
Abstract
Acute lung injury and its more severe form, acute respiratory distress syndrome, are major challenges in critically ill patients. Activation of circulating neutrophils and transmigration into the alveolar airspace are associated with development of acute lung injury, and inhibitors of neutrophil recruitment attenuate lung damage in many experimental models. The molecular mechanisms of neutrophil recruitment in the lung differ fundamentally from those in other tissues. Distinct signals appear to regulate neutrophil passage from the intravascular into the interstitial and alveolar compartments. Entry into the alveolar compartment is under the control of CXC chemokine receptor (CXCR)2 and its ligands (CXC chemokine ligand [CXCL]1–8). The mechanisms that govern neutrophil sequestration into the vascular compartment of the lung involve changes in the actin cytoskeleton and adhesion molecules, including selectins, β2 integrins and intercellular adhesion molecule-1. The mechanisms of neutrophil entry into the lung interstitial space are currently unknown. This review summarizes mechanisms of neutrophil trafficking in the inflamed lung and their relevance to lung injury.
Similar content being viewed by others


Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are characterized by increased permeability of the alveolar–capillary barrier, resulting in influx of protein-rich edema fluid and consequently impairment in arterial oxygenation. Although mortality has decreased over recent decades, it remains high (30–40%), and pulmonary and nonpulmonary morbidity in ARDS survivors is significant [1].
Although ALI has been described in neutropenic patients, activation and transmigration of circulating neutrophils (polymorphonuclear leukocytes [PMNs]) are thought to play a major role in the early development of ALI [2]. In most animal models, elimination of PMNs markedly decreases the severity of ALI [3]. In addition, recovery from neutropenia in some patients with lung injury is associated with a deterioration in pulmonary function [4].
Various animal models have been developed to study the molecular basis of PMN trafficking in the lung (Table 1), but each mimics only some aspects of the clinical situation. Pre-existing pulmonary or nonpulmonary diseases, fluid resuscitation, and mechanical ventilation significantly influence the course of ALI but are not considered in most animal models. In addition, experimental methods with which to study PMN recruitment are limited; for example, intravital microscopy has produced great insight into leukocyte–endothelial interactions in many organs, but it is still technically challenging in the lung.
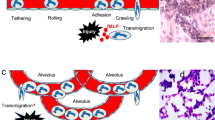
The specific architecture of the lung leads to unique properties of the pulmonary microcirculation. Even under physiologic conditions, neutrophils must stop several times and change their shape to traverse the small pulmonary capillaries (2–15 μm [5]). This leads to an increased transit time through the pulmonary capillary bed and a significant 40- to 100-fold PMN accumulation ('marginated pool') in the lungs (Fig. 1) [6].

Neutrophil trafficking in the lung. Neutrophils (polymorphonuclear leukocytes [PMNs], colored blue) enter a pulmonary capillary (left). Because of the small diameter of the capillary, neutrophils must deform, which increases transit time ('margination') even under resting conditions (inset A: margination). In venules, adhesion molecule (AM)-dependent rolling can occur. In response to an inflammatory stimulus (red arrow), neutrophils adhere to the capillary endothelium (inset B: sequestration). AMs and chemokines (not shown) might be involved in this process. Alveolar macrophages and type II pneumocytes produce CXC chemokines, which attract neutrophils to migrate through the endothelium (inset C1: transendothelial migration), interstitial space, and epithelium (inset C2: transepithelial migration) to reach the alveolar space. The requirement of AMs for the different steps is dependent on the stimulus and the used model (see text for details). Arrows indicate directions of flow, and dashed lines indicate endothelial and epithelial basement membrane.
In the systemic microcirculation, PMN recruitment from blood into tissue at sites of inflammation usually occurs in post-capillary venules and requires capture, rolling, and firm adhesion on activated endothelial cells. Selectin, integrin and immunoglobulin adhesion molecules, cytokines, chemokines, and other chemoattractants participate in this sequential process in a variety of vascular beds [7–9]. In contrast, the principal site of leukocyte migration in the lung is the capillary bed. PMN migration into the different lung compartments (intravascular, interstitial, and intra-alveolar) is differentially regulated because PMNs can enter the pulmonary interstitium without advancing to the alveolar airspace. However, crossing the epithelial barrier seems to be pivotal for inducing lung damage and it is associated with an increase in mortality [10]. Bacterial endotoxin (lipopolysaccharide [LPS]) is known to induce a large influx of PMNs into the alveolar airspace, but only when it is given intratracheally. In contrast, systemic LPS leads to PMN sequestration in the pulmonary vasculature, but most of these cells never appear in bronchoalveolar lavage (BAL) fluid [11].
This review summarizes experimental findings that provide insight into the mechanisms of PMN recruitment in the pulmonary microvasculature, including the migration steps from blood to alveolar airspace. The interrelation between lung inflammation and coagulation provides a target for potential future pharmacological interventions, and we critically discuss the clinical relevance of these experimental findings.
Sequestration of neutrophils in the inflamed lung
In contrast to physiologic margination, neutrophil sequestration reflects the process of neutrophil accumulation in the pulmonary vasculature in response to an inflammatory stimulus. Inflammatory challenge results in a rapid depression in circulating neutrophils in the blood, mainly due to a dramatic increase in PMN sequestration in the pulmonary microvasculature. Altered biomechanical properties of neutrophils are thought to be important for PMN sequestration in the lung in response to various mediators such as tumor necrosis factor (TNF)-α, IL-8, platelet-activating factor, and N-formylmethionyl-leucyl-phenylalanine (fMLP) [12, 13]. Activated neutrophils lose their ability to deform mainly because of intracellular polymerization of actin filaments. Actin filaments are redistributed from the central, perinuclear regions to the peripheral regions in response to chemoattractants, forming actin stress fibers, lamellipodia, ruffles, and filopodia. As a result of these changes in cell shape in response to a chemoattractant, transit time through the pulmonary vasculature is prolonged, resulting in an increased concentration of PMNs [14]. Inhibition of cellular actin organization with cytochalasin D prevents fMLP-induced PMN sequestration [15]. Circulating neutrophils in ARDS patients were found to be less deformable [16], emphasizing the key role played by structural changes in PMN entrapment in the lung microvasculature.
