
Abstract
Valproic acid (VPA) is a broad-spectrum antiepileptic drug and is usually well tolerated, but rare serious complications may occur in some patients receiving VPA chronically, including haemorrhagic pancreatitis, bone marrow suppression, VPA-induced hepatotoxicity (VHT) and VPA-induced hyperammonaemic encephalopathy (VHE). Some data suggest that VHT and VHE may be promoted by carnitine deficiency. Acute VPA intoxication also occurs as a consequence of intentional or accidental overdose and its incidence is increasing, because of use of VPA in psychiatric disorders. Although it usually results in mild central nervous system depression, serious toxicity and even fatal cases have been reported. Several studies or isolated clinical observations have suggested the potential value of oral L-carnitine in reversing carnitine deficiency or preventing its development as well as some adverse effects due to VPA. Carnitine supplementation during VPA therapy in high-risk patients is now recommended by some scientific committees and textbooks, especially paediatricians. L-carnitine therapy could also be valuable in those patients who develop VHT or VHE. A few isolated observations also suggest that L-carnitine may be useful in patients with coma or in preventing hepatic dysfunction after acute VPA overdose. However, these issues deserve further investigation in controlled, randomized and probably multicentre trials to evaluate the clinical value and the appropriate dosage of L-carnitine in each of these conditions.
Similar content being viewed by others
Introduction
Valproic acid (VPA) is a broad-spectrum antiepileptic drug (AED) that has been used for more than 30 years and is effective in the treatment of many different types of partial and generalized epileptic seizure. It is also prescribed to treat bipolar and schizoaffective disorders, social phobias and neuropathic pain, as well as for prophylaxis or treatment of migraine headache. VPA is a branched chain carboxylic acid (2-propylpentanoic acid or di-n-propylacetic acid), with a chemical structure very similar to that of short chain fatty acids (Fig. 1) [1].

Chemical structure of valproic acid.
It is usually well tolerated. Indeed, VPA has fewer common side effects than do other AEDs, especially on behaviour and cognitive functions. Moreover, its adverse effects can often be minimized by initiating the drug slowly. However, rare serious complications may occur in some patients receiving VPA chronically, including fatal haemorrhagic pancreatitis, bone marrow suppression, VPA-induced hepatotoxicity (VHT) and VPA-induced hyperammonaemic encephalopathy (VHE). Some data suggest that VHT and VHE may be promoted either by a pre-existing carnitine deficiency or by deficiency induced by VPA per se.
Acute VPA intoxication also occurs as a consequence of intentional or accidental overdose. Its incidence is increasing [2–5], probably because of the use of VPA in psychiatric disorders. It usually results in mild and self-limited central nervous system (CNS) depression. However, serious toxicity and even deaths have been reported [2, 6, 7].
This paper reviews clinical evidence concerning the use of carnitine supplementation in the management of VHT, VHE and acute VPA poisoning. The potential benefit of carnitine supplementation in the prevention of VHT of VHE in the setting of chronic VPA dosing is also briefly discussed.
Pharmacology of valproic acid
VPA potentiates γ-aminobutyric acid (GABA)ergic functions in some specific brain regions that are thought to be involved in the control of seizure generation and propagation by increasing both GABA synthesis and release [8]. Furthermore, VPA reduces the release of the epileptogenic γ-hydroxybutyric acid and attenuates the neuronal excitation induced by N-methyl-D-aspartate (NMDA)-type glutamate receptors [9]. Finally, VPA could also exert direct effects on excitable membranes, and alter dopaminergic and serotoninergic neurotransmissions [10].
VPA is available as oral immediate-release, enteric-coated and delayed-release preparations, and as an intravenous formulation. Therapeutic daily doses range from 1 to 2 g in adults, and from 15 to 60 mg/kg in children [11].
Non-enteric-coated preparations of VPA are rapidly and nearly completely absorbed from the gastrointestinal tract, with peak plasma concentrations occurring 1–4 hours after ingestion [12]. Peak plasma concentrations occur only 4–5 hours after therapeutic doses of enteric-coated tablets. Peak plasma concentrations may be markedly delayed following acute overdose [11–14].
Therapeutic serum concentrations range from 50 to 125 μg/ml [11, 15]. At such therapeutic concentrations VPA is 80–90% bound to plasma proteins, but the percentage decreases at higher VPA levels. VPA has a small volume of distribution (0.13–0.23 l/kg) [11, 15, 16].
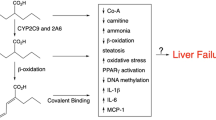
VPA is extensively metabolized by the liver via glucuronic acid conjugation, mitochondrial β – and cytosolic (endoplasmic reticulum) ω-oxidation to produce multiple metabolites, some of which may be biologically active (Fig. 2). However, because of their low plasma and brain concentrations, it is unlikely that they contribute significantly to the anticonvulsant effects of VPA [10]. Nevertheless, some of them may be involved in toxic effects of VPA, either in patients on chronic dosing or after an acute overdose. For example, 2-propyl-2-pentenoic acid (2-en-VPA), a byproduct of β-oxidation, and 2-propyl-4-pentenoic acid (4-en-VPA), a byproduct of ω-oxidation, have been incriminated in the development of cerebral oedema and in the hepatotoxicity of VPA, respectively [17–24]. 4-en-VPA and propionic acid metabolites resulting from ω-oxidation could also promote hyperammonaemia [19, 24]. Other metabolites, such as 3-cetoVPA or 4-cetoVPA, may produce a false-positive urine ketone determination [25].

Liver metabolism of valproic acid. See text for further details. VPA, valproic acid.
Mitochondrial β-oxidation of VPA involves its transport within 2 the mitochondrial matrix, using the same pathway as do long-chain fatty acids. This pathway consists of several steps and is sometimes called the 'carnitine shuttle' (Fig. 3). First, in the cytosol, VPA is activated and links with reduced acetyl coenzyme A (CoA-SH) to form valproyl-CoA (by the ATP-dependent medium-chain acyl-CoA synthetase, located on the outer side of the mitochondrial membrane). Valproyl-CoA then crosses the outer mitochondrial membrane. Under the effect of the palmityl carnitine transferase (PCT)1, valproylcarnitine is formed; this step is needed because the inner mitochondrial membrane is not permeable to acylcarnitines. Valproylcarnitine is then exchanged for free carnitine by carnitine translocase. In the mitochondrial matrix, PCT2 transformes valproylcarnitine into valproyl-CoA, which is able to enter a slow β-oxidation process [26]. Carnitine also helps to prevent valproyl-CoA accumulation [27].

The 'carnitine shuttle'. See text for further details. ACoAS, acyl-CoA synthetase; CoA, coenzyme A; CPT, carnitine palmityl transferase; CT, carnitine translocase.
The ω-oxidation is normally responsible for only a small component of VPA metabolism (Fig. 2). However, during long-term or high-dose VPA therapy, or after acute VPA overdose, a greater degree of ω-oxidation occurs, potentially increasing the risk for toxicity.
