PGC-1. Am J Physiol Heart Circ Physiol 2006, 290: H416-H423. 10.1152/ajpheart.00865.2005
Mukamolova GV, Kaprelyants AS, Young DI, Young M, Kell DB: A bacterial cytokine. Proc Natl Acad Sci USA 1998, 95: 8916-8921. 10.1073/pnas.95.15.8916
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
MS receives research funding from the Medical Research Council and Wellcome Trust to undertake basic science research on mitochondria.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Protti, A., Singer, M. Bench-to-bedside review: Potential strategies to protect or reverse mitochondrial dysfunction in sepsis-induced organ failure. Crit Care 10, 228 (2006). https://doi.org/10.1186/cc5014
Published:
DOI: https://doi.org/10.1186/cc5014