The role of adhesion molecules in this process is not clear. L-selectin-deficient mice exhibit attenuation of prolonged (5 min after complement injection) capillary sequestration, whereas rapid PMN accumulation is only reduced in noncapillary vessels [17]. L-selectin is required for the sequestration of neutrophils in the lung in response to the formyl peptide fMLP, but not C5a, as shown by blocking antibodies and L-selectin-deficient mice [18]. In a model of sepsis-induced ARDS, antibodies to E-selectin and L-selectin did not affect PMN sequestration [19]. Deficiency in either E-selectin and P-selectin or CD18 alone did not affect sequestration [20]. However, blocking CD18, α4, and α5 integrin in combination resulted in a significant attenuation of fMLP-induced sequestration into the lung. Additional inhibition of E-selectin and L-selectin further attenuated PMN sequestration, whereas inhibition of these selectins alone had no effect [21]. Interestingly, these blocking antibodies did not inhibit the physical deformation properties of PMNs, suggesting that neutrophil deformability is not the only regulator of sequestration.
Transendothelial migration of neutrophils
Transendothelial PMN migration in response to inflammatory stimuli occurs in the pulmonary capillary bed, mainly by penetrating interendothelial junctions or at bicellular or tricellular corners of endothelial cells, although there is an alternative, transcellular route [22]. Sequestered and adherent neutrophils induce cytoskeletal changes in the endothelial cells. Adhesive interactions between leukocytes and endothelial cells, leading to intracellular signaling through transmembrane proteins in the area of tight junctions (e.g. platelet endothelial cell adhesion molecule [PECAM]-1, CD99, VE-cadherin), might trigger transient remodeling of the junction [23, 24]. It has been suggested that neutrophil proteases may digest the subendothelial matrix. However, inhibition of these proteases does not affect neutrophil transendothelial migration [25] or migration through the basement membrane [26].
Role of adhesion molecules
The initial steps of the leukocyte adhesion cascade include capture and rolling of circulating leukocytes and require E-, L- and P-selectin [27], whereas integrins – heterodimeric transmembrane glycoproteins – mediate firm adhesion by interacting with intercellular adhesion molecules (e.g. intercellular adhesion molecule [ICAM]-1, ICAM-2, vascular cell adhesion moleculre [VCAM]-1) [28]. Selectins and β2 integrins (CD18) are the most studied adhesion molecules in ALI.
Integrins
PMN migration in the lung can occur in both CD18-dependent and CD18-independent pathways, depending on the stimulus (Table 2). Neutrophil recruitment requires CD18 when induced by Escherichia coli, Pseudomonas aeruginosa, IL-1, or IgG immune complexes, whereas migration in response to Gram-positive bacteria, hyperoxia, and complement factor C5a is CD18 independent. Aspiration of hydrochloric acid induces a CD18-independent PMN migration at the site of instillation but CD18-dependent PMN recruitment in the contralateral lung [29]. The way of application also influences the CD18 dependency of PMN migration. Intratracheal instillation of LPS leads to a CD18 dependent recruitment into the alveolar airspace [30]. However, the same endotoxin given intraperitoneally [20] results in a neutrophil sequestration that is independent of CD18 and attenuates PMN recruitment in response to intratracheal LPS [31].
Most stimuli inducing a CD18-dependent PMN migration upregulate ICAM-1 – a major ligand for CD18 – on endothelial cells. Endothelial ICAM-1 expression was increased following E. coli LPS, but not Streptococcus pneumoniae challenge [32]. The fact that IL-1 and TNF-α, both of which are nuclear factor-κB-dependent proinflammatory cytokines that regulate expression of ICAM-1, are required for CD18/ICAM-1-dependent pathways supports this hypothesis. In addition, members of the β1 integrin family (very late antigen-4 and -5) may mediate CD18-independent PMN migration [22].
The CD18 integrin Mac-1 (CD11b/CD18) appears to be very important in PMN recruitment in the lung because antibodies to Mac-1, but not to lymphocyte function-associated antigen-1 (LFA-1, CD11a/CD18), inhibited neutrophil migration significantly in an inhaled LPS model [33]. Neutrophil inhibiting factor, a hookworm-derived protein that binds to and blocks CD11b, also prevents PMN recruitment into the lung [34].
PECAM-1, a member of the immunoglobulin superfamily, localizes close to interendothelial clefts and has been suggested to regulate PMN migration in the pulmonary vasculature. In an early study [35], antibodies to PECAM-1 attenuated neutrophil emigration into the lung in response to IgG immune complex deposition. However, PECAM-1 expression does not change in response to inflammatory changes [36, 37], and in a more recent study [38] blocking PECAM-1 did not prevent E. coli or S. pneumoniae induced lung injury in rats or mice.
Selectins
Although selectins are essential for initiating the rolling process in the systemic vasculature, their role for PMN transmigration in the pulmonary microcirculation is less clear and depends on the inflammatory stimlus. LPS-induced migration into the alveolar airspace was not inhibited by blocking all three selectins [39]. Neutrophil emigration is unaffected by Streptococcus pneumoniae in mice lacking E-selectin and P-selectin with L-selectin function blocked [40]. In contrast, all selectins have been shown to participate in the development of lung injury induced by complement or intratracheal deposition of IgG complexes [41] or bacterial LPS [42], suggesting that the involvement of selectins may be stimulus-dependent. L-selectin function is necessary for sustained intracapillary accumulation of neutrophils, but not for emigration of neutrophils [43].