Less than 3% of VPA is excreted unchanged in the urine [10, 14, 15], much of which is in the form of valproylcarnitine [27, 28].
Elimination of VPA follows first-order kinetics, with a half-life ranging from 5 to 20 hours (mean 11 hours). However, following overdose the half-life may be prolonged to as long as 30 hours [11, 16, 17].
Carnitine
Carnitine (3-hydroxy-4-trimethylamino-butyric acid or β-hydroxy-gamma-N-trimethylamino-butyrate) thus appears essential to ensure proper metabolism of VPA. This amino acid derivative is an important nutrient; 75% comes from the diet, particularly in red meat and dairy products. It is not a true vitamin because it is also biosynthesized endogenously from dietary amino acids (methionin, lysine), especially in the liver and in the kidneys [29, 30].
Most body carnitine is stored in skeletal muscles, but it is also stored in other tissues with high energy demands (myocardium, liver, suprarenal glands; 2.5-4 μmol/g tissue) [31]. Plasma carnitine represents less than 0.6% of total body stores. Indeed, the total plasma concentration (free carnitine + acylcarnitine) is only 45–85 μmol/l.
The two main metabolic functions of carnitine are to facilitate fatty acyl group transport into mitochondria and to maintain the ratio of acyl-CoA to free CoA in the mitochondria [32].
Transport of long-chain fatty acids
Carnitine facilitates transport of long-chain fatty acids from the cytosol compartment of the muscle fibre into the mitochondria, where they undergo β-oxidation and produce acetyl-CoA, which enters the Krebs cycle [27]. Indeed, esterification as acylcarnitine is indispensable for transport of long-chain fatty acids through the mitochondrial membrane [33]. This transport process includes several steps ('carnitine shuttle'), which are described above for VPA [34, 35].
Prevention of the intramitochondrial accumulation of acyl-CoA
Carnitine facilitates prevention of intramitochondrial accumulation of acyl-CoA by transforming acyl-CoA into acylcarnitine. In this way, carnitine protects the cell from the membrane-destabilizing effects of toxic acyl groups, as well as their restraining effects on several enzymes that participate in intermediary metabolism and energy production in the mitochondria. Carnitine thus plays a central role in the metabolism of fatty acids and energy by regulating the mitochondrial ratio of free CoA to acyl-CoA.
Formulations of L-carnitine
L-Carnitine is available in some countries as an oral preparation (1 g/10 ml solution, 330 mg tablets) or as an injectable drug (intramuscular or intravenous, 1 g/5 ml solution; e.g. Levocarnil® [Sigma-Tau, Ivry-sur-Seine, France] and Carnitor® [Sigma-Tau, Gaithersburg, MD, USA]). It has been administered in senile dementia, metabolic nerve diseases, HIV infection, tuberculosis, myopathies, cardiomyopathies, renal failure and anaemia, and has been included in baby foods and milk [31]. Carnitine supplementation has also been advocated in chronic VPA treatment, but data are limited (see below).
Carnitine deficiency
A typical, well balanced omnivorous diet contains significant amounts of carnitine (20–200 mg/day for a 70 kg person) as well as the essential amino acids and micronutrients needed for carnitine biosynthesis. Even in strict vegetarian diets (as little as 1 mg/day exogenous carnitine for a 70 kg person), endogenous synthesis combined with the high tubular reabsorption rate is enough to prevent deficiency in generally healthy people. Thus, carnitine deficiency is an unusual problem in the healthy, well nourished adult population [36].
Primary carnitine deficiency is rare and is caused by a genetic defect in membrane carnitine transporter in muscle and/or other organs. Both the myopathic and systemic forms are inherited autosomal and recessive.
Secondary carnitine deficiency is associated with several inborn errors of metabolism and acquired medical conditions [29, 36]. Preterm neonates develop carnitine deficiency because of impaired proximal renal tubule carnitine reabsorption and immature carnitine biosynthesis. The final step in carnitine synthesis that occurs in liver and kidney depends on the enzyme γ-butyrobetaine hydroxylase, which may be deficient in children. An increasing number of problems are reported in relation to carnitine metabolism in preterm infants not receiving an exogenous source of carnitine. Children with various forms of organic acidaemia have carnitine requirements that exceed their dietary intake and biosynthetic capability, in order to permit excretion of accumulating organic acids.
In cirrhosis and chronic renal failure, endogenous carnitine biosynthesis is impaired. Patients with renal disease also appear to lose carnitine via haemodialysis treatment – a loss that cannot be repleted simply by endogenous biosynthesis and dietary intake. Other chronic conditions such as malabsorption, Fanconi syndrome, diabetes mellitus, heart failure and Alzheimer's disease have also been associated with carnitine deficiency. Carnitine deficiency is also observed in critical conditions that involve increased catabolism, such as trauma, sepsis and organ failure, which result in increased need for exogenous carnitine.
Finally, several drugs, especially VPA but also anti-HIV nucleoside analogues, pivalic acid-containing antibiotics and some chemotherapy agents (e.g. ifosfamide, cisplatin and doxorubicin), are associated with decreased carnitine levels and occasionally with true carnitine deficiency [36, 37].
With respect to VPA, this agent depletes carnitine stores, especially during long-term or high-dose therapy, through various synergistic mechanisms [22, 38–41]. First, as a branched chain fatty acid, VPA combines with carnitine to form valproylcarnitine, which is excreted in urine [42]. However, because this excretion accounts for less than 1% of total acylcarnitine elimination in urine [43], it is unlikely that excretion of VPA alone is sufficient to produce carnitine deficiency in well nourished patients [44]. Second, a reduction in tubular reabsorption of both free carnitine and acylcarnitine has been reported during VPA treatment [45, 46], Third, VPA reduces endogenous synthesis of carnitine by blockade of the enzyme butyrobetaine hydroxylase. Fourth, valproylcarnitine inhibits the membrane carnitine transporter, thereby decreasing the transport of extracellular carnitine into the cell and the mitochondria. VPA also induces reversible inhibition of plasmalemmal carnitine uptake in vitro in cultured human skin fibroblasts [47]. Fifth, VPA metabolites combine with mitochondrial CoA-SH. The pool of free CoA-SH decreases, so that free mitochondrial carnitine stores cannot be restored from acylcarnitine (including valproylcanitine) under the action of CPT2. Finally, the mitochondrial depletion of CoA-SH impairs β-oxidation of fatty acids (and VPA) and ATP production. ATP depletion further impairs the function of the ATP-dependent membrane carnitine transporter.
Although systematic assessment of carnitine status has been recommended in VPA-treated patients [27, 29], hypocarnitin-aemia has not been confirmed in all studies. For example, in a recent cross-sectional surveillance study conducted in 43 paediatric patients taking VPA [48], only two were found to have carnitine levels below the normal limit, suggesting that routine carnitine level checking is not justified. Indeed, VPA-treated patients may be carnitine depleted despite having normal carnitine serum levels [49].
Risks factors for carnitine depletion include age under 24 months, the presence of concomitant neurologic or metabolic disorders, and receipt of multiple AEDs. Measurement of carnitine levels is probably warranted in those patients who are at risk for carnitine deficiency in order to identify those who need carnitine supplementation.
Carnitine depletion has several adverse effects. First, it can impair the transport of long-chain fatty acids into the mitochondrial matrix, with subsequent decrease in β-oxidation, acetyl-CoA and ATP production. In turn, the impairment in β-oxidation can shift the metabolism of VPA toward predominantly peroxisomal ω-oxidation, resulting in excessive production and accumulation of ω-oxidation products, including 4-en-VPA – a metabolite that is incriminated in VPA-induced hepatotoxicity. Carnitine depletion can also result in intracellular accumulation of toxic acyl-CoA, resulting in impairment in several enzymatic processes (α-ketoacid oxidation and gluconeogenesis, among others). Finally, carnitine depletion can impair the urea cycle, resulting in accumulation of ammonia (Fig. 4) [4, 38]. This effect may be due either to inhibition of carbamyl phosphate synthase (CPS) I by ω-oxidation metabolites (CPS I is the first mitochondrial enzymatic step of the urea cycle) or to decreased synthesis of N-acetyl glutamic acid (NAGA) from acetyl-CoA and glutamate by NAGA synthetase (NAGA is an important cofactor of CPS I).

Effects of decreased β-oxidation and increased ω-oxidation of fatty acids and VPA on the urea cycle. See text for further details. NAGA, N-acetyl glutamic acid; CoA, coenzyme A; CPS, carbamyl phosphate synthetase; OTC, ornithine transcarbamylase; 4-en-VPA, 2-propyl-4-pentenoic acid.
Carnitine supplementation in valproic acid induced toxicity
Because VPA-induced hyperammonaemia and VHT could be mediated at least in part by carnitine deficiency, it has been hypothesized that L-carnitine supplementation may prevent, correct, or attenuate these adverse effects. Although strong recommendations were made by the Paediatric Neurology Advisory Committee in 1996 and reproduced in many textbooks, the role of carnitine remains ill defined. Three conditions – VHT, VHE and acute VPA overdose – receive separate focus below.
Valproate-induced hepatotoxicity
In up to 44% of patients chronic dosing with VPA may be associated with elevation in transaminases [23] during the first months of therapy. Usually, it resolves completely when the drug is discontinued. Severe VHT in association with hepatic failure is rare, but it may develop as an idiosyncratic reaction that is often fatal. It usually occurs during the first 6 months of VPA therapy and is commonly but not always preceded by minor elevations in transaminases. Reports of severe VHT following acute VPA overdose are rare [50].
The most common clinical presentation consists of lethargy, jaundice, nausea, vomiting, haemorrhage, worsening seizures and anorexia [23]. Histological changes are similar to those observed in the Reye's syndrome, with early production of microvesicular steatosis followed by development of centri-lobular necrosis [23].
Risks factors include age under 24 months (especially those with organic brain disease), developmental delay, coincident congenital metabolic disorders, previous liver dysfunction, or severe epilepsy treated with polytherapy or ketogenic diets [23, 51, 52]. Although the overall incidence is estimated at 1/5000 to 1/50,000, the occurrence of fatal hepatotoxicity could be as high as 1/800 to 1/500 in these high-risk groups [11].
The mechanisms of both subacute and idiosyncratic VHT remain incompletely understood, but it has been believed since the early 1980s – based on limited experimental and clinical evidence [53–56] – that hypocarnitinaemia, subsequent imbalance between β-oxidation and ω-oxidation, and accumulation of 4-en-VPA are involved. Additionally, carnitine deficit may result in disruption of mitochondrial functions due to depletion in CoA-SH [23, 27, 51].
Reduced serum free carnitine as well as reduced levels of 3-keto-VPA, the main metabolite of β-oxidation of VPA, was first reported in 1982 by Bohles and coworkers [53] in a 3-year-old girl who developed acute liver disease with typical features of Reye's syndrome after treatment with VPA for 6 months. Reduced free carnitine and increased serum and urine acylcarnitine levels were also demonstrated in patients with VPA-induced Reye-like syndrome [45]. In a patient with fatal VHT, Krahenbuhl and coworkers [57] demonstrated a reduction in free and total carnitine in plasma and liver.
Laub and coworkers [58] prospectively examined the influence of VPA on carnitinaemia, as well as the possible aetiological role of carnitine in fatal VHT. Total carnitine, free carnitine and acylcarnitine were measured in the serum of 21 paediatric patients receiving VPA therapy, 21 healthy matched control individuals, and 21 patients receiving various AEDs other than VPA. The free carnitine level was the lowest (P < 0.05) and the short-chain acylcarnitine/free carnitine ratio was the highest (P < 0.01) in the VPA group. Moreover, patients receiving polytherapy including VPA had lower total carnitine values than did patients receiving VPA monotherapy (P < 0.05). However, the authors suggested that carnitine deficiency cannot be the only reason for fatal VHT, because a 3.5-year-old girl developed hepatic failure under VPA therapy despite normal serum carnitine values, and died despite oral L-carnitine supplementation.
Other mechanisms such as VPA-induced lipid peroxidation and glutathione depletion could also contribute to hepatotoxicity [59]. Indeed, 4-en-VPA is transformed through β-oxidation to reactive intermediates such as 2-propyl-2, 4-pentadienoic acid (2, 4-dien-VPA) that are capable of depleting mitochondrial GSH, as suggested by rat studies [60]. Unsaturated VPA metabolites (4-en-VPA and 2, 4-dien-VPA) are potent inducers of microvesicular steatosis in rats, whereas VPA itself failed to induce discernible liver lesions at near lethal doses [60]. Studies in rats [61] also suggested that both VPA and its unsaturated metabolites inhibit β-oxidation through different mechanisms, such as sequestration of CoA-SH and direct inhibition of specific enzymes in the β-oxidation sequence by CoA esters, particularly 4-en-VPA-CoA.
Role of carnitine
The common mild elevation in aminotransferases is usually reversible when VPA therapy is discontinued or the dose reduced. Even in severe VHT, the prognosis seems to be improved if VPA therapy is promptly discontinued [62]. Some experimental and clinical evidence also suggests that the early administration of intravenous L-carnitine could further improve survival in severe VHT. Intravenous rather than oral supplementation is recommended because it is likely to ensure higher levels of carnitine in the blood (L-carnitine has poor gastrointestinal bioavailability, which is further compromised by digestive dysfunction).
In an experimental study conducted in rats treated with therapeutic and toxic doses of VPA, Shakoor [63] found that carnitine supplementation was able to prevent fatty infiltration and liver necrosis induced by VPA. No animal study has evaluated the effect of L-carnitine when hepatotoxicity has already developed. There is also a lack of human controlled studies. Among cases of severe hepatotoxicity occurring during VPA therapy, survival has been reported mainly in those patients treated with carnitine [64–67], and this approach is likely to be biased. However, failures of carnitine therapy have occasionally been reported [58].