The impact of adhesion molecule mediated leukocyte–endothelium interaction in patients with ALI is yet to be elucidated. In lung tissue samples from patients who had died from ARDS, a strong upregulation of ICAM-1, VCAM-1 and E-selectin was found, suggesting that these adhesion molecules play a role in human lung injury [36]. In an ex vivo study [44], human PMNs were found to express higher levels of CD18 after incubation with BAL fluid from ARDS patients who received a conventional as opposed to a lung protective ventilation strategy. It was suggest that this, in addition to lower mechanical stress, could explain the beneficial effect of mechanical ventilation using low tidal volumes [45].
Chemokines
Chemokines are a group of approximately 40 small chemoattractant proteins (70–125 amino acids; 6–14 kDa) that bind to G-protein coupled receptors [46]. In humans, CXC chemokine ligand (CXCL)1–CXCL3 and CXCL5–CXCL8 bind to CXC chemokine receptor (CXCR)1 and CXCR2, and are potent chemotactic factors for neutrophils. In mice, CXCL1–3, CXCL5 and CXCL6 bind to CXCR2. CXCL1 and CXCL2 have been shown to induce rapid integrin activation, causing arrest from rolling and chemotaxis [47]. Chemotaxis to chemokines or other soluble chemoattractants such as C5a, platelet-activating factor, leukotriene B4, or fMLP might be the most important trigger for PMN recruitment into the lung.
Chemokines are produced by activated macrophages, monocytes, neutrophils, endothelium, epithelium, platelets, and various parenchymal cells [48]. After having been secreted, some chemokines are immobilized by specific glycosaminoglycans (GAGs) on target cells. GAG-bound chemokines may be able to activate neutrophils, or they must first dissociate from GAGs to interact with their receptors [49]. Neutrophils stimulated by chemokines generate a (chemokine receptor-rich) pseudopod at the leading edge and a tail-like uropod, allowing for a directional movement toward the chemokine gradient [50]. Actin polymerization and depolymerization required for this cell remodeling are regulated by Rho, Rac, and Cdc42 proteins, which are members of the Rho family of small G proteins [51]. Chemokines are able to activate these small G proteins and thereby induce locomotion (Rac), unidirectional movement (Cdc42), and uropod retraction (Rho) [49]. Lack of Rac2, the predominant Rac isoform in human neutrophils, results in a severe immunodeficiency with impaired neutrophil polarization and chemotaxis [52].
The best studied CXC chemokine in humans is CXCL8 (IL-8), which has significant relevance to lung injury. High concentrations of IL-8 in BAL fluid from ARDS patients are associated with increased neutrophil influx into the airspace, and in vitro chemotactic activity of BAL fluid can be attenuated by removing IL-8 [53]. Intratracheal instillation of IL-8 leads to a PMN influx in models of ALI, and blocking IL-8 has been shown to ameliorate lung damage in models of acid aspiration [54], pancreatitis [55], and reperfusion injury [56]. Recent studies suggest that IL-8 in BAL fluid from ARDS patients is bound to an anti-IL-8 autoantibody. This immune complex exhibits chemotactic and proinflammatory activity [57] and its concentration might be an important prognostic factor for the development and outcome of ARDS [58, 59].
In rodents, the two most important chemokines for PMN recruitment into the lung are keratinocyte-derived chemokine (KC) and macrophage inflammatory protein (MIP)-2 [46]. Both bind to and activate CXCR2, but differ in their biological potency and affect PMN migration into the lung in different ways [60]. Only KC is selectively transported from the lung to the blood, whereas MIP-2 is retained in the lung compartment [61]. Circulating KC may be able to 'prime' circulating PMNs to migrate into the lung in response to MIP-2. After an intraperitoneal LPS challenge in mice, mRNA for both KC and MIP-2 is increased in lung tissue [62]. MIP-2 and CINC (the rat orthologue of KC) are upregulated in a hindlimb ischemia/reperfusion-induced lung injury model [63]. Neutralization of either chemokine significantly decreases neutrophil recruitment into the lung [64]. Similarly, absence or blockade of CXCR2 attenuates neutrophil influx into the lung [65]. In a murine model of ventilator-induced lung injury, mechanical ventilation with high peak pressures resulted in an increase in both KC and MIP-2, and their level correlated with lung injury and neutrophil sequestration [66].
These chemokines may be released by alveolar macrophages, alveolar type II cells, and endothelial cells [20, 67, 68]. After an intratracheal LPS challenge, both KC and MIP-2 are found in the alveolar airspace. KC is synthesized, secreted, and deposited on syndecan-1 (a cell-bound proteoglycan) molecules. Matrilysin, a matrix metalloproteinase, cleaves this KC–syndecan-1 complex and thereby may create a chemotactic gradient. Matrilysin-deficient mice lack the ability to create such a chemotactic gradient, and transepithelial efflux of neutrophils is attenuated [10]. In the setting of ALI, the p38 mitogen-activated protein kinase pathway appears to play a major role. P38 has been shown to stimulate the nuclear factor-κB mediated production of various cytokines, such as IL-1β and TNF-α, and to affect chemotaxis, adhesion and oxygen release, particularly in neutrophils [2]. P38 is activated in neutrophils after endotoxin exposure, and inhibition of p38 attenuates intratracheal LPS-induced neutrophil migration into the airspace without affecting the PMN accumulation in the lung and, even more interesting, without affecting the alveolar expression of chemokines KC and MIP-2 [69].