In a series of 92 patients (most of whom had features of chronic illness or were malnourished children) with severe, symptomatic VHT, Bohan and coworkers [68] observed that 48% of the 42 patients treated with L-carnitine survived, whereas only 10% of the 50 (historical) patients treated solely with aggressive supportive care survived (P < 0.001). Moreover, the 10 patients who were diagnosed within 5 days and treated with intravenous L-carnitine survived. Although these observations are interesting, the comparison with historical control individuals is a serious limitation in the interpretation of these results.
Valproate-induced hyperammonaemic encephalopathy
In chronic VPA dosing hyperammonaemia occurs in nearly 50% of patients, but this remains asymptomatic in almost 50% of cases [39]. VHE is a rare phenomenon in adults, especially when VPA is used as monotherapy. VHE is typically characterized by acute onset of impaired consciousness, focal neurologic symptoms and increased seizure frequency [69]. It may occur after both 'acute on chronic' overdosage and regular chronic use of VPA [16–19, 24, 25, 38, 70]. Very high ammonia levels have been reported, even with normal liver function tests [71].
Various mechanisms have been implicated in the development of VPA-induced hyperammonaemia. Matsuda and coworkers [45] demonstrated a considerable reduction in serum free carnitine concentration in five patients with hyperammonaemia associated with VPA therapy (of whom three had a Reye-like syndrome). Various authors have shown that serum ammonia concentrations directly correlate with the dose or serum concentrations of VPA, and inversely with serum concentrations of carnitine [22, 38, 39]. Lokrantz and coworkers [72] recently reported the case of an old woman taking VPA monotherapy for her partial epilepsy in whom a typical hyperammonaemic encephalopathy was precipitated by treatment for a urinary tract infection with pivmecillinam – an antibiotic known to decrease the serum concentration of carnitine. Also, metabolites of VPA ω-oxidation (including propionic derivatives and 4-en-VPA) inhibit the mitochondrial CPS I, which is the first enzyme necessary for ammonia elimination via the urea cycle in the liver [19, 24]. This effect appears related to the dose of VPA [73]. As acetyl-CoA stores are depleted, the synthesis of NAGA – an important cofactor of CPS I – from acetyl-CoA and glutamate by NAGA synthetase is decreased.
VHE is more frequently observed in patients with congenital defects of the urea enzymatic cycle or with carnitine deficiency [69]. It may also be precipitated by a protein-rich diet [74, 75] or catabolism induced by fasting [76, 77].
An increase in the renal production of ammonia could be another factor that contributes to the development of hyperammonaemia in VPA-treated patients. Indeed, VPA promotes the transport of glutamine through the mitochondrial membrane, thereby enhancing glutaminase activity. Ammonia is released as a result of the transformation of glutamine into glutamate [78, 79]. Both animal [76] and human [77] studies suggest that VPA-induced hyperammonaemia may be enhanced via renal rather than hepatic mechanisms.
The pathogenesis of hyperammonaemic encephalopathy is still incompletely understood, and a detailed discussion of the topic is beyond the scope of this review. Ammonia readily crosses the blood-brain barrier and is thought to inhibit glutamate uptake, thereby increasing extracellular glutamate concentrations in the brain and resulting in activation of NMDA receptors. NMDA receptor activation is associated with a decrease in phosphorylation by protein kinase C, activation of Na+-K+ ATPase and ATP depletion. Activation of the NMDA receptors is a major factor in the pathogenesis of hyperammonaemic encephalopathy and is probably the cause of seizures. However, other factors may be involved, including accumulation of lactate, pyruvate, glutamine and free glucose, and depletion of glycogen, ketone bodies and glutamate.
With respect to VHE, Verrotti and coworkers [69] demonstrated an increase in glutamine production in astrocytes whereas glutamine release was inhibited. Glutamine accumulation increases intracellular osmolarity, promoting an influx of water with resultant astrocytic swelling, cerebral oedema and increased intracranial pressure [68]. The VPA β-oxidation metabolite 2-en-VPA is another agent that can promote cerebral oedema when it accumulates in brain and plasma. Although β-oxidation is impaired in the setting of VPA toxicity, this metabolite has a prolonged elimination half-life and could be responsible for the prolonged coma that is sometimes observed despite the normalization in plasma VPA concentrations [17, 18].
Conversely, there is no evidence for accumulation of valproyl-CoA in brain tissue, suggesting that the effects of VPA in the CNS are independent of the formation of this metabolite [80]. Development of cerebral oedema is not clearly correlated with the dose of VPA ingested [20].
Role of carnitine
Carnitine supplementation (50 mg/kg per day) for 4 weeks was shown to correct both carnitine deficiency and hyperammonaemia in 14 VPA-treated patients [38]. Administration of exogenous carnitine is thought to decrease ammonia levels by binding to VPA, thereby enhancing the β-oxidation process and production of acetyl-CoA, and relieving the inhibition of urea synthesis.
Bohles and coworkers [81] investigated the effects of carnitine supplementation in 69 children and young adults treated with VPA monotherapy. Their mean plasma ammonia concentration was within the normal range, but 24 patients (35.3%) with ammonia concentrations above 80 μg/dl were considered hyperammonaemic and 15 of these 24 (22.1%) had ammonia concentrations above 100 μg/dl. Total plasma carnitine concentrations were determined in 48 out of 69 patients and were found to be rather low, as was the percentage of free carnitine. Fourteen hyperammonaemic and one normoammonaemic patients were supplemented with L-carnitine (500 mg/m2, twice daily). Prolonged L-carnitine supplementation was associated with normalization in plasma ammonia concentrations and marked increase in carnitine concentration in all 15 patients. The plasma ammonia concentrations were significantly correlated with the percentage of free plasma carnitine in plasma (r = -0.67, P < 0.0001). These findings indicate that carnitine supplementation allows normalization of elevated plasma ammonia concentrations. However, a correlation between ammonia levels and clinical condition is not always observed.
Borbath and coworkers [82] reported the case of a 51-year-old woman who received 10 mg/kg VPA daily to prevent seizures after a neurosurgical procedure, and who developed VHE (ammonia concentration 234 μmol/l) without any sign of hepatic dysfunction. VPA was stopped and L-carnitine supplementation (100 mg/kg) was administered intravenously. Ammonia levels rapidly decreased within 10 hours (to 35 μmol/l), the neurological condition improved and triphasic waves on the electroencephalogram disappeared. However, the initial plasma carnitine level was normal in this patient.
Conversely, Hantson and coworkers [83] recently reported the case of a 47-year-old epileptic man in whom parenteral VPA therapy was associated with a severe hyperammonaemic encephalopathy (peak ammonia concentration 411 μmol/l) without any biological signs of hepatotoxicity. VPA treatment was discontinued and L-carnitine supplementation (100 mg/kg per day) was initiated. Although subsequent normalization in the blood arterial ammonia level was observed within 4 days, the patient remained comatose for 3 weeks. The clinical course was correlated with magnetic resonance imaging and multimodal evoked potential findings, but not with ammonia levels.
Acute valproic acid overdose
Acute VPA intoxication is an increasing problem and this topic was recently reviewed [4]. The clinical and biological manifestations that may be encountered reflect both exaggerated therapeutic effect and impairment in metabolic pathways.
CNS depression is the most common manifestation of toxicity, ranging in severity from mild drowsiness to profound coma and fatal cerebral oedema [20, 50]. However, the majority of patients only experience mild to moderate lethargy and recover uneventfully with only supportive care [4, 84, 85]. Although there is no close relationship between plasma VPA concentrations and the severity of CNS toxicity [11, 86], patients who ingest more than 200 mg/kg VPA and/or have plasma concentrations greater than 180 μg/ml usually develop severe CNS depression. In such severe cases, cerebral oedema becomes clinically apparent 12 hours to 4 days after the overdose [18, 20, 50, 87], although CNS depression may be delayed if a slow-release preparation has been ingested [12, 50].
Other clinical findings include respiratory depression, nausea, vomiting, diarrhoea, hypothermia or fever, hypotension, tachycardia, miosis, agitation, hallucinations, tremors, myoclonus and seizures. In contrast to poisoning with phenytoin or carbamazepine, nystagmus, dysarthria and ataxia are rarely noted following VPA overdose. Other recognized but rare complications of overdose include heart block, pancreatitis, acute renal failure, alopecia, leucopenia, thrombocytopenia, anaemia, optic nerve atrophy and acute respiratory distress syndrome [18, 50]. Acute VPA poisoning is rarely associated with a minor and reversible elevation in transaminases [17, 18]. Hyperammonaemia, anion gap metabolic acidosis, hyperosmolality, hypernatraemia and hypocalcaemia [18, 20, 50] may also develop.
Management of acute VPA intoxication is largely supportive. Patients who present early may benefit from gastrointestinal decontamination with a single dose of activated charcoal. Other interventions may involve blood pressure support with intravenous fluids and vasopressors, and correction of electrolyte abnormalities or acid-base disorders (commonly an anion gap metabolic acidosis). Mechanical ventilation may be necessary in patients who require airway protection or who develop cerebral oedema or respiratory depression.
In patients with renal dysfunction, refractory hypotension, severe metabolic abnormalities, active seizure, or persistent coma, extracorporeal removal by haemodialysis or haemofiltration may be considered, although there are no controlled trials that demonstrate an improvement in outcome with these measures [88–90]. Similarly, there is no evidence that multiple doses of activated charcoal do increase the elimination of VPA or toxic metabolites.
Role of carnitine
Although data in this setting are sparse, consisting of anecdotal case reports, it has been suggested that carnitine supplementation could hasten resolution of coma, prevent development of hepatic dysfunction and reverse mitochondrial metabolic abnormalities in patients with acute VPA intoxication [22, 70]. For example, a healthy, nonepileptic, 16-month-old child ingested a massive overdose (approximately 4 g) of VPA [22]. Upon admission to the hospital he was in a deep coma and had generalized hypotonicity and no response to pain. His serum and urinary concentrations of VPA were 1316.2 and 3289.5 μg/ml, respectively. Urinary concentrations of the β-oxidation metabolites of VPA were low, whereas concentrations of ω-oxidation metabolites were high. Moreover, the hepatotoxic compound 4-en-VPA was detected in urine. Gastric lavage and general supportive measures were undertaken, including intravenous infusion of saline to increase urine output, and oral L-carnitine was administered for 4 days to correct hypocarnitinaemia. Subsequently, the β-oxidation metabolites increased, the ω-oxidation metabolites decreased and 4-en-valproate was no longer detected in urine. However, the child only regained consciousness on day 4, when his serum VPA concentration reached therapeutic levels. The patient completely recovered and was discharged from hospital on day 8 without any sequelae.
In another child who accidentally ingested 400 mg/kg VPA, decreased β-oxidation and markedly increased ω-oxidation were also observed, and the concentration of 4-en-VPA was markedly increased, although there was neither hyperammonaemia nor signs of liver dysfunction [70]. After L-carnitine supplementation for 3 days, VPA metabolism returned to normal. Once again the child remained comatose until day 3. The level of valproylcarnitine was not increased and was not affected by L-carnitine supplements.
Minville and coworkers [91] recently reported a case of severe VPA poisoning in a 36-year-old man. Haemodialysis was initiated to decrease the high serum VPA concentration and L-carnitine therapy (50 mg/kg per day for 4 days) was empirically started. Despite this treatment, cerebral oedema appeared on the third day. With usual neuroprotective measures, the patient improved after 4 days and finally recovered without sequelae.
Because hepatotoxicity is rare after acute overdose, the lack of transaminase elevation following prophylactic carnitine administration does not demonstrate its hepatoprotective properties. As far as CNS depression is concerned, the clinical observations do not suggest that carnitine is able to hasten the recovery of consciousness. Nevertheless, in 1996 the Paediatric Neurology Advisory Committee recommended carnitine supplementation for children with VPA overdoses. Subsequent, more restrictive recommendations limit carnitine supplements to those children with overdoses above 400 mg/kg.
Carnitine supplements in the prevention of valproic acid toxicity
Raskind and El-Chaar [41] extensively reviewed the patho-physiology and significance of VPA-induced carnitine deficiency and evaluated the literature pertaining to carnitine supplementation during VPA therapy in children. Despite the lack of prospective, randomized clinical trials, a few studies have shown carnitine supplementation in patients receiving VPA to result in subjective and objective improvements and to prevent VHT, in parallel with increases in carnitine serum levels. The Pediatric Neurology Advisory Committee in 1996 and some textbooks and manuals strongly recommended carnitine supplementation (50–100 mg/kg per day) during VPA therapy for children at risk for developing a carnitine deficiency, in VPA overdose and in VHT [33, 92, 93]. Carnitine supplementation has been classified 'grade C' (may be useful) in patients treated with VPA for seizure disorders [94]. There is no clear evidence that appreciable toxic effects are associated with use of carnitine. Moreover, when it is used to prevent carnitine deficiency, carnitine did not seem to alter the anticonvulsant properties of VPA in an experimental model in mice [95].
Until further data become available, L-carnitine supplementation may be recommended in those children on VPA therapy at greatest risk for hepatotoxicity (<2 years of age, more than one anticonvulsant, poor nutritional status, ketogenic diet). In older children or adults it may be considered if there are clinical symptoms suggestive of carnitine deficiency (hypotonia, lethargy), a significant decrease in the serum free carnitine levels, an impairment in hepatic function tests, or hyperammonaemia, even in the absence of VHE.
Conclusion
The potential value of oral L-carnitine in reversing carnitine deficiency and preventing adverse effects due to VPA-induced dysfunction of β-oxidation is suggested by several studies and isolated observations. Carnitine supplementation is now recommended in acute overdose, VHE and VHT by some scientific committees and textbooks, especially in high-risk paediatric patients.