Although KC and MIP-2 are the most studied chemokines in rodent models of ALI, other CXC chemokines might be involved. Epithelial neutrophil-activating peptide (ENA)-78 (CXCL5) has been measured in the airspace of ARDS patients and correlated with neutrophil counts in BAL fluid [70], although the chemotactic potency appeared to be less when compared with IL-8 [71]. Increased ENA-78 expression was also found in a model of lung injury in rats induced by hepatic ischemia/reperfusion [72]. In contrast, LPS-induced CXC chemokine (LIX; the murine orthologue of ENA-78) expression in the lung was not affected in a model of abdominal sepsis [73].
Lungkine (CXCL15) is exclusively expressed in lung epithelial cells and is upregulated in various lung inflammation models. In a Klebsiella pneumoniae infection model, lungkine-deficient mice exhibited reduced PMN migration into the alveolar space, whereas recruitment from the blood into the lung parenchyma appeared to be unaffected, suggesting a role for this chemokine in migration through the alveolar epithelium [74].
Neutrophil recruitment: crosstalk between inflammation and coagulation
More than 80 years ago, the deposition of fibrin strands in the inflamed lung tissue was suggested to be responsible for the emigration of neutrophils into the alveolar airspace [75]. Using modern methods, these observations have been confirmed [22]. There is clear evidence that fibrin deposition and microvascular thrombosis are early events in the development of ALI/ARDS, although the mechanisms remain unclear. Several attempts were made to regulate the crosstalk between coagulation and inflammation in clinical trials. Although not focused on the treatment of ARDS, human recombinant protein C has been shown to increase survival in patients with severe sepsis [76].
Because of its central role in triggering blood coagulation with extensive consequences for both fibrin formation and inflammatory response, the modulation of tissue factor (TF)-dependent pathways has gained interest in animal and human studies. TF is the cellular transmembrane receptor for factor VIIa. It is expressed by circulating monocytes and, at least under inflammatory conditions, by endothelial cells. In the lung, TF is expressed by alveolar macrophages and alveolar epithelial cells [77]. During inflammation, TF expression and activity are increased in lung, brain, and kidney [78]. Beyond its importance for hemostasis and thrombosis, TF has direct and indirect effects on the inflammatory system, mainly via production of thrombin, which activates proteinase activated receptor-1 to -4 and cleaves fibrinogen [79]. Activated by the binding of factor VIIa, TF induces the expression of several proinflammatory genes (e.g. IL-1β, IL-6, IL-8) [80] that may be involved in the development of ARDS [81]. TF levels in patients with ARDS are correlated with lung injury score, suggesting that blocking TF-dependent inflammation might be promising for the development of new therapeutic strategies.
TF pathway inhibitor (TFPI)-1 is an endogenous inhibitor of the TF–factor VIIa complex. However, its physiological role in controlling excessive TF-induced coagulation and inflammation in the clinical setting of sepsis and lung injury might be limited [82]. Although treatment with recombinant TFPI-1 in animal models of lung injury was promising, it failed to improve survival of patients with severe sepsis in a phase III clinical trial [83]. TFPI-1 does not affect the internalization of factor VIIa, which is pivotal for further TF-dependent signaling [84], and requires factor Xa for the formation of the inhibitory quaternary complex [85]. Therefore, TFPI-1 strategies might be less effective than blocking TF and/or factor VIIa directly. Blocking TF might interrupt a self-amplifying loop in which further signaling promotes inflammation [86]. Experimental findings showed that inhibition of the TF–factor VIIa complex increased survival in endotoxin-induced sepsis and ALI [87, 88], even in a state of established sepsis [89].
Conclusion
Patients with ALI still have a poor prognosis in terms of survival and long-term morbidity. The decrease in ARDS-related mortality is mainly based on improvement in supportive therapies such as protective ventilatory strategies [45]. Other supportive approaches such as restrictive fluid management are currently being investigated. Despite great effort, our understanding of the molecular and cellular mechanisms of ALI and ARDS were recently found to be 'embryonic at best' [90].
The excessive activation and migration of circulating neutrophils from blood to the alveolar airspace is one of the key events in the early development of ALI. Blocking Mac-1, ICAM-1, or combinations of adhesion molecules has been shown to protect against lung injury in many experimental studies. In animal models, reliable methodologic approaches to assess PMN recruitment in the lung can elucidate the different mechanisms of PMN trafficking in the intravascular, interstitial, and alveolar compartments. However, the clinical relevance of these animal models remains unclear, and translation into clinical studies is difficult. These limitations include the absence of comorbidity, mechanical ventilation, fluid management, antibiotic treatment, nutrition, and other factors that may have an impact on outcome in humans. In addition, classical ARDS criteria are usually not tested in animal models. Therefore, clinical studies are required to obtain definitive answers [90].
Finally, neutrophil recruitment into the lung is essential for host defense against bacterial infections [91, 92]. The dual role of neutrophils in the lung – defending against infection and mediating lung injury – is not well understood, but must be considered in evaluation of therapeutic approaches.
Abbreviations
- ALI:
-
acute lung injury
- ARDS:
-
acute respiratory distress syndrome
- BAL:
-
bronchoalveolar lavage
- CXCL:
-
CXC chemokine ligand
- CXCR:
-
CXC chemokine receptor
- ENA:
-
epithelial neutrophil-activating peptide
- fMLP:
-
N-formylmethionyl-leucyl-phenylalanine
- GAG:
-
glycosaminoglycan
- ICAM:
-
intercellular adhesion molecule
- IL:
-
interleukin
- KC:
-
keratinocyte-derived chemokine
- LPS:
-
lipopolysaccharide
- MIP:
-
macrophage inflammatory protein
- PECAM:
-
platelet endothelial cell adhesion molecule
- PMN:
-
polymorphonuclear leukocyte
- TF:
-
tissue factor
- TFPI:
-
tissue factor pathway inhibitor
- TNF:
-
tumor necrosis factor
- VCAM:
-
vascular cell adhesion molecule.
References
Ware LB, Matthay MA: The acute respiratory distress syndrome. N Engl J Med. 2000, 342: 1334-1349. 10.1056/NEJM200005043421806.
Abraham E: Neutrophils and acute lung injury. Crit Care Med. 2003, Suppl: S195-S199. 10.1097/01.CCM.0000057843.47705.E8.
Abraham E, Carmody A, Shenkar R, Arcaroli J: Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2000, 279: L1137-L1145.
Azoulay E, Darmon M, Delclaux C, Fieux F, Bornstain C, Moreau D, Attalah H, Le Gall JR, Schlemmer B: Deterioration of previous acute lung injury during neutropenia recovery. Crit Care Med. 2002, 30: 781-786. 10.1097/00003246-200204000-00010.
Doerschuk CM, Beyers N, Coxson HO, Wiggs B, Hogg JC: Comparison of neutrophil and capillary diameters and their relation to neutrophil sequestration in the lung. J Appl Physiol. 1993, 74: 3040-3045.
Doerschuk CM, Allard MF, Martin BA, MacKenzie A, Autor AP, Hogg JC: Marginated pool of neutrophils in rabbit lungs. J Appl Physiol. 1987, 63: 1806-1815.
Butcher EC: Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991, 67: 1033-1036. 10.1016/0092-8674(91)90279-8.
Springer TA: Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994, 76: 301-314. 10.1016/0092-8674(94)90337-9.
Ley K: Integration of inflammatory signals by rolling neutrophils. Immunol Rev. 2002, 186: 8-18. 10.1034/j.1600-065X.2002.18602.x.
Li Q, Park PW, Wilson CL, Parks WC: Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002, 111: 635-646. 10.1016/S0092-8674(02)01079-6.
Wagner JG, Harkema JR, Roth RA: Pulmonary leukostasis and the inhibition of airway neutrophil recruitment are early events in the endotoxemic rat. Shock. 2002, 17: 151-158. 10.1097/00024382-200202000-00012.
Drost EM, MacNee W: Potential role of IL-8, platelet-activating factor and TNF-alpha in the sequestration of neutrophils in the lung: effects on neutrophil deformability, adhesion receptor expression, and chemotaxis. Eur J Immunol. 2002, 32: 393-403. 10.1002/1521-4141(200202)32:2<393::AID-IMMU393>3.0.CO;2-5.
Tanaka H, Nishino M, Dahms TE: Physiologic responses to small emboli and hemodynamic effects of changes in deformability of polymorphonuclear leukocytes in isolated rabbit lung. Microvasc Res. 2002, 63: 81-90. 10.1006/mvre.2001.2368.
Doerschuk CM: The role of CD18-mediated adhesion in neutrophil sequestration induced by infusion of activated plasma in rabbits. Am J Respir Cell Mol Biol. 1992, 7: 140-148.
Worthen GS, Schwab B, Elson EL, Downey GP: Mechanics of stimulated neutrophils: cell stiffening induces retention in capillaries. Science. 1989, 245: 183-186.
Skoutelis AT, Kaleridis V, Athanassiou GM, Kokkinis KI, Missirlis YF, Bassaris HP: Neutrophil deformability in patients with sepsis, septic shock, and adult respiratory distress syndrome. Crit Care Med. 2000, 28: 2355-2359.
Doyle NA, Bhagwan SD, Meek BB, Kutkoski GJ, Steeber DA, Tedder TF, Doerschuk CM: Neutrophil margination, sequestration, and emigration in the lungs of L-selectin-deficient mice. J Clin Invest. 1997, 99: 526-533.
Olson TS, Singbartl K, Ley K: L-selectin is required for fMLP-but not C5a-induced margination of neutrophils in pulmonary circulation. Am J Physiol Regul Integr Comp Physiol. 2002, 282: R1245-R1252.
Carraway MS, Welty-Wolf KE, Kantrow SP, Huang YC, Simonson SG, Que LG, Kishimoto TK, Piantadosi CA: Antibody to E- and L-selectin does not prevent lung injury or mortality in septic baboons. Am J Respir Crit Care Med. 1998, 157: 938-949.
Andonegui G, Bonder CS, Green F, Mullaly SC, Zbytnuik L, Raharjo E, Kubes P: Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest. 2003, 111: 1011-1020. 10.1172/JCI200316510.
Burns JA, Issekutz TB, Yagita H, Issekutz AC: The beta2, alpha4, alpha5 integrins and selectins mediate chemotactic factor and endotoxin-enhanced neutrophil sequestration in the lung. Am J Pathol. 2001, 158: 1809-1819.
Burns AR, Smith CW, Walker DC: Unique structural features that influence neutrophil emigration into the lung. Physiol Rev. 2003, 83: 309-336.
Furuse M, Itoh M, Hirase T, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S: Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol. 1994, 127: 1617-1626. 10.1083/jcb.127.6.1617.
Su WH, Chen HI, Jen CJ: Differential movements of VE-cadherin and PECAM-1 during transmigration of polymorphonuclear leukocytes through human umbilical vein endothelium. Blood. 2002, 100: 3597-3603. 10.1182/blood-2002-01-0303.
Mackarel AJ, Cottell DC, Russell KJ, FitzGerald MX, O'Connor CM: Migration of neutrophils across human pulmonary endothelial cells is not blocked by matrix metalloproteinase or serine protease inhibitors. Am J Respir Cell Mol Biol. 1999, 20: 1209-1219.
Betsuyaku T, Shipley JM, Liu Z, Senior RM: Neutrophil emigration in the lungs, peritoneum, and skin does not require gelatinase B. Am J Respir Cell Mol Biol. 1999, 20: 1303-1309.
Patel KD, Cuvelier SL, Wiehler S: Selectins: critical mediators of leukocyte recruitment. Semin Immunol. 2002, 14: 73-81. 10.1006/smim.2001.0344.