Carnitine supplementation does not appear to be harmful and could be beneficial in patients with VHT or hyperammonaemia, regardless of whether the exposure was acute, chronic, or both. Conversely, although carnitine appears to normalize the metabolic pathways of VPA in acute overdose, the few clinical data that are available do not support the use of carnitine in patients with VPA-induced CNS depression. Finally, prophylactic supplementation with L-carnitine seems reasonable in high risk patients.
However, better delineation of the therapeutic and prophylactic roles of L-carnitine in these conditions will require further investigations in controlled, randomized and probably multicentre trials to evaluate the clinical value and the appropriate dosage of L-carnitine in each of these conditions.
Abbreviations
- AED:
-
antiepileptic drug
- CNS:
-
central nervous system
- CoA:
-
coenzyme A
- CPS:
-
carbamyl phosphate synthase
- GABA:
-
γ-aminobutyric acid
- NAGA:
-
N-acetyl glutamic acid
- NMDA:
-
N-methyl-D-aspartate
- PCT:
-
palmityl carnitine transferase
- VHE:
-
VPA-induced hyperammonaemic encephalopathy
- VHT:
-
VPA-induced hepatotoxicity
- VPA:
-
valproic acid.
References
Johannessen CU, Johannessen SI: Valproate: past, present, and future. CNS Drug Rev 2003, 9: 199-216.
Spiller HA, Krenzelok EP, Klein-Schwartz W, Winter ML, Weber JA, Sollee DR, Bangh SA, Griffith JR: Multicenter case series of valproic acid ingestion: serum concentrations and toxicity. J Toxicol Clin Toxicol 2000, 38: 755-760. 10.1081/CLT-100102388
Watson WA, Litovitz TL, Klein-Schwartz W, Rodgers GC Jr, Youniss J, Reid N, Rouse WG, Rembert RS, Borys D: 2003 annual report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med 2004, 22: 335-404. 10.1016/j.ajem.2004.06.001
Sztajnkrycer MD: Valproic acid toxicity: overview and management. J Toxicol Clin Toxicol 2002, 40: 789-801. 10.1081/CLT-120014645
Bedry R, Parrot F: Severe valproate poisoning [in French]. Réanimation 2004, 13: 324-333. 10.1016/j.reaurg.2004.03.014
Ellenhorn M: Diagnosis and treatment of human poisoning. In Medical Toxicology. Edited by: Ellenhorn M. Baltimore: Willians and Wilkins; 1997:609-610.
Leikin JPF: Poisoning and Toxicology Handbook. 3rd edition. Hudson: Lexi-Comp Inc; 2002.
Bolanos JP, Medina JM: Effect of valproate on the metabolism of the central nervous system. Life Sci 1997, 60: 1933-1942. 10.1016/S0024-3205(96)00687-X
Loscher W: Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs 2002, 16: 669-694.
Loscher W: Valproate: a reappraisal of its pharmacodynamic properties and mechanisms of action. Prog Neurobiol 1999, 58: 31-59. 10.1016/S0301-0082(98)00075-6
McNamara JO: Drugs effective in the therapy of the epilepsies. In Goodman & Gilman's The Pharmacological Basis of Therapeutics. Edited by: Hardman J, Limbird LE, Molinoff PB, Ruddon RW, Gilman AG. New York: McGraw-Hill; 1996:476.
Graudins A, Aaron CK: Delayed peak serum valproic acid in massive divalproex overdose: treatment with charcoal hemoperfusion. J Toxicol Clin Toxicol 1996, 34: 335-341.
Ingels M, Beauchamp J, Clark RF, Williams SR: Delayed valproic acid toxicity: a retrospective case series. Ann Emerg Med 2002, 39: 616-621. 10.1067/mem.2002.124443
Brubacher JR, Dahghani P, McKnight D: Delayed toxicity following ingestion of enteric-coated divalproex sodium (Epival). J Emerg Med 1999, 17: 463-467. 10.1016/S0736-4679(99)00008-6
Chadwick DW: Concentration-effect relationships of valproic acid. Clin Pharmacokinet 1985, 10: 155-163.
Gugler R, von Unruh GE: Clinical pharmacokinetics of valproic acid. Clin Pharmacokinet 1980, 5: 67-83.
Gram L, Bentsen KD: Valproate: an updated review. Acta Neurol Scand 1985, 72: 129-139.
Tennison MB, Miles MV, Pollack GM, Thorn MD, Dupuis RE: Valproate metabolites and hepatotoxicity in an epileptic population. Epilepsia 1988, 29: 543-547.
Coulter DL, Allen RJ: Secondary hyperammonaemia: a possible mechanism for valproate encephalopathy. Lancet 1980, 1: 1310-1311. 10.1016/S0140-6736(80)91773-0
Khoo SH, Leyland MJ: Cerebral edema following acute sodium valproate overdose. J Toxicol Clin Toxicol 1992, 30: 209-214.
Breum L, Astrup A, Gram L, Andersen T, Stokholm KH, Chris-tensen NJ, Werdelin L, Madsen J: Metabolic changes during treatment with valproate in humans: implication for untoward weight gain. Metabolism 1992, 41: 666-670. 10.1016/0026-0495(92)90061-E
Ishikura H, Matsuo N, Matsubara M, Ishihara T, Takeyama N, Tanaka T: Valproic acid overdose and L-carnitine therapy. J Anal Toxicol 1996, 20: 55-58.
Bryant AE 3rd, Dreifuss FE: Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology 1996, 46: 465-469.
Coulter DM: Study of reasons for cessation of therapy with perhexiline maleate, sodium valproate and labetalol in the intensified adverse reaction reporting scheme. N Z Med J 1981, 93: 81-84.
Mortensen PB: Dicarboxylic acids and the lipid metabolism. Dan Med Bull 1984, 31: 121-145.
Li J, Norwood DL, Mao LF, Schulz H: Mitochondrial metabolism of valproic acid. Biochemistry 1991, 30: 388-394. 10.1021/bi00216a012
Coulter DL: Carnitine, valproate, and toxicity. J Child Neurol 1991, 6: 7-14.
Hiraoka A, Arato T, Tominaga I: Reduction in blood free carnitine levels in association with changes in sodium valproate (VPA) disposition in epileptic patients treated with VPA and other anti-epileptic drugs. Biol Pharm Bull 1997, 20: 91-93.
Borum PR, Bennett SG: Carnitine as an essential nutrient. J Am Coll Nutr 1986, 5: 177-182.
Jacobi G, Thorbeck R, Ritz A, Janssen W, Schmidts HL: Fatal hepatotoxicity in child on phenobarbitone and sodium valproate. Lancet 1980, 1: 712-713.