Diamond MS, Springer TA: The dynamic regulation of integrin adhesiveness. Curr Biol. 1994, 4: 506-517.
Motosugi H, Quinlan WM, Bree M, Doerschuk CM: Role of CD11b in focal acid-induced pneumonia and contralateral lung injury in rats. Am J Respir Crit Care Med. 1998, 157: 192-198.
Mizgerd JP, Horwitz BH, Quillen HC, Scott ML, Doerschuk CM: Effects of CD18 deficiency on the emigration of murine neutrophils during pneumonia. J Immunol. 1999, 163: 995-999.
Wagner JG, Driscoll KE, Roth RA: Inhibition of pulmonary neutrophil trafficking during endotoxemia is dependent on the stimulus for migration. Am J Respir Cell Mol Biol. 1999, 20: 769-776.
Burns AR, Takei F, Doerschuk CM: Quantitation of ICAM-1 expression in mouse lung during pneumonia. J Immunol. 1994, 153: 3189-3198.
Moreland JG, Fuhrman RM, Pruessner JA, Schwartz DA: CD11b and intercellular adhesion molecule-1 are involved in pulmonary neutrophil recruitment in lipopolysaccharide-induced airway disease. Am J Respir Cell Mol Biol. 2002, 27: 474-480.
Ohno S, Malik AB: Polymorphonuclear leucocyte (PMN) inhibitory factor prevents PMN-dependent endothelial cell injury by an anti-adhesive mechanism. J Cell Physiol. 1997, 171: 212-216. 10.1002/(SICI)1097-4652(199705)171:2<212::AID-JCP12>3.3.CO;2-0.
Vaporciyan AA, DeLisser HM, Yan HC, Mendiguren II, Thom SR, Jones ML, Ward PA, Albelda SM: Involvement of platelet-endothelial cell adhesion molecule-1 in neutrophil recruitment in vivo. Science . 1993, 262: 1580-1582.
Muller AM, Cronen C, Muller KM, Kirkpatrick CJ: Heterogeneous expression of cell adhesion molecules by endothelial cells in ARDS. J Pathol. 2002, 198: 270-275. 10.1002/path.1186.
Eppihimer MJ, Russell J, Langley R, Vallien G, Anderson DC, Granger DN: Differential expression of platelet-endothelial cell adhesion molecule-1 (PECAM-1) in murine tissues. Microcirculation. 1998, 5: 179-188. 10.1038/sj.mn.7300012.
Tasaka S, Qin L, Saijo A, Albelda SM, DeLisser HM, Doerschuk CM: Platelet endothelial cell adhesion molecule-1 in neutrophil emigration during acute bacterial pneumonia in mice and rats. Am J Respir Crit Care Med. 2003, 167: 164-170. 10.1164/rccm.2202011.
Burns JA, Issekutz TB, Yagita H, Issekutz AC: The alpha 4 beta 1 (very late antigen (VLA)-4, CD49d/CD29) and alpha 5 beta 1 (VLA-5, CD49e/CD29) integrins mediate beta 2 (CD11/CD18) integrin-independent neutrophil recruitment to endotoxin-induced lung inflammation. J Immunol. 2001, 166: 4644-4649.
Mizgerd JP, Meek BB, Kutkoski GJ, Bullard DC, Beaudet AL, Doerschuk CM: Selectins and neutrophil traffic: margination and Streptococcus pneumoniae-induced emigration in murine lungs. J Exp Med. 1996, 184: 639-645.
Mulligan MS, Miyasaka M, Ward PA: Protective effects of combined adhesion molecule blockade in models of acute lung injury. Proc Assoc Am Physicians. 1996, 108: 198-208.
Hayashi H, Koike H, Kurata Y, Imanishi N, Tojo SJ: Protective effects of sialyl Lewis X and anti-P-selectin antibody against lipopolysaccharide-induced acute lung injury in rabbits. Eur J Pharmacol. 1999, 370: 47-56. 10.1016/S0014-2999(99)00068-0.
Kubo H, Doyle NA, Graham L, Bhagwan SD, Quinlan WM, Doerschuk CM: L- and P-selectin and CD11/CD18 in intracapillary neutrophil sequestration in rabbit lungs. Am J Respir Crit Care Med. 1999, 159: 267-274.
Zhang H, Downey GP, Suter PM, Slutsky AS, Ranieri VM: Conventional mechanical ventilation is associated with bronchoalveolar lavage-induced activation of polymorphonuclear leukocytes: a possible mechanism to explain the systemic consequences of ventilator-induced lung injury in patients with ARDS. Anesthesiology. 2002, 97: 1426-1433.
The Acute Respiratory Distress Syndrome Network: Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000, 342: 1301-1308. 10.1056/NEJM200005043421801.
Olson TS, Ley K: Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002, 283: R7-R28.
Ley K: Arrest chemokines. Microcirculation. 2003, 10: 289-295. 10.1038/sj.mn.7800194.
Rollins BJ: Chemokines. Blood . 1997, 90: 909-928.
Rot A, Von Andrian UH: Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004, 22: 891-928. 10.1146/annurev.immunol.22.012703.104543.
Nieto M, Frade JM, Sancho D, Mellado M, Martinez A, Sanchez-Madrid F: Polarization of chemokine receptors to the leading edge during lymphocyte chemotaxis. J Exp Med. 1997, 186: 153-158. 10.1084/jem.186.1.153.
Takai Y, Sasaki T, Matozaki T: Small GTP-binding proteins. Physiol Rev. 2001, 81: 153-208.
Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, Gonzalez-Aller C, Hiester A, deBoer M, Harbeck RJ, Oyer R, Johnson GL, Roos D: Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci USA. 2000, 97: 4654-4659. 10.1073/pnas.080074897.
Miller EJ, Cohen AB, Nagao S, Griffith D, Maunder RJ, Martin TR, Weiner-Kronish JP, Sticherling M, Christophers E, Matthay MA: Elevated levels of NAP-1/interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am Rev Respir Dis. 1992, 146: 427-432.
Folkesson HG, Matthay MA, Hebert CA, Broaddus VC: Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest. 1995, 96: 107-116.
Osman MO, Kristensen JU, Jacobsen NO, Lausten SB, Deleuran B, Deleuran M, Gesser B, Matsushima K, Larsen CG, Jensen SL: A monoclonal anti-interleukin 8 antibody (WS-4) inhibits cytokine response and acute lung injury in experimental severe acute necrotising pancreatitis in rabbits. Gut. 1998, 43: 232-239.
Sekido N, Mukaida N, Harada A, Nakanishi I, Watanabe Y, Matsushima K: Prevention of lung reperfusion injury in rabbits by a monoclonal antibody against interleukin-8. Nature. 1993, 365: 654-657. 10.1038/365654a0.
Krupa A, Kato H, Matthay MA, Kurdowska AK: Proinflammatory activity of anti-IL-8 autoantibody:IL-8 complexes in alveolar edema fluid from patients with Acute Lung Injury. Am J Physiol Lung Cell Mol Physiol. 2004, 286: L1105-L1113. 10.1152/ajplung.00277.2003.
Kurdowska A, Noble JM, Steinberg KP, Ruzinski JT, Hudson LD, Martin TR: Anti-interleukin 8 autoantibody: interleukin 8 complexes in the acute respiratory distress syndrome. Relationship between the complexes and clinical disease activity. Am J Respir Crit Care Med. 2001, 163: 463-468.
Kurdowska A, Noble JM, Grant IS, Robertson CR, Haslett C, Donnelly SC: Anti-interleukin-8 autoantibodies in patients at risk for acute respiratory distress syndrome. Crit Care Med. 2002, 30: 2335-2337. 10.1097/00003246-200210000-00024.
Lomas JL, Chung CS, Grutkoski PS, LeBlanc BW, Lavigne L, Reichner J, Gregory SH, Doughty LA, Cioffi WG, Ayala A: Differential effects of macrophage inflammatory chemokine-2 and keratinocyte-derived chemokine on hemorrhage-induced neutrophil priming for lung inflammation: assessment by adoptive cells transfer in mice. Shock. 2003, 19: 358-365. 10.1097/00024382-200304000-00011.
Quinton LJ, Nelson S, Zhang P, Boe DM, Happel KI, Pan W, Bagby GJ: Selective transport of cytokine-induced neutrophil chemoattractant from the lung to the blood facilitates pulmonary neutrophil recruitment. Am J Physiol Lung Cell Mol Physiol. 2004, 286: L465-L472. 10.1152/ajplung.00153.2003.
Aoki K, Ishida Y, Kikuta N, Kawai H, Kuroiwa M, Sato H: Role of CXC chemokines in the enhancement of LPS-induced neutrophil accumulation in the lung of mice by dexamethasone. Biochem Biophys Res Commun . 2002, 294: 1101-1108. 10.1016/S0006-291X(02)00573-9.
Bless NM, Warner RL, Padgaonkar VA, Lentsch AB, Czermak BJ, Schmal H, Friedl HP, Ward PA: Roles for C-X-C chemokines and C5a in lung injury after hindlimb ischemia-reperfusion. Am J Physiol. 1999, 276: L57-L63.
Greenberger MJ, Strieter RM, Kunkel SL, Danforth JM, Laichalk LL, McGillicuddy DC, Standiford TJ: Neutralization of macrophage inflammatory protein-2 attenuates neutrophil recruitment and bacterial clearance in murine Klebsiella pneumonia. J Infect Dis. 1996, 173: 159-165.
Goncalves AS, Appelberg R: The involvement of the chemokine receptor CXCR2 in neutrophil recruitment in LPS-induced inflammation and in Mycobacterium avium infection. Scand J Immunol. 2002, 55: 585-591. 10.1046/j.1365-3083.2002.01097.x.
Belperio JA, Keane MP, Burdick MD, Londhe V, Xue YY, Li K, Phillips RJ, Strieter RM: Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J Clin Invest . 2002, 110: 1703-1716. 10.1172/JCI200215849.
Johnson MC, Kajikawa O, Goodman RB, Wong VA, Mongovin SM, Wong WB, Fox-Dewhurst R, Martin TR: Molecular expression of the alpha-chemokine rabbit GRO in Escherichia coli and characterization of its production by lung cells in vitro and in vivo. J Biol Chem. 1996, 271: 10853-10858. 10.1074/jbc.271.18.10853.
Vanderbilt JN, Mager EM, Allen L, Sawa T, Wiener-Kronish J, Gonzalez R, Dobbs LG: CXC chemokines and their receptors are expressed in type II cells and upregulated following lung injury. Am J Respir Cell Mol Biol. 2003, 29: 661-668. 10.1165/rcmb.2002-0227OC.
Nick JA, Young SK, Brown KK, Avdi NJ, Arndt PG, Suratt BT, Janes MS, Henson PM, Worthen GS: Role of p38 mitogen-activated protein kinase in a murine model of pulmonary inflammation. J Immunol. 2000, 164: 2151-2159.
Goodman RB, Strieter RM, Martin DP, Steinberg KP, Milberg JA, Maunder RJ, Kunkel SL, Walz A, Hudson LD, Martin TR: Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1996, 154: 602-611.
Wiedermann FJ, Mayr AJ, Kaneider NC, Fuchs D, Mutz NJ, Schobersberger W: Alveolar granulocyte colony-stimulating factor and alpha-chemokines in relation to serum levels, pulmonary neutrophilia, and severity of lung injury in ARDS. Chest. 2004, 125: 212-219. 10.1378/chest.125.1.212.