Evangeliou A, Vlassopoulos D: Carnitine metabolism and deficit: when supplementation is necessary? Curr Pharm Biotechnol 2003, 4: 211-219. 10.2174/1389201033489829
Roe CRM, Kahler SG, Kodo N, Norwood DL: Carnitine homeostatis in the organic acidurias. In Fatty Acid Oxidation: Clinical, Biochemical, and Molecular Aspects. Edited by: Tanaka KC. New York: Alan R Liss; 1990:382-402.
Lehninger AL, Nelson DL, Cox MM: Oxidation of fatty acids [in French]. In Principes de Biochimie. New York: Médecine–Sciences Flammarion; 1994:479-505.
DiMauro S, DiMauro PM: Muscle carnitine palmityltransferase deficiency and myoglobinuria. Science 1973, 182: 929-931.
Roe CC, Coates PM: Mitochondrial fatty acid oxidation disorders. In The Metabolic and Molecular Basic of Inherited Disease. Edited by: Scriver CR, Beaudet AL, Sly WS, Valle D. New York: McGraw-Hill; 1995:1501-1533.
Scaglia F: Carnitine deficiency. eMedicine 2004. [http://www.emedicine.com/ped/topic321.htm]
Murphy JV, Marquardt KM, Shug AL: Valproic acid associated abnormalities of carnitine metabolism. Lancet 1985, 1: 820-821. 10.1016/S0140-6736(85)91481-3
Ohtani Y, Endo F, Matsuda I: Carnitine deficiency and hyperammonemia associated with valproic acid therapy. J Pediatr 1982, 101: 782-785.
Gidal BE, Inglese CM, Meyer JF, Pitterle ME, Antonopolous J, Rust RS: Diet- and valproate-induced transient hyperammonemia: effect of L-carnitine. Pediatr Neurol 1997, 16: 301-305. 10.1016/S0887-8994(97)00026-X
Matsumoto J, Ogawa H, Maeyama R, Okudaira K, Shinka T, Kuhara T, Matsumoto I: Successful treatment by direct hemop-erfusion of coma possibly resulting from mitochondrial dysfunction in acute valproate intoxication. Epilepsia 1997, 38: 950-953.
Raskind JY, El-Chaar GM: The role of carnitine supplementation during valproic acid therapy. Ann Pharmacother 2000, 34: 630-638. 10.1345/aph.19242
Millington DS, Bohan TP, Roe CR, Yergey AL, Liberato DJ: Valproylcarnitine: a novel drug metabolite identified by fast atom bombardment and thermospray liquid chromatographymass spectrometry. Clin Chim Acta 1985, 145: 69-76. 10.1016/0009-8981(85)90020-8
Sugimoto T, Muro H, Woo M, Nishida N, Murakami K: Valproate metabolites in high-dose valproate plus phenytoin therapy. Epilepsia 1996, 37: 1200-1203.
Hirose S, Mitsudome A, Yasumoto S, Ogawa A, Muta Y, Tomoda Y: Valproate therapy does not deplete carnitine levels in otherwise healthy children. Pediatrics 1998, 101: E9. 10.1542/peds.101.5.e9
Matsuda I, Ohtani Y: Carnitine status in Reye and Reye-like syndromes. Pediatr Neurol 1986, 2: 90-94. 10.1016/0887-8994(86)90062-7
Camina MF, Rozas I, Castro-Gago M, Paz JM, Alonso C, Rodriguez-Segade S: Alteration of renal carnitine metabolism by anticonvulsant treatment. Neurology 1991, 41: 1444-1448.
Tein I, Xie ZW: Reversal of valproic acid-associated impairment of carnitine uptake in cultured human skin fibroblasts. Biochem Biophys Res Commun 1994, 204: 753-758. 10.1006/bbrc.1994.2523
Fung EL, Tang NL, Ho CS, Lam CW, Fok TF: Carnitine level in Chinese epileptic patients taking sodium valproate. Pediatr Neurol 2003, 28: 24-27. 10.1016/S0887-8994(02)00460-5
Shapiro YG, Gutman A: Muscle carnitine deficiency in patients using valproic acid. J Pediatr 1991, 118: 646-649.
Andersen GO, Ritland S: Life threatening intoxication with sodium valproate. J Toxicol Clin Toxicol 1995, 33: 279-284.
Zimmerman HJ, Ishak KG: Valproate-induced hepatic injury: analyses of 23 fatal cases. Hepatology 1982, 2: 591-597.
Antoniuk SA, Bruck I, Honnicke LR, Martins LT, Carreiro JE, Cat R: Acute hepatic failure associated with valproic acid in children. Report of 3 cases [in Portuguese]. Arq Neuropsiquiatr 1996, 54: 652-654.
Bohles H, Richter K, Wagner-Thiessen E, Schafer H: Decreased serum carnitine in valproate induced Reye syndrome. Eur J Pediatr 1982, 139: 185-186. 10.1007/BF01377353
Coulter DL: Carnitine deficiency: a possible mechanism for valproate hepatotoxicity. Lancet 1984, 1: 689. 10.1016/S0140-6736(84)92209-8
Stumpf DA, Parker WD, Haas R: Carnitine deficiency with valproate therapy. J Pediatr 1983, 103: 175-176.
Ohtani Y, Matsuda I: Valproate treatment and carnitine deficiency. Neurology 1984, 34: 1128-1129.
Krahenbuhl S, Mang G, Kupferschmidt H, Meier PJ, Krause M: Plasma and hepatic carnitine and coenzyme A pools in a patient with fatal, valproate induced hepatotoxicity. Gut 1995, 37: 140-143.
Laub MC, Paetzke-Brunner I, Jaeger G: Serum carnitine during valproic acid therapy. Epilepsia 1986, 27: 559-562.
Raza M, Al-Bekairi AM, Ageel AM, Qureshi S: Biochemical basis of sodium valproate hepatotoxicity and renal tubular disorder: time dependence of peroxidative injury. Pharmacol Res 1997, 35: 153-157. 10.1006/phrs.1997.0134
Tang W, Borel AG, Fujimiya T, Abbott FS: Fluorinated analogues as mechanistic probes in valproic acid hepatotoxicity: hepatic microvesicular steatosis and glutathione status. Chem Res Toxicol 1995, 8: 671-682. 10.1021/tx00047a006
Kesterson JW, Granneman GR, Machinist JM: The hepatotoxicity of valproic acid and its metabolites in rats. I. Toxicologic, biochemical and histopathologic studies. Hepatology 1984, 4: 1143-1152.
Goldfrank LR, Flomenbaum NE, Lewin NA, Howland MA, Hoffman RS, Nelson LS, (editors): Goldfrank's Toxicological Emergencies. 7th edition. New York: McGraw-Hill; 2002.
Shakoor KAK: Valproic acid induced hepatotoxicitv and protective role of carnitine. Pakistan J Pathol 1997, 133-134.
Siemes H, Nau H, Schultze K, Wittfoht W, Drews E, Penzien J, Seidel U: Valproate (VPA) metabolites in various clinical conditions of probable VPA-associated hepatotoxicity. Epilepsia 1993, 34: 332-346.