Colletti LM, Green M: Lung and liver injury following hepatic ischemia/reperfusion in the rat is increased by exogenous lipopolysaccharide which also increases hepatic TNF production in vivo and in vitro. Shock. 2001, 16: 312-319.
Neumann B, Zantl N, Veihelmann A, Emmanuilidis K, Pfeffer K, Heidecke CD, Holzmann B: Mechanisms of acute inflammatory lung injury induced by abdominal sepsis. Int Immunol. 1999, 11: 217-227. 10.1093/intimm/11.2.217.
Chen SC, Mehrad B, Deng JC, Vassileva G, Manfra DJ, Cook DN, Wiekowski MT, Zlotnik A, Standiford TJ, Lira SA: Impaired pulmonary host defense in mice lacking expression of the CXC chemokine lungkine. J Immunol. 2001, 166: 3362-3368.
Miller WS: A study of the factors underlying the formation of alveolar pores in pneumonia. J Exp Med. 1923, 38: 707-711.
Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ, Recombinant human protein C Worldwide Evaluation in Severe Sepsis (PROWESS) study group: Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001, 344: 699-709. 10.1056/NEJM200103083441001.
Welty-Wolf KE, Carraway MS, Ortel TL, Piantadosi CA: Coagulation and inflammation in acute lung injury. Thromb Haemost. 2002, 88: 17-25.
Erlich J, Fearns C, Mathison J, Ulevitch RJ, Mackman N: Lipopolysaccharide induction of tissue factor expression in rabbits. Infect Immun. 1999, 67: 2540-2546.
Coughlin SR: Thrombin signalling and protease-activated receptors. Nature. 2000, 407: 258-264. 10.1038/35025229.
Taylor FB, Chang AC, Peer G, Li A, Ezban M, Hedner U: Active site inhibited factor VIIa (DEGR VIIa) attenuates the coagulant and interleukin-6 and -8, but not tumor necrosis factor, responses of the baboon to LD100 Escherichia coli. Blood. 1998, 91: 1609-1615.
Meduri GU, Kohler G, Headley S, Tolley E, Stentz F, Postlethwaite A: Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest. 1995, 108: 1303-1314.
Gando S, Kameue T, Matsuda N, Hayakawa M, Morimoto Y, Ishitani T, Kemmotsu O: Imbalances between the levels of tissue factor and tissue factor pathway inhibitor in ARDS patients. Thromb Res. 2003, 109: 119-124. 10.1016/S0049-3848(03)00151-8.
Abraham E, Reinhart K, Opal S, Demeyer I, Doig C, Rodriguez AL, Beale R, Svoboda P, Laterre PF, Simon S, Light B, Spapen H, Stone J, Seibert A, Peckelsen C, De Deyne C, Postier R, Pettila V, Artigas A, Percell SR, Shu V, Zwingelstein C, Tobias J, Poole L, Stolzenbach JC, Creasey AA, OPTIMIST Trial Study Group: Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003, 290: 238-247. 10.1001/jama.290.2.238.
Hansen CB, Pyke C, Petersen LC, Rao LV: Tissue factor-mediated endocytosis, recycling, and degradation of factor VIIa by a clathrin-independent mechanism not requiring the cytoplasmic domain of tissue factor. Blood. 2001, 97: 1712-1720. 10.1182/blood.V97.6.1712.
Doshi SN, Marmur JD: Evolving role of tissue factor and its pathway inhibitor. Crit Care Med. 2002, Suppl: S241-S250. 10.1097/00003246-200205001-00012.
Ruf W, Riewald M: Tissue factor-dependent coagulation protease signaling in acute lung injury. Crit Care Med. 2003, Suppl: S231-S237. 10.1097/01.CCM.0000057848.27456.04.
Welty-Wolf KE, Carraway MS, Miller DL, Ortel TL, Ezban M, Ghio AJ, Idell S, Piantadosi CA: Coagulation blockade prevents sepsis-induced respiratory and renal failure in baboons. Am J Respir Crit Care Med . 2001, 164: 1988-1996.
Miller DL, Welty-Wolf K, Carraway MS, Ezban M, Ghio A, Suliman H, Piantadosi CA: Extrinsic coagulation blockade attenuates lung injury and proinflammatory cytokine release after intratracheal lipopolysaccharide. Am J Respir Cell Mol Biol. 2002, 26: 650-658.
Carraway MS, Welty-Wolf KE, Miller DL, Ortel TL, Idell S, Ghio AJ, Petersen LC, Piantadosi CA: Blockade of tissue factor: treatment for organ injury in established sepsis. Am J Respir Crit Care Med . 2003, 167: 1200-1209. 10.1164/rccm.200204-287OC.
Matthay MA, Zimmerman GA, Esmon C, Bhattacharya J, Coller B, Doerschuk CM, Floros J, Gimbrone MA, Hoffman E, Hubmayr RD, Leppert M, Matalon S, Munford R, Parsons P, Slutsky AS, Tracey KJ, Ward P, Gail DB, Harabin AL: Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med. 2003, 167: 1027-1035. 10.1164/rccm.200208-966WS.
Tateda K, Moore TA, Newstead MW, Tsai WC, Zeng X, Deng JC, Chen G, Reddy R, Yamaguchi K, Standiford TJ: Chemokine-dependent neutrophil recruitment in a murine model of Legionella pneumonia: potential role of neutrophils as immunoregulatory cells. Infect Immun. 2001, 69: 2017-2024. 10.1128/IAI.69.4.2017-2024.2001.
Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK: Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001, 194: 519-527. 10.1084/jem.194.4.519.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Reutershan, J., Ley, K. Bench-to-bedside review: Acute respiratory distress syndrome – how neutrophils migrate into the lung. Crit Care 8, 453 (2004). https://doi.org/10.1186/cc2881
Published:
DOI: https://doi.org/10.1186/cc2881