DeVivo DC: Effect of L-carnitine treatment for valproate-induced hepatotoxicity. Neurology 2002, 58: 507-508.
Konig SA, Siemes H, Blaker F, Boenigk E, Gross-Selbeck G, Hanefeld F, Haas N, Kohler B, Koelfen W, Korinthenberg R, et al.: Severe hepatotoxicity during valproate therapy: an update and report of eight new fatalities. Epilepsia 1994, 35: 1005-1015.
Romero-Falcon A, de la Santa-Belda E, Garcia-Contreras R, Varela JM: A case of valproate-associated hepatotoxicity treated with L-carnitine. Eur J Intern Med 2003, 14: 338-340. 10.1016/S0953-6205(03)00104-3
Bohan TP, Helton E, McDonald I, Konig S, Gazitt S, Sugimoto T, Scheffner D, Cusmano L, Li S, Koch G: Effect of L-carnitine treatment for valproate-induced hepatotoxicity. Neurology 2001, 56: 1405-1409.
Verrotti A, Trotta D, Morgese G, Chiarelli F: Valproate-induced hyperammonemic encephalopathy. Metab Brain Dis 2002, 17: 367-373. 10.1023/A:1021918104127
Murakami K, Sugimoto T, Woo M, Nishida N, Muro H: Effect of L-carnitine supplementation on acute valproate intoxication. Epilepsia 1996, 37: 687-689.
Barrueto F Jr, Hack JB: Hyperammonemia and coma without hepatic dysfunction induced by valproate therapy. Acad Emerg Med 2001, 8: 999-1001.
Lokrantz CM, Eriksson B, Rosen I, Asztely F: Hyperammonemic encephalopathy induced by a combination of valproate and pivmecillinam. Acta Neurol Scand 2004, 109: 297-301. 10.1046/j.1600-0404.2003.00227.x
Marini AM, Zaret BS, Beckner RR: Hepatic and renal contributions to valproic acid-induced hyperammonemia. Neurology 1988, 38: 365-371.
Laub MC: Nutritional influence on serum ammonia in young patients receiving sodium valproate. Epilepsia 1986, 27: 55-59.
Gidal BE, Inglese CM, Meyer JF, Pitterle ME, Antonopolous J, Rust RS: Diet- and valproate-induced transient hyperammonemia: effect of L-carnitine. Pediatr Neurol 1997, 16: 301-305. 10.1016/S0887-8994(97)00026-X
Warter JM, Imler M, Marescaux C, Chabrier G, Rumbach L, Micheletti G, Krieger J: Sodium valproate-induced hyperammonemia in the rat: role of the kidney. Eur J Pharmacol 1983, 87: 177-182. 10.1016/0014-2999(83)90327-8
Warter JM, Brandt C, Marescaux C, Rumbach L, Micheletti G, Chabrier G, Krieger J, Imler M: The renal origin of sodium valproate-induced hyperammonemia in fasting humans. Neurology 1983, 33: 1136-1140.
Collins RM Jr, Zielke HR, Woody RC: Valproate increases glutaminase and decreases glutamine synthetase activities in primary cultures of rat brain astrocytes. J Neurochem 1994, 62: 1137-1143.
Vittorelli A, Gauthier C, Michoudet C, Baverel G: Metabolic viability and pharmaco-toxicological reactivity of cryopreserved human precision-cut renal cortical slices. Toxicol In Vitro 2004, 18: 285-292. 10.1016/j.tiv.2003.08.010
Becker CM, Harris RA: Influence of valproic acid on hepatic carbohydrate and lipid metabolism. Arch Biochem Biophys 1983, 223: 381-392. 10.1016/0003-9861(83)90602-1
Bohles H, Sewell AC, Wenzel D: The effect of carnitine supplementation in valproate-induced hyperammonaemia. Acta Paediatr 1996, 85: 446-449.
Borbath I, de Barsy T, Jeanjean A, Hantson P: Severe hyperammonemic encephalopathy shortly after valproic acid prescription and rapid awakening following levocarnitine. J Toxicol Clin Toxicol 2000, 38: 219-220.
Hantson P, Grandin C, Duprez T, Nassogne MC, Guérit JM: Comparison of clinical, magnetic resonance and evoked potentials data in a case of valproic-acid-related hyperammonemic coma. Eur Radiol 2005, 15: 59-64. 10.1007/s00330-004-2338-9
Garnier R, Boudignat O, Fournier PE: Valproate poisoning. Lancet 1982, 2: 97. 10.1016/S0140-6736(82)91713-5
Berkovic SF, Bladin PF, Jones DB, Smallwood RA, Vajda FJ: Hepatotoxicity of sodium valproate. Clin Exp Neurol 1983, 19: 192-197.
Mortensen PB, Hansen HE, Pedersen B, Hartmann-Andersen F, Husted SE: Acute valproate intoxication: biochemical investigations and hemodialysis treatment. Int J Clin Pharmacol Ther Toxicol 1983, 21: 64-68.
Zafrani ES, Berthelot P: Sodium valproate in the induction of unusual hepatotoxicity. Hepatology 1982, 2: 648-649.
Van Keulen JGV, Gemke RJ, Van Wijk JAE, Touw DJ: Treatment of valproic acid overdose with continuous arteriovenous hemofiltration [abstract]. J Toxicol Clin Toxicol 2000, 38: 219.
Hicks LK, McFarlane PA: Valproic acid overdose and haemodialysis. Nephrol Dial Transplant 2001, 16: 1483-1486. 10.1093/ndt/16.7.1483
Guillaume CP, Stolk L, Dejagere TF, Kooman JP: Successful use of hemodialysis in acute valproic acid intoxication. J Toxicol Clin Toxicol 2004, 42: 335-336.
Minville V, Roche Tissot C, Samii K: Haemodialysis, L-carnitine therapy and valproic acid overdose [in French]. Ann Fr Anesth Reanim 2004, 23: 357-360.
De Vivo DC, Bohan TP, Coulter DLDreifuss FE, Greenwood RS, Nordli DR Jr, Shields WD, Stafstrom CE, Tein I: L-carnitine supplementation in childhood epilepsy: current perspectives. Epilepsia 1998, 39: 1216-1225.
Samuels M: Manual of Neurologic Therapeutics. Philadelphia: Lippincott, Williams & Wilkins; 1999.
AACE Nutrition Guidelines Task Force: American association of clinical endocrinologists medical guidelines for the clinical use of dietary supplements and nutraceuticals. Endocr Pract 2003, 9: 417-470.
Lammert F, Matern S: Hepatic diseases caused by drugs [in German]. Schweiz Rundsch Med Prax 1997, 86: 1167-1171.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Lheureux, P.E., Penaloza, A., Zahir, S. et al. Science review: Carnitine in the treatment of valproic acid-induced toxicity – what is the evidence?. Crit Care 9, 431 (2005). https://doi.org/10.1186/cc3742
Published:
DOI: https://doi.org/10.1186/cc